

Abstract
Iterative fungal polyketide synthases (PKSs) use a unique set of biochemical rules in the synthesis of complex polyketides. These rules dictate polyketide starter unit selection, chain length control, and post-PKS processing. We have demonstrated the E. coli expression and reconstitution of an iterative, unreduced fungal PKS. The Gibberella fujikuroi PKS4 was expressed at high levels, purified to homogeneity and functionally characterized. In the presence of malonyl-CoA, PKS4 was able to synthesize the nonaketide 3,8,10,11-tetrahydroxy-1-methyl-12H-benzo[b]xanthen-12-one (2) as the predominant product. PKS4 selectively used octanoyl-CoA as the starter unit and synthesized two novel benzopyrone-containing polyketides. Our work sets the stage for a comprehensive characterization of the intact PKS and its domains, and offers significant opportunity towards the enzymatic synthesis of additional compounds.
Iterative fungal polyketide synthases (PKSs) use a unique set of biochemical rules in the synthesis of complex polyketides.1, 2 These rules dictate polyketide starter unit selection,3 chain length control,4 and post-PKS processing.5 In contrast to the bacterial PKSs, the biosynthetic mechanisms of fungal PKSs are not well understood and their potential for combinatorial biosynthesis has not yet been realized.6 This is largely due to difficulties associated with manipulating these megasynthases in their native or related fungal hosts, and with obtaining intact enzymes for biochemical analysis.7 In this work, we demonstrate the enzymatic synthesis of novel polyketides using a fungal PKS that was expressed from Escherichia coli and reconstituted in vitro. These results set the stage for detailed characterization and protein engineering of the megasynthase, and offer opportunities towards the enzymatic synthesis of new compounds.
PKS4 from Gibberella fujikuroi is an unreduced nonaketide synthase8 involved in the biosynthesis of the anticancer fungal polyketide bikaverin9 (1, Figure 1). The domain architecture (Figure 1A) of the megasynthase is typical for PKSs in this family, which includes the linear arrangement of starter unit acyltransferase (SAT),3 ketosynthase (KS), malonyl-CoA:acyl carrier protein (ACP) transacylase (MAT), product template (PT),10 ACP and thioesterase/Claisen cyclase (TE/CLC) domains.5 We used splice by overlap extension PCR to amplify the uninterrupted pks4 gene from the gDNA of G. fujikuroi. The gene (6.1 kB) was inserted into the expression vector pSMa76 and protein expression was performed using E. coli strain BL21(DE3) at 16°C. Soluble PKS4 (219 kDa) was purified using nickel-NTA affinity chromatography to near homogeneity (Figure S1, Supporting Information) with a final yield of ∼ 8 mg/L. The purified enzyme can be strongly labelled with [2-14C]malonyl-CoA, indicating that the MAT domain is active. The ACP domain was verified to be in the holo form (Figure S2), likely phosphopantethienylated by the E. coli holo-ACP synthase.
Figure 1.
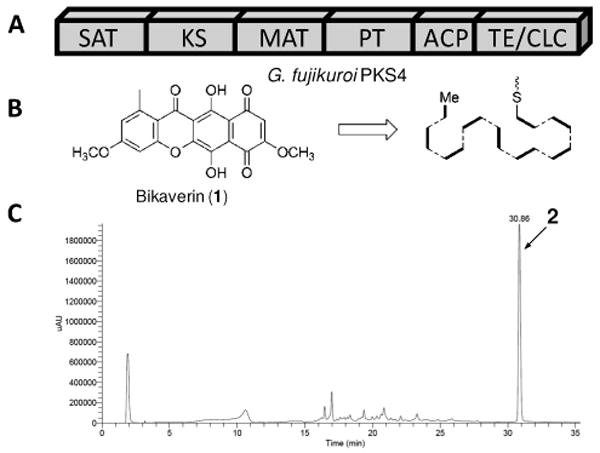
(A) PKS4 megasynthase and its domains; (B) Bikaverin (1) and its polyketide backbone folding pattern as determined from acetate feeding;13 (C) Polyketide products synthesized by PKS4 (10 μM) using malonyl-CoA (2 mM). The LC chromatogram (280 nm) shows PKS4 produced predominantly a compound with RT = 30.86 min. The compound was verified to be SMA76a (2).
We then assayed whether the purified PKS4 (10 μM) enzyme can turnover a polyketide compound in vitro using malonyl-CoA (2 mM). LC-MS analysis of the organic extract revealed the presence of a predominant compound SMA76a (Figure 1C, RT=30.8 min). SMA76a has a parent ion peak [M-H]- at m/z 323, which is in agreement with the mass of a nonaketide that has been subjected to two dehydration reactions. The molecular weight of SMA76a corresponds to that of 2 (Scheme 1), the unoxidized and unmethylated precursor of 1. PKS4 synthesized SMA76a with a turnover of 0.1 per min, which is within the kinetic range of bacterial type II minimal PKSs, such as those from actinorhodin and tetracenomycin pathways.11
Scheme 1.

To confirm the identity of SMA76a is indeed 2, we scaled up the in vitro PKS4 reaction, using the malonyl-CoA synthase MatB from Rhizobium trifolii to generate the malonyl-CoA molecules in situ (Supporting Information).12 The yield of SMA76a from the in vitro reaction was approximately 1 mg/10 mL. SMA76a was purified with a single reverse-phase HPLC step and was characterized by extensive NMR spectroscopy. 1H NMR revealed five aromatic proton signals and one methyl signal (δH−18 = 2.72 ppm). Eighteen carbon signals were observed in 13C NMR, including the characteristic ketone (δC−11 = 183 ppm). All of the NMR signals and correlations are consistent with the structure of 2.
The robust synthesis of 2 using PKS4 demonstrates the megasynthase purified from E. coli is functional, and its entire repertoire of catalytic activities can be reconstituted. PKS4 was able to maintain near complete chain length control during polyketide elongation. The nonaketide product 2 was the dominant polyketide produced in this assay. Using authentic samples and LC-MS ion extraction, we detected the presence of truncated compounds in the reaction mixture, such as the triketide 4-hydroxy-6-methyl-2-pyrone (TAL) and the pentaketide 1,3,6,8-tetrahydroxynaphthalene (THN). However, these compounds were only present in trace amounts (< 1%) and were not characterized further. In addition, PKS4 alone maintained precise control of cyclization regioselectivity. We did not detect any nonaketides that contain alternative cyclization patterns. A proposed poly-β-keto processing pathway is shown in Scheme 1. Cyclization of the first ring is facilitated by the aldol condensation reaction between C-2 and C-7, which requires synthesis of the full length polyketide chain. This is followed by TE/CLC catalyzed Claisen-like condensation between C-10 and C-1 to generate the bicyclic THN scaffold.5 Attack of C-13 carbonyl by the nucleophilic C-9 phenol forms the tricyclic naphthopyrone. These three steps are identical to those catalyzed by Aspergillus nidulans wA synthase during YWA1 synthesis.14 Finally, intramolecular C-12/C-17 cyclization and dehydration affords 2.
Equipped with the reconstituted enzyme, we probed the starter unit specificity of PKS4 by incubating PKS4 and [2-14C]malonyl-CoA with various alkylacyl-CoAs and assaying for the synthesis of new products by radio-TLC. PKS4 did not utilize any of the short chain acyl-CoAs (C2-C6) as the starter unit. The lack of recognition by PKS4 towards acetyl-CoA is consistent with that of Colletotrichum lagenarium PKS1, which uses malonyl-CoA as the starter unit.15 Surprisingly, PKS4 produced an entirely different set of polyketide products when octanoyl-CoA (C8) was included in the reaction mixture. HPLC analysis of the reaction extract showed the presence of two new major compounds with nearly identical UV absorption spectra. The level of 2 was decreased ten-fold when compared to that from the reaction without octanoyl-CoA. LC-MS indicated the molecular weights of the two compounds eluting at 30.4 min (SMA76b, 3) and 33 min (SMA76c, 4) were 318 and 276, respectively. Addition of octanoyl-S-NAC and other long chain alkylacyl-CoAs (C10, C12) to PKS4 and malonyl-CoA did not yield new products.
To examine how chain length control and polyketide cyclization may be affected by the octanoyl starter unit, a scaled-up reaction containing octanoyl-CoA (1 mM) was performed. 3 and 4 were purified by HPLC and their structures were determined by NMR spectroscopy (Scheme 2, Supporting Information). Both 3 and 4 are benzopyrones primed with the octanoyl starter unit. The major compound 3 (0.6 mg/10 mL) was derived from a hexaketide backbone that contained eighteen carbons, the same length as that of 2. This is reminiscent of the bacterial type II minimal PKS, where increasing the starter unit size led to a corresponding decrease in the number of chain-extension cycles to maintain the length of the polyketide backbone.16 Hence, the overall size of the polyketide product and the number of iterative condensations are likely controlled by the cavity volume of the PKS4 KS active site. Interestingly, synthesis of pentaketide 4 (0.3 mg/10 mL) suggests chain length control was incomplete when octanoyl starter unit was incorporated into the polyketide backbone, possibly due to the flexibility of the long alkyl chain in the KS active site.
Scheme 2.

The synthesis of 3 demonstrates that modification in polyketide backbone composition derails certain programmed cyclization steps shown in Scheme 1. While PKS4 was able to maintain the C-2/C-7 connectivity to afford the first aromatic ring, it was unable to perform the expected C-10/C-1 Claisen-like condensation to synthesize the naphthalene-containing compound 5. Instead, the benzopyrone-containing compound 3 was synthesized through the likely spontaneous O-9/C-1 esterification. The benzopyrone moiety was previously observed in citreoisocoumarin, which was synthesized by the wA synthase with an inactivated TE/CLC domain.5 Therefore, synthesis of 3 may be similarly due to the inability of the PKS4 TE/CLC domain to catalyze regioselective cyclization of the second ring in the presence of the octanoyl starter. This suggests that the hexaketide portion (C-1 ∼ C-11) in 3 is insufficient for recognition by the TE/CLC domain and additional poly-β-keto units may be required for anchoring the polyketide in the TE/CLC active site. Alternatively, the octanoyl starter unit may interfere with binding of the nascent polyketide in the TE/CLC active site, leading to spontaneous benzopyrone formation.
PKS4 preferentially selected octanoyl-CoA over malonyl-CoA as the starter unit and synthesized predominantly 3 and 4 in vitro. This was surprising, considering PKS4 is normally primed with malonyl-CoA and synthesizes 2 in G. fujikuroi, which is further modified into the metabolite bikaverin. Hence, there exists a previously unknown mechanism by which PKS4 can be specifically primed by an octanoyl starter unit. The octanoyl unit can be selected through transthioesterification either by the SAT-like domain at the N-terminus of PKS4 or directly by the KS. Interestingly, the well-conserved SAT active site sequence GXCXG,3 in which the cysteine is the site of starter unit attachment, is absent from the PKS4 SAT domain. Therefore, the PKS4 SAT domain may be inactive and the octanoyl starter unit is instead directly captured by the KS domain to prime polyketide synthesis.
Supplementary Material
Experimental details, and NMR spectroscopy data. This material is available free of charge via the internet at http://pubs.acs.org.
Acknowledgments
This work was supported by the American Heart Association (0535069N) and NIH RO1-GM75857(CCCW).
References
- 1.Thomas R. Chembiochem. 2001;2:612–627. doi: 10.1002/1439-7633(20010903)2:9<612::AID-CBIC612>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 2.Cox RJ. Org Biomol Chem. 2007;5:2010–2026. doi: 10.1039/b704420h. [DOI] [PubMed] [Google Scholar]
- 3.Crawford JM, Dancy BC, Hill EA, Udwary DW, Townsend CA. Proc Natl Acad Sci U S A. 2006;103:16728–16733. doi: 10.1073/pnas.0604112103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu X, Yu F, Li XC, Du L. J Am Chem Soc. 2007;129:36–37. doi: 10.1021/ja0672122. [DOI] [PubMed] [Google Scholar]
- 5.Fujii I, Watanabe A, Sankawa U, Ebizuka Y. Chem Biol. 2001;8:189–197. doi: 10.1016/s1074-5521(00)90068-1. [DOI] [PubMed] [Google Scholar]
- 6.Hoffmeister D, Keller NP. Nat Prod Rep. 2007;24:393–416. doi: 10.1039/b603084j. [DOI] [PubMed] [Google Scholar]
- 7.Schumann J, Hertweck C. J Biotechnol. 2006;124:690–703. doi: 10.1016/j.jbiotec.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 8.Linnemannstons P, Schulte J, del Mar Prado M, Proctor RH, Avalos J, Tudzynski B. Fungal Genet Biol. 2002;37:134–148. doi: 10.1016/s1087-1845(02)00501-7. [DOI] [PubMed] [Google Scholar]
- 9.Zhan J, Burns AM, Liu MX, Faeth SH, Gunatilaka AA. J Nat Prod. 2007;70:227–232. doi: 10.1021/np060394t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Udwary DW, Merski M, Townsend CA. J Mol Biol. 2002;323:585–598. doi: 10.1016/s0022-2836(02)00972-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang Y, Lee TS, Kobayashi S, Khosla C. Biochemistry. 2003;42:6588–6595. doi: 10.1021/bi0341962. [DOI] [PubMed] [Google Scholar]
- 12.An JH, Kim YS. Eur J Biochem. 1998;257:395–402. doi: 10.1046/j.1432-1327.1998.2570395.x. [DOI] [PubMed] [Google Scholar]
- 13.McInnes AG, Smith DG, Walter JA. J C S Chem Comm. 1975:66–68. [Google Scholar]
- 14.Watanabe A, Fujii I, Sankawa U, Mayorga ME, Timberlake WE, Ebizuka Y. Tetrahedron Lett. 1999;40:91–94. [Google Scholar]
- 15.Fujii I, Mori Y, Watanabe A, Kubo Y, Tsuji G, Ebizuka Y. Biochemistry. 2000;39:8853–8858. doi: 10.1021/bi000644j. [DOI] [PubMed] [Google Scholar]
- 16.Tang Y, Lee TS, Khosla C. PLoS Biol. 2004;2:227–238. doi: 10.1371/journal.pbio.0020031. b) Xu, Z.; Schenk, A. Hertweck, C. 2007, 129, 6022-6030. c) Nicholson, T. P.; Winfield, C.; Westcott, J.; Crosby, J.; Simpson, T. J.; Cox, R. J. 2004, 686-687.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details, and NMR spectroscopy data. This material is available free of charge via the internet at http://pubs.acs.org.