

Abstract
Statistical tools enable unified analysis of data from multiple global proteomic experiments, producing unbiased estimates of normalization terms despite the missing data problem inherent in these studies. The modeling approach, implementation and useful visualization tools are demonstrated via case study of complex biological samples assessed using the iTRAQ™ relative labeling protocol.
Keywords: Proteomics, ANOVA, iTRAQ™, Normalization, relative labeling protocol, Missing data, Gauss-Siedel, Backfitting, Fixed effects model, Mixed effects model
A. INTRODUCTION
The objective of global proteomics via mass spectrometry is to detect and quantify all proteins present in a biological sample. Proteins that exhibit an increase/decrease in abundance between two or more groups of interest, (e.g., diseased and non-diseased) are considered candidate biomarkers. However, experimental factors such as differences in sample collection, sample characteristics such as cellular concentration, variations in sample processing and the experimental process add variability to the observed abundances. Experimental variability hinders the comparison of effects of interest, and if not accounted for during the design and analysis stages, can lead the researcher down an erroneous path of discovery.
Several mass spectrometry (MS) techniques have been developed that allow greater control over experimental factors that introduce variability and ultimately decrease the quality of the data. Recently, focus has centered on the ability to assess multiple samples within a single MS experiment. Binary sample labeling techniques such as 16O/18O(1), ICAT™(2), and SILAC(3) were developed to evaluate paired samples whereas iTRAQ™(4) was developed to simultaneously analyze four, and more recently, eight samples(5). The binary labeling techniques add complexity to the acquired spectra and to their interpretation by introducing additional peaks into the mass spectra. Furthermore, overlapping isotopic clusters require further analytical techniques to deconvolute the resulting spectrum and the associated protein/peptide abundances(4, 6, 7). The iTRAQ™ labeling system overcomes this to some extent since the labeled species are isobaric and protein abundances are measured only in the resulting MS/MS fragmentation spectra.
Although sample labeling techniques allow greater control over experimental variability within an MS experiment, the analysis of multiple MS experiments remains difficult. Within an experiment, it is important that equal amounts of total protein are labeled under each labeling condition to ensure that the observed abundances are not influenced by total protein concentration. Once the samples are labeled and mixed together for MS analysis, labeling methods naturally control for instrument variability. The same principles apply when performing multiple experiments; with the introduction of additional experiments, the sources of experimental variability increase. Prakash et al. (8) evaluated data produced by multiple laboratories and instruments and found definite systematic experiment-to-experiment effects, even in well controlled experiments with technical replicates.
The adverse effects of experimental variability are well known and most vendor software packages (e.g., ProQuant software for iTRAQ™) routinely correct for experimental error via some form of normalization. For example, ProQuant applies a bias correction to ratios within an experiment in order to correct for systematic within MS experiment variability(9). They recommend not combining data across MS experiments unless the bias corrections are similar across experiments since the software is unable to adjust for across experiment variability. However, it is desirable to be able to analyze data across multiple MS experiments since it allows studies to incorporate larger sample sizes, obtaining more accurate estimates of biological effects and thus having more power to detect meaningful differences. The issues associated with comparing relative measurements and the method for controlling for sources of experimental variation (hereafter, referred to as external factors) date back to the early 1900s(10) in agricultural experiments and have been utilized extensively in the gene expression microarray literature(11,12). In this issue, Hill et al.(13) discuss how classical analysis of variance (ANOVA) methodology can be used to simultaneously correct for experimental variability and how such a model may be built for iTRAQ™.
In addition to increased sources of variability in a study with multiple experiments, due to the nature of current instrument technologies, overlap in protein and peptide identification between experiments is less than ideal, leading to a large amount of missing data(14,15,16,17). The abundance distribution of proteins is nearly geometric. This means that if n proteins are present at abundance x, then 2n proteins are present at abundance x/2, and so forth. Identification of the lower abundant peptides is problematic due to the data-dependent acquisition of the mass analysis process. Thus, the probability of missing data for a protein is not random; it is related to abundance(14,15). This is a pervasive issue in global proteomic studies. Wang et al. (15) performed replicate mass analysis experiments of two simple spike-in studies on consecutive days and found that the total number of features identified in an experiment decreased over time by 49%–73%. Liu et al. (14) conducted a controlled study with nine technical replicate global proteomic experiments of yeast lysate. Of a total 1751 proteins identified in the entire study, only 35.4% were found in every experiment and 24% were found in only one experiment. Liu and colleagues developed a model to predict protein detection rates depending on sample complexity and conclude that ten replicates are required to identify 95% of proteins present in yeast lysate; more technical replicates would be required for human samples due to the greater complexity of the human proteome. In marker discovery studies a common strategy is to remove the most abundant proteins, i.e., those that would be detected in every experiment yet are thought to be uninteresting from a biomarker point of view (e.g., albumin). In addition, due to larger sample sizes, time and financial resources generally preclude the ability to perform technical replicates of all biological replicates. Wang et al. (15) point out that with such severe non-random missing data rates, most global normalization practices used for microarrays will result in severe bias in global proteomic data.
Here, we extend the discussion of Hill et al.(13) by applying analysis of variance (ANOVA) methodology to analyze iTRAQ™ data from complex biological samples across multiple MS experiments. We discuss the model components that are necessary due to missing data and the practical implementation due to the large size of global proteomic data sets. Particularly, we discuss the importance of stage-wise and iterative regression to adequately normalize and analyze large datasets from complex biological samples. Even though we focus on iTRAQ™ labeling, the principles discussed herein are applicable to any labeling or label-free platform. Thus, we begin with a description of the cardiomyopathy study, the case study we utilize to describe the methodologies, and follow with a discussion of how to apply ANOVA methodology to complex samples using iterative regression.
For generality, throughout this paper we define an experiment to indicate all labeled specimens that are mixed together and subjected to mass analysis simultaneously. We define a tag to denote the label attached to a particular specimen for use in distinguishing it from other samples within the same experiment. Lastly, we define study to indicate the collection of MS experiments utilized together in addressing a particular research question.
B. MATERIALS AND METHODS
Sample Preparation
Highly abundant proteins were depleted using Biotech’s Seppro Microbead-conjugated avian IgY antibodies to specifically remove human serum albumin, IgG, IgA, IgM, transferrin, fibrinogen, apolipoprotein A-I, apolipoprotein A-II, haptoglobin, α-1 antitrypsin, α-1 acid glycoprotein and α-2 macroglobulin (www.beckmancoulter.com). After washing, the bound protein was eluted, and 100 μg protein was reduced, denatured, cysteine-blocked and digested with trypsin. The sample was then labeled with one of the specific isobaric iTRAQ™ tags (containing a reporter fragment of mass 114, 115, 116, or 117) (iTRAQ™ reagents; ABI). Four samples (each differentially labeled with one of the four iTRAQ™ tags) were combined for LC/MS/MS analysis.
MS Methods
Peptides were fractionated on a strong cation exchange column, and thirteen fractions were then analyzed by capillary reverse-phase LC (Ultimate, LC packings)/MS/MS (Quadrupole-Time of Flight; QSTAR, ABI). Independent data acquisition for each fraction was by Analyst QS software (version 1.1; ABI). MS analysis consisted of 1 s survey scan from 400 to 1600 m/z, and MS/MS of 2 s scan. After fragmentation of three most intense ions in survey scan, they were excluded for 60 s.
Quantification
The ion exchange fractions were analyzed separately and then grouped using ProQuant (version 1.0; ABI). Quantification (relative contribution) of the four samples is determined from the areas of the four iTRAQ™ signature ions in the MS/MS spectrum. For each MS/MS scan, ProQuant identifies precursor m/z and charge in the TOF MS scan for that cycle. The program checks the next ten cycles for precursor m/z and charge. Spectra that are matched for m/z and charge are flagged as part of a merged set. The program finds the peaks in the spectrum (or summed spectra) for the four signature ions derived from the iTRAQ™ reagents (114, 115, 116, 117). Peaks for the signature ions are integrated and reported as “Area 114,” etc. Areas are adjusted according to the isotope correction factors for each lot of iTRAQ™ reagents.
Peptide/Protein identification
Results from the MS/MS are used to search the protein sequence database (KBMS human; March 2005; Celera) with the Interrogator™ Algorithm in ProQuant(18). Interrogator compares the fragment ion masses and precursor molecular weight to theoretical fragment ions in the search database. The accession number(s) for the protein database entry(-ies) containing the identified peptide is recorded along with peptide sequence and area counts (see above). Because the KBMS sequence source database is a compilation of several databases (NCBI RefSeq; SwissProt; Celera; EMBL, and others), identical sequences can be included under different headers (each with a different accession number). A separate database record is generated for each match of the peptide in the source database, including multiple entries for the same protein species. Thus, the resulting database contains replicate records that differ only with respect to the accession numbers.
C. CASE STUDY
The case study consists of six iTRAQ™ experiments that compared serum protein profiles in patients across three histologic subtypes of acute cardiomyopathy. The histologic subtypes were idiopathic dilated cardiomyopathy (DCM), giant cell myocarditis (GCM), and lymphocytic myocarditis (LM). GCM is a rare and fulminant form of autoimmune myocarditis with a greater than 90% rate of death or transplantation. Conversely, LM and DCM are more common and less lethal disorders; however, LM and DCM can present similarly to the more lethal GCM subtype. The objective of this study was to develop a non-invasive diagnostic test for GCM as timely institution of appropriate immunosuppressive therapy for GCM significantly increases heart transplantation-free survival. Currently, the gold standard for the diagnosis of myocarditis requires an endomyocardial biopsy; however, endomyocardial biopsy is severely limited by the risks of cardiac perforation and death. Thus, a non-invasive serum diagnostic test is desirable. This study was approved by the Mayo Clinic Institutional Review Board.
Experimental Design
The experimental design utilized for the case study is provided in Table 1. In addition to the three histologic subtypes, nine normal control samples were pooled to obtain a single pooled control sample giving four disease groups of interest in the cardiomyopathy study (Pooled Control, DCM, GCM, and LM). Since the present discussion is meant to be a demonstration of the utility of ANOVA rather than a definitive analysis of these data, the group identities are masked. The letters A, B, C, and D in Table 1 denote the four disease groups of interest and numbers 1, …, 6 denote the six independent specimens that were sampled from each of DCM, GCM, LM and the six aliquots of the control pool.
Table 1.
Experimental Design of Cardiomyopathy Study.
| Experimental Run Order | Tag | |||
|---|---|---|---|---|
| 114 | 115 | 116 | 117 | |
| 1 | A1 | B1 | C1 | D1 |
| 2 | B2 | D2 | A2 | C2 |
| 3 | D3 | C3 | A3 | B3 |
| 4 | C4 | A4 | D4 | B4 |
| 5 | B5 | A5 | D5 | C5 |
| 6 | D6 | B6 | C6 | A6 |
The letters A, B, C and D denote the four treatment groups under investigation. The numbers denote independent samples. For example, A1 is the first sample in group A. The actual labels of the disease groups are masked for the purposes of this manuscript.
Data Characteristics
There were a total of 992 unique proteins and 2637 unique peptides identified across the six experiments. Of the 992 proteins identified, 743 and 127 were observed in only one or two experiments respectively and 16 proteins were observed in all six experiments. Similarly, of the 2637 peptides identified, 1386 and 396 were observed in only one or two experiments respectively and 172 peptides were observed in all six experiments. Thus, as found by other authors (15,14,16,17), due to the data-dependent acquisition induced thresholding that generally occurs during mass analysis for global proteomic studies, there is a large amount of missing data. Figure 1 depicts the protein coverage across all six experiments. A protein present in all six experiments would show as a black horizontal line across the entire plot in Figure 1. Experiment 4 clearly has more black lines than the other five experiments indicating that the samples used for the 4th experiment had much higher protein concentration than any of the other five experiments.
Figure 1.
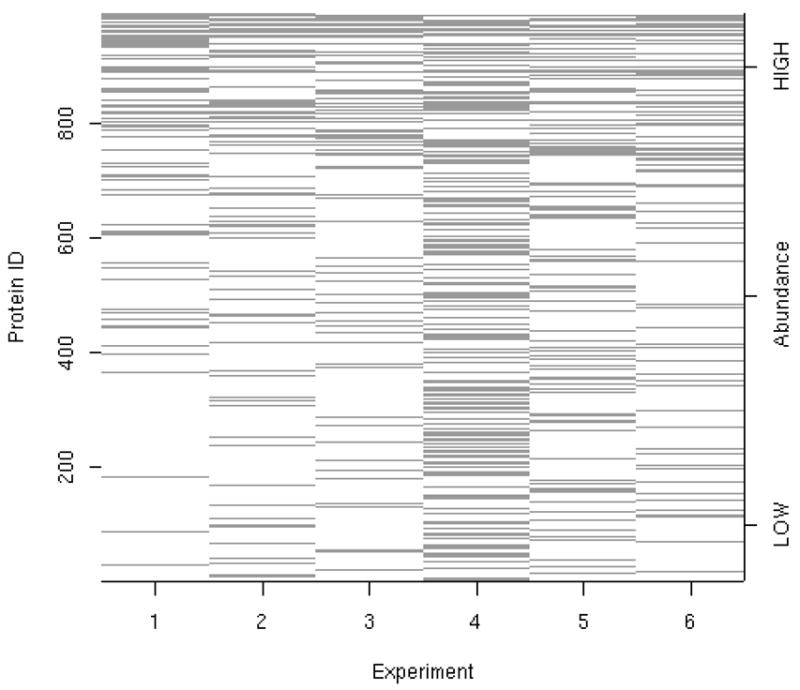
Protein coverage across all six iTRAQ™ experiments. The horizontal axis indicates experiment run order. The vertical axis indicates proteins, where proteins are ordered by their average abundance rank across each experiment. Each horizontal line represents the presence of a protein within each experiment. The proteins at the top of the plot have highest average abundance and those at the bottom of the plot have the lowest average abundance. A protein present in all six experiments would show as a black horizontal line across the entire plot. Note that many more proteins were detected in experiment 4 than in the other five experiments. Due to thresholding which occurs during mass analysis, not all proteins will be observed in all experiments.
D. Constructing the ANOVA Model
As discussed in Hill et al.(13) in this issue, an ANOVA model can be constructed that accounts for the known sources of variation present in a study. In particular, Hill and colleagues discuss the importance of accounting for variability due to differential iTRAQ™ tagging efficiencies, experiment-to-experiment variability, and variability in total protein concentration. ANOVA is a versatile tool that allows data from multiple experiments to be analyzed collectively and has the capability to correctly account for the missing data that is inherent to proteomic data from complex biological samples.
In constructing the ANOVA model for the cardiomyopathy data, we include both factors that account for experimental variability, and thus serve as normalization terms, as well as factors that assess the research questions of interest. The fact that researchers think in terms of fold change (ratios) for this type of data reflects the belief that effects are multiplicative in nature on the raw scale. Since ANOVA models operate in terms of additive effects, we transform the data to the log scale. We use log base 2 where a value of 1.0 indicates a 2-fold change for ease of interpretation. Thus, let y be an observed log2(abundance). Then we assume that the observed value can be decomposed as
Following the notation of Hill et al.(13), we write this more formally for the cardiomyopathy study as
| (1) |
For computational reasons that we discuss in detail in section E, we have arranged the terms into three groups which we will refer to as I, II and III, set apart by parentheses. That is, yi,j(i),c,q,s,l = (group I) + (group II) + (group III) + hi,j(i),c,q,s,l, where group I = (u + vq,l + bq), etc. As in Hill et al.(13), u is the overall mean, bq describes the effect due to a given iTRAQ™ experiment, and vq,l describes the experimental effects of loading, mixing, and other sample handling effects. If a given patient sample is used in only one experiment, then vq,l could be considered the ‘patient’ effect, since all later effects of sample handling are indistinguishable from a baseline difference in the patient’s total protein. Overall we can think of the terms in group I as the ‘experimental’ effects; they are aspects of the experiment that would not exist in an ideal world of perfectly reproducible hardware, procedures, and subjects. The second set of terms, group II, are the differential effects of protein (pi) and peptide (fj(i)). It has been observed that if a single purified protein is trypsinized and the results subjected to mass spectrometry, the reported peptide abundances may differ by two-to-three orders of magnitude. The group III effects are those of actual interest, i.e., they describe why the experiment was run; they answer the question “Which proteins and/or peptides are differentially expressed across the conditions rc indicates the group effect, ri,c denotes the proteins differentially expressed of interest?” Specifically, between groups, and gj(i),c denotes the peptides differentially expressed between groups. Lastly, hi,j(i),c,q,s,l is the residual error for each observation. We refer the reader to Hill et al.(13) in this issue for a more in depth discussion of the rationale for including these effects in the model and for their interpretation.
In the analysis below we removed the gj(i),c term. That is, we fit a model that assumes that there will be differential expression of certain proteins between the conditions of interest, but that any increase in protein expression will affect all of the peptides for that protein equally. This is done largely to simplify the presentation. Although one expects this to be the common case, there are certainly biological conditions where a change to the levels of one or more peptides, but not the protein as a whole, will occur; for example a post-translational modification that involved a peptide substitution.
E. Practical Implementation of the ANOVA Model for Complex Samples
The effects in group I of model (1) are global effects. That is, the experimental effects (e.g., experiment, tag, etc.) are expected to be the same for all proteins and peptides observed. Thus, it is desirable to use all observed data to obtain one overall estimate of these effects. For the cardiomyopathy study, there were nearly 1000 identified proteins and over 2500 identified peptides, making it difficult to estimate all of the parameters in model (1) simultaneously using current software and computers. The threshold of what is “too large” for a computer changes continuously, of course, but this is likely to remain an issue for proteomic analyses for quite some time as the total amount of storage needed for the analysis is proportional to the number of experiments times the number of peptides, and both of these dimensions are increasing continuously. Thus, below we detail different strategies to confront this computationally challenging problem, including subsetting, stagewise regression, and iterative regression.
Subsetting
One approach to estimating the parameters in model (1) is to subset the data and estimate the parameters separately for each identified protein. Although this solves the memory issue, the resulting fits are, unfortunately, incorrect. For example, model (1) accounts for possible iTRAQ™ experimental effects; each experiment may have a larger or smaller total amount of protein loaded due to imperfect pipetting, which would lead to all of the proteins for that experiment having a larger (or smaller) intensity. When each protein is fit separately, then the experimental effect is re-estimated multiple times (e.g. estimated separately for every protein), giving an inconsistent solution that marks a given experiment as sometimes high, sometimes low. Thus, estimating the experimental effects individually for each protein rather than globally results in inefficient and incorrect normalization.
Stagewise Regression
In section D we arranged the model (1) terms into three groups I, II, and III, corresponding to experimental, protein/peptide, and differential expression portions of the model. If one fits the model to the entire data set in a stagewise fashion, i.e., first group I, then group II, then group III, each of the individual fits may be simple enough to fit into memory. However, the choice of partition is critical to obtaining valid parameter estimates. This is, in fact, exactly what is done in most microarray experiments, where group II contains the differential binding effects for each probe (Affymetrix) or region (spotted arrays), and groups I and III are again the experimental and differential effects, respectively. Wolfinger et al. (12) for instance first fit group I, and then groups II+III together. Ballman et al. (19) show that cyclic lowess normalization is a stagewise fit of groups I+II, using an iterative algorithm, followed by group III. The popular RMA(20) and GCRMA(21) algorithms first deal with groups I+II using a quantile based method, and then group III using outlier resistant regression.
When the data is fit in two stages, the first stage is usually referred to as normalization where global estimates of experimental effects are obtained, particularly in the gene-expression microarray arena. For the stagewise approach to give correct answers, however, it is necessary that the parameter estimates from the multiple stages are uncorrelated, or more technically, that the portions of the linear model design matrix corresponding to the multiple stages be orthogonal. It turns out that if the fraction of differentially expressed proteins is small, then group III is nearly orthogonal to the group I and II model parts. Thus, estimating the differential expression terms in group III separately from the terms in groups I and II is likely to be reliable for most research studies, including the cardiomyopathy study shown here, but not for a designed experiment such as in Hill et al.(13) where nearly all of the proteins are differentially expressed.
For microarray data, where there are a fixed number of probes and each probe has a measured value for each sample, the data are fully balanced. In such a case, groups I and II are orthogonal and the stagewise algorithm is correct. This is not the case for proteomics data, unfortunately, due to the imbalance (missing observations) in the data. Each global proteomic experiment will have different sets of proteins detected as discussed previously, resulting in an unbalanced data set for which the experimental and protein/peptide parameters are not uncorrelated. For a very simple linear model with group I consisting only of an experiment effect, stagewise estimation of group I is equivalent to globally normalizing the data by the mean peptide intensity of each experiment. Wang et al.(15) show that for proteomics data the estimation bias from this procedure can be extreme due to missing data and they provide a nice graphical display (their figure 2) of exactly how the underlying imbalance leads to this problem. Stagewise fits of model (1) are subject to the same concern. Wang et al.(15) proposed computing the experiment and loading effects only on the balanced subset of peptides that appear in all experiments as one approach to avoid this bias. We propose that using all of the data in an ANOVA model is much more efficient. Due to the imbalance in the data across multiple iTRAQ™ experiments, the group II effects must be estimated together with the group I effects for correct estimation of group I terms. However, estimation of groups I and II simultaneously is still too large for current computational resources.
Figure 2.
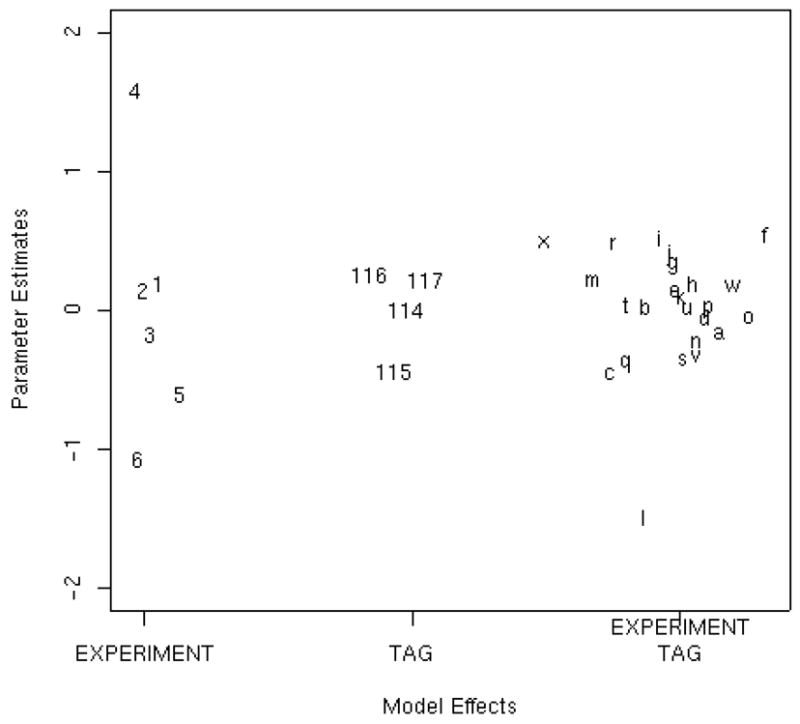
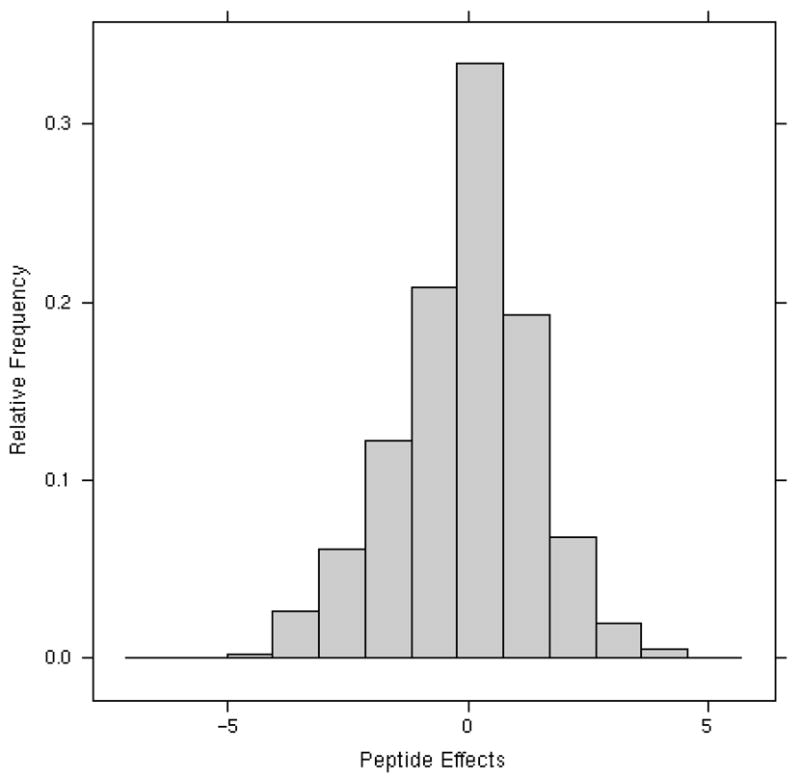
Estimated parameter effects from the normalization section of the model. Figure 2A: Estimated group I parameter effects of experiment bq, and loading effects vq,l decomposed into the tag and experiment-by-tag effects. Since the data were analyzed on log base 2 scale, a coefficient of 1.0 represents a 2-fold effect. Figure 2B: Histogram showing the distribution of the peptide effects fj(i) relative to the average. These effects span a wide range, nearly 10-fold.
Iterative Regression
An alternative approach to stagewise regression is a numerical procedure referred to as iterative refinement. The Gauss-Siedel algorithm for instance, also known as backfitting, is one iterative technique; for a good overview see Hastie and Tibshirani(23), who use backfitting to estimate parameters for generalized additive models.
For the linear models problem here, the backfitting or Gauss-Seidel algorithm is closely related to stagewise regression. The difference is that the algorithm cycles through the stages, so that each stage is repeatedly re-fit given the solution to the previous stages. For the cardiomyopathy data, a fixed effects model was too large for either R or SAS. Thus, we used backfitting to iteratively solve for parameters in groups I and II, the experimental and protein/peptide terms. The final result of this iterative fit is then used to normalize the data, before proceeding to estimate the differential expression terms in group III. That is, the normalized data are
the residuals from the fits of groups I and II with the systematic bias factors subtracted out. These normalized values are used as inputs for estimating the effects in group III. With the peptide effects included in the normalization stages of the model fitting, the group III parameters are separable and can be estimated one protein at a time. Thus, as in Wolfinger et al. (12), the normalized data are used as inputs to the differential expression model which was fit separately for each of the 992 proteins. Thus, the normalization terms are estimated globally, whereas the differential protein effects are not. Fitting group III parameters on a protein-by-protein basis assumes that each protein has a different variance parameter, rather than a global variance parameter. Jain et al.(22) give a nice discussion of the pros and cons of estimating a variance for each protein and a summary of other approaches. We focus here on estimation of the normalization terms.
Ideally, all data, including that for unidentified peptides could be used to estimate the group I and II normalization effects. However, per the above, normalization needs to involve both the group I and II terms, which means that the peptides must have been matched in some way, i.e., we know that peptide “x” in one sample is the same as peptide “x” in another. For this matching, an identification is not needed, merely the knowledge of matching. However, if identification and matching are the same process, as is the case for many iTRAQ™ data pipelines, the matching information will not be available for the unidentified peptides. Thus, the list of detected peptides is first subsetted to the set with identification information before proceeding with data analysis.
Mixed Effects Models
An extension of the linear model approach outlined above (also called a fixed effects model) is a mixed effects model which contains both fixed and random effects. Random effects are an extension which align naturally with certain aspects of proteomics data and thus may be preferred. Declaring an effect, e.g., peptide, in the model as random adds a constraint to that effect in the form of an a priori Gaussian distribution with mean 0 and variance(s) τ. Fitting the model involves estimation of both the fixed effect coefficients and the variance(s) τ.
Computationally, mixed and fixed effects models have similar issues. The best method is to fit the entire model to all data simultaneously. This may be computationally challenging for large data sets, however, due to computational restrictions. As described previously for fixed effects models, fitting separate models for each protein remains an invalid approach with respect to global terms; global estimates of both experimental effects and the variance parameter(s) τ are needed. For models containing random effects in groups I or II, the stagewise approach has exactly the same requirements for balance as described previously for the fixed effects case. For balanced data, the approach in Wolfinger et al. (12), with stagewise fits of both fixed and random group I effects followed by subset fits of both fixed and random group II and III effects is valid; however, it would not estimate group I effects correctly in unbalanced proteomic data. As with fixed effects models, the parameters from groups I and II must be estimated together when imbalance is present in the data in order to correctly estimate the group I effects. Unfortunately, the standard iterative regression methods available for fixed effects models are not applicable to mixed effects models, and a solution remains an open problem.
For the cardiomyopathy data, we were able to perform a single global fit of group I and II terms where group I effects were fixed effects and group II effects were random effects. This was accomplished with the counsel of the fifth author (RDW), who is a principal architect of the SAS mixed effects model software. Note that due to the abundance dependent nature of the imbalance in these data, the peptide variance effect will be underestimated. As in Hill et al.(13) the fixed versus mixed effects model had little impact on the normalization effects (data not shown). In the microarray domain both mixed effects(12) and fixed effects approaches(20,21) have been proposed, but lacking case studies where the results are substantially different the latter has become the dominant approach.
F. Results
For the cardiomyopathy study we used backfitting to estimate the group I and II parameters in model (1). The parameter estimates for the experiment bq and loading vq,l decomposed into tag and experiment by tag normalization effects are displayed in Figure 2A; the figure is helpful for identifying peculiarities in the data at the experiment or specimen level. Note that there are six parameter estimates associated with experiment (bq) since the cardiomyopathy study contained six MS experiments. Likewise, the experiment-by-tag loading parameter (vq,l) can be decomposed into four tag effects since iTRAQ™ technology has four tags and 24 experiment-by-tag effects, one for each experiment-by-tag combination. Recall that the data were analyzed on the log base 2 scale, so a coefficient of 1.0 represents a 2-fold effect. The experiment (bq) parameter estimates indicate the mean shift of each experiment from the overall mean abundance. From Figure 2A it is clear that experiment 4 had abundance values that were about 2.8-fold larger than the average. This phenomenon is also apparent in Figure 1 by the presence of more bars for experiment 4; Figure 1 is a simple present/absent plot of detected proteins in each experiment. The small amount of spread in the estimates of the tag effects in Figure 2A indicates that there is only a minor tag effect in this study. Additionally, the sample denoted by l (experiment 6, tag 115) in the experiment-by-tag effects stands out with an abundance approximately 2.8 fold lower than the average. While the peptide term (fj(i)) in model (1) does not represent an experimental effect, it is needed in the model in order to obtain correct estimates of the experimental effects as discussed in Section E. This is due to the missing values that are a direct result of thresholding. The peptide effects shown in Figure 2B have a much larger range than the three experimental effects shown in Figure 2A; about 5% of the peptides have coefficients less than −1.8, indicating that they are 6-fold less “abundant” than the average.
Figure 3 shows the distribution of peptide abundance pre- and post normalization, i.e., before and after subtracting the group I and II estimated effects shown in Figures 2A and 2B. Prior to normalization, there is clear variability from tag-to-tag and experiment-to-experiment, which is reduced by 56% after applying the normalization model. Interestingly, Table 2 shows the estimated normalization constants of Figure 2 with and without concurrent adjustment for the peptide and protein effects; the naïve estimates have zero digits of computational accuracy and are biased towards zero by 19%–48%. The scatter plot in Figure 4 displays the normalized values relative to the estimated mean abundances from the normalization portion of the model for tag 114 and experiment 4. This plot is analogous to the MVA (minus versus average) plots in the microarray literature. The smoother shows the moving average as a function of predicted abundance; if the normalization has been successful, the data points should be clustered around the y=0 line as is the case in Figure 4. Thus, we are reassured that the linear ANOVA model adequately normalized the cardiomyopathy data. If abundance-dependent biases were evident in Figure 4 via a non-linear smoother that is not parallel to the y=0 reference line, then the adjustment for experimental factors would require a more complex model.
Figure 3.
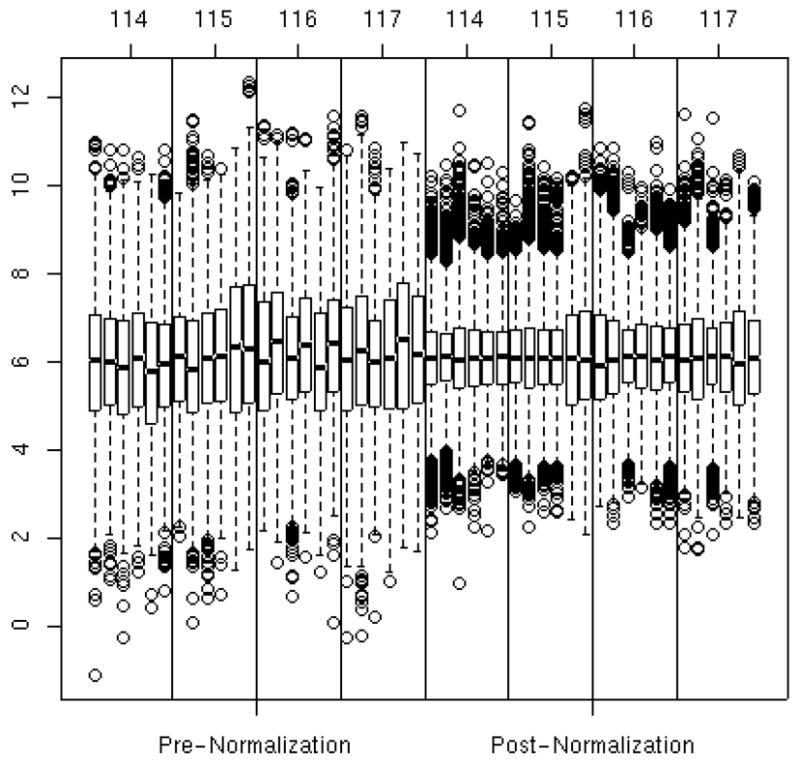
Box and whisker plots of peptide abundance on the log base 2 scale for each experiment-by-tag combination pre-normalization (left 4 panels) and post-normalization (right 4 panels). The vertical axis is the log base 2 abundance values. The horizontal axis corresponds to experiment and tag combination. The sort order of the box plots are first by tag and then by experiment. For example, the first six box plots are from tag 114 in experiments 1 through 6, the next six are from tag 115 in experiments 1 through 6, etc. The top and bottom of the box represent the 75th and 25th percentiles of the distribution, respectively, and the line inside the box denotes the median. The ‘whiskers’, or dashed lines and dots indicate data points on the extremes of the distribution. Much of the variation between experiment-by-tag combinations was removed via the normalization model.
Table 2.
Comparison of normalization parameter estimates with and without accounting for thresholding.
| Experiment | ||||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |
| Correct | 0.12 | 0.09 | −0.13 | 1.09 | −0.43 | −0.75 |
| Naive | 0.15 | 0.13 | −0.10 | 0.56 | −0.35 | −0.39 |
Estimates of normalization effects with (correct) and without (naïve) adjusting for protein and peptide. The comparisons between groups of interest, i.e., statistical contrasts, are generally underestimated when the model does not account for the thresholding present in the data. The naïve estimates have zero digits of computational accuracy. The most notable difference in estimates is seen for experiment 4, in which many more peptides were detected than the other experiments and the naïve estimate is biased towards zero by 48%.
Figure 4.
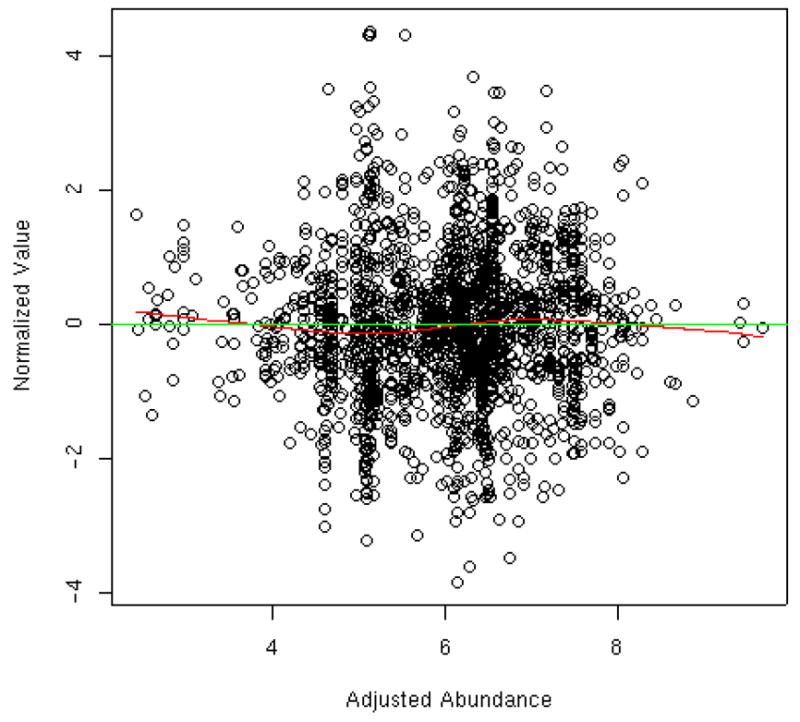
Normalized abundance values vs. predicted abundance from the normalization portion of the model for tag 114 in experiment 4. The horizontal line at y=0 represents the expected zero mean of the normalized values. The scatter plot smoother represents the local average (relative to the predicted abundance) of the normalized values. Data points would cluster about the y=0 line in the absence of systematic bias. The slight nonlinearity in the smoother indicates that nonlinear biases are possible, but they are not severe in this data set.
Of the 992 unique proteins identified in the cardiomyopathy study, a total of 562 proteins could be assessed for differential expression. A test could not be constructed for the remaining 430 proteins because they were identified in only one MS experiment and by a single peptide. The test statistics or the corresponding p-values can be used for ranking the proteins according to statistical significance. Because of the large number of proteins being tested, the value of 0.05 should not be viewed as an absolute cut-off value for significance, but rather as a screening threshold. There is a rich literature on multiple testing and the appropriate corrections(24) which we will not go into here. Rather, we show some selected results.
Figure 5, known as a volcano plot in the microarray literature, shows an overall view of the differences between groups A and D. The volcano plot is useful for assessing significance together with fold change for the entire study. The vertical axis corresponds to statistical significance (−log base 10 (p value)) and the horizontal axis denotes average fold change between disease groups. Data points in the top right and left rectangles correspond to proteins with both small p-values and large fold changes. This information together with biological information about each protein is useful for determining the proteins of most interest for further investigation.
Figure 5.
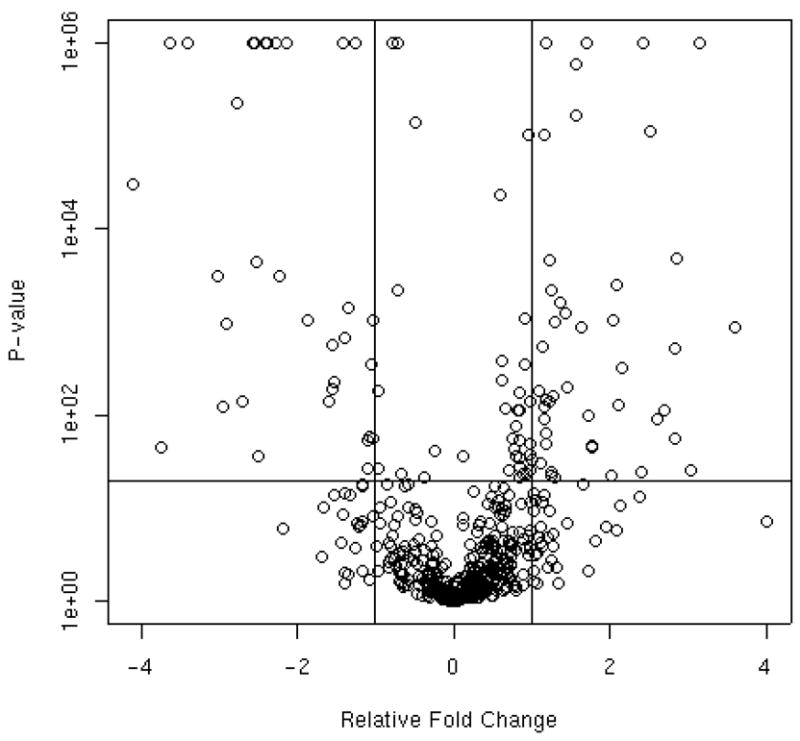
Statistical significance versus fold change for groups A and D, is commonly called a volcano plot in the gene expression microarray literature. The vertical axis denotes –log base 10 of the p-value and the horizontal axis denotes average fold change on the log base 2 scale. The horizontal reference line corresponds to a p-value cut-off of 0.0083, the Bonferroni adjusted significance criteria. The two vertical lines correspond to a 2-fold change.
Normalized data for all peptides associated with one protein selected from the list of all proteins with data across multiple experiments is shown in Figure 6 and is referred to as a dot plot. Each point represents the normalized expression value of each peptide mapped to that protein within that experiment and group where the horizontal line represents the overall group mean. Figure 6 is helpful in determining if patterns of expression are consistent across experiments and subjects and whether the overall significance may be influenced by a set of potential outlying peptide values. This protein was observed in three of the six experiments and these data indicate that it is present at higher abundance in Group D in comparison to the other three groups. A single outlying data point approximately 16-fold higher than the Group C average is apparent in experiment 4. We refer the reader to Hill et al.(13) in this issue for further guidance in computing point and interval estimates. The analyses and results presented here are not meant to be definitive. Further work is needed to more fully understand potential sources of systematic biases that may affect data produced by iTRAQ™ and other multi-labeling proteomic platforms. Furthermore, we presented a protein level analysis, with focus on comparing mean protein abundance levels between disease groups for all peptides mapped to a given protein. It is possible to examine individual peptide abundance profiles within a protein as well. Additionally, it may be desirable to ignore the mapping of peptides to proteins all together and assess differences across disease groups separately for each individual peptide. This can be done by deleting the effects containing protein from the ANOVA model and directly evaluating the peptide effects.
Figure 6.
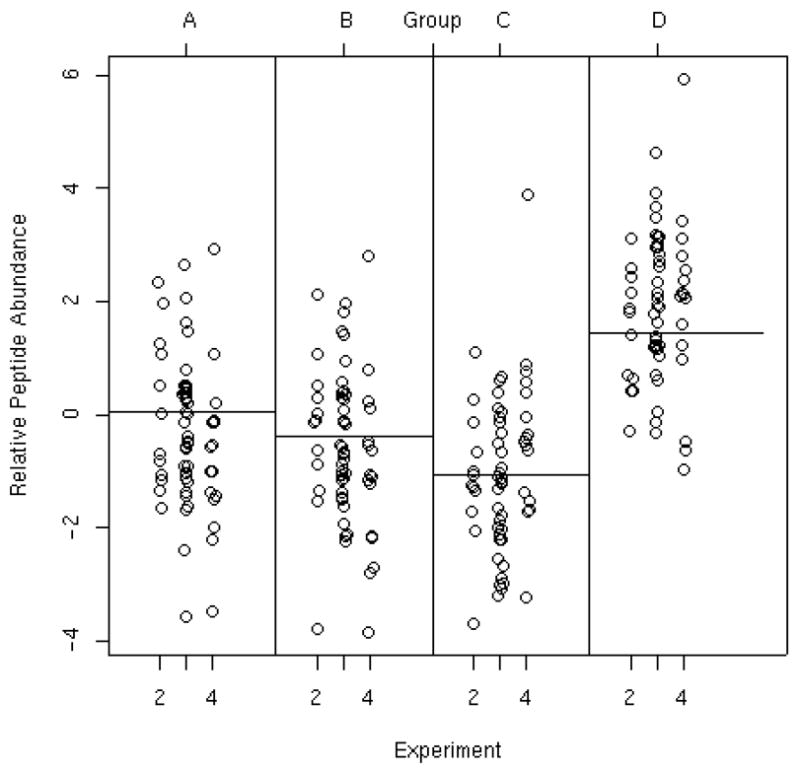
Normalized peptide abundance by disease group and experiment run order for one protein selected from the list of all proteins with data across multiple experiments. The vertical axis represents abundance on the log base 2 scale. The horizontal axis corresponds to experiment number. The four panels indicate the four disease groups which are indicated along the top of the plot. The horizontal lines indicate the estimated mean in the respective disease groups. This protein appeared in only three experiments.
G. Discussion
Global proteomic studies show great promise and are being exploited more frequently in the search for biomarkers of disease status. As current MS technologies are pushed to the limits, efficient and understandable analysis methods are needed for proper inference. Analysis methods for data that are relative in nature, rather than absolute, have been studied since the early 1900s and are well understood in the statistical community. Hill et al.(13) in this issue discuss application of ANOVA to a simple sample. In this manuscript, we extend this and demonstrate how ANOVA can be used to analyze data from multiple experiments on proteomic platforms utilizing relative labeling procedures by presenting a case study analysis of an iTRAQ™ study. ANOVA methods afford wide flexibility in experimental design, techniques to declare significance of results, efficient use of the data (the smallest possible standard errors of the results), and guidance in sample size calculations. ANOVA models can be used to incorporate normalization and differential expression into a single unified model.
Researchers are accustomed to interpreting results from relative labeling platforms on the fold change (ratio) scale. Results from an ANOVA analysis can be interpreted either on the fold change scale or log scale. Using ANOVA tools on individual abundance data rather than strictly fold change ratios from one experiment at a time has the added advantage of using all information available as well as providing standard errors and statistical tests, an objective measure of significance. ANOVA tools can easily accommodate any experimental design, simple or complex. We note that most vendor software exports only protein level ratios for proteins from one MS experiment. The individual abundance values may be obtained from some, but only with much effort. We appeal here to vendors to make the individual abundance values more readily available for their users. This will enable more efficient analyses.
The size of global proteomic data sets resulting from studies involving even moderate numbers of complex samples from biological replicates demands non-standard model fitting algorithms. These algorithms generally partition the modeling process into a normalization portion and a differential expression portion. Such techniques have been well worked out in the microarray literature for balanced data. The severe imbalance due to missing observations in global proteomic data sets(14,15,16,17) prevents the direct extension of these methods to global proteomic studies(15). Thus, a peptide effect must be included in the normalization portion of the model for proper estimation of the experimental effects.
The use of sound experimental design is the foundation of successful interpretation of study results. Many designs are available, and the best choice for a particular study depends on the particular objectives and resources available. When combining data across multiple MS experiments, experimental design becomes even more important and deserves appropriate consideration. In multi-channel platforms it is important that the allocation of subjects to channels (tags in iTRAQ™) be made with care. The experimental design in Table 1 is referred to as a complete block design. In a complete block design every treatment/disease group of interest appears in each block; here, an MS experiment denotes a block. In the agricultural studies that motivated ANOVA in the 1930s, “blocks” referred to plots of land, each of which might have a unique influence on the outcomes; an experiment is the natural analog of this in an iTRAQ™ study. Balancing the treatment/disease groups across blocks, as much as possible, gives the greatest precision for treatment comparisons.
Due to feasibility, the number of treatment groups may be too large (e.g., >4 in an iTRAQ™ experiment) and only a subset of the treatments may be applied to each block leading to an incomplete block design. In an incomplete block design, each block contains a subset of the possible treatments. Another experimental design that is often utilized in high-dimensional genomic and proteomic studies is the reference design. Here, a pooled reference sample is allocated to every block using the same labeling tag and the treatments of interest are allocated to the remaining tag(s). The motivation behind implementing a reference design is the ability to create ratios relative to a common sample and to quantify and correct for experimental variability across MS experiments. However, it has been shown in the gene-expression microarray literature that block designs are typically more efficient(11,25). Ultimately, the optimal experimental design, and therefore the particular ANOVA model used, depends on the research question of interest and the constraints of the platform. However, it is typically important to balance the design as much as possible, i.e., each treatment group should be represented the same number of times and each pair of treatments should have the same number of within-experiment pairings. For a more detailed discussion on experimental designs see(26).
We acknowledge that improvements can be made to the analysis discussed here by more appropriately handling the incomplete observations present in global proteomic data. Due to the abundance-dependant nature of missing observations, fitting the ANOVA model described in equation (1) using a censoring mechanism is a natural next step. We have performed initial work in using censoring mechanisms to fit the model described here. It is clear that the mechanism by which iTRAQ™ data are censored is not adequately captured by standard censoring software causing severe biases in parameter estimates. We are currently pursuing this further.
Supplementary Material
SAS programs that perform the backfitting algorithm and mixed effects models described in the text are provided. This material is available free at http://pubs.acs.org.
Acknowledgments
We greatly appreciate funding from the Kemper Foundation and the University of Minnesota Biomedical Informatics and Computational Biology Program. In addition, the project described was supported by Grant Number 1 UL1 RR024150-01* from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and the NIH Roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH. Information on NCRR is available at http://www.ncrr.nih.gov/. Information on Reengineering the Clinical Research Enterprise can be obtained from http://nihroadmap.nih.gov/clinicalresearch/overviewtranslational.asp. We are grateful to Ms. Rhonda Larsen for assistance with this manuscript.
References
- 1.Yao X, Freas A, Ramirez J, Demirev PA, Fenselau C. Proteolytic 18O labeling for comparative proteomics: model studies with two serotypes of adenovirus. Anal Chem. 2001;73(13):2836–42. doi: 10.1021/ac001404c. [DOI] [PubMed] [Google Scholar]
- 2.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nature Biotechnology. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 3.Oda Y, Huang K, Cross FR, Cowburn D, Chait BT. Accurate quantitation of protein expression and site-specific phosphorylation. Proc Natl Acad Sci U S A. 1999;96(12):6591–6. doi: 10.1073/pnas.96.12.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3(12):1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 5.Lau KW, Jones AR, Swainston N, Siepen JA, Hubbard SJ. Capture and analysis of quantitative proteomic data. Proteomics. 2007 doi: 10.1002/pmic.200700127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eckel-Passow JE, Oberg AL, Therneau TM, Mason CJ, Mahoney DW, Johnson KL, Olson JE, Bergen HR., 3rd Regression analysis for comparing protein samples with 16O/18O stable-isotope labeled mass spectrometry. Bioinformatics. 2006;22(22):2739–45. doi: 10.1093/bioinformatics/btl464. [DOI] [PubMed] [Google Scholar]
- 7.Mason CJ, Therneau TM, Eckel-Passow JE, Johnson KL, Oberg AL, Olson JE, Nair KS, Muddiman DC, Bergen HR., 3rd A method for automatically interpreting mass spectra of 18O-labeled isotopic clusters. Mol Cell Proteomics. 2007;6(2):305–18. doi: 10.1074/mcp.M600148-MCP200. [DOI] [PubMed] [Google Scholar]
- 8.Prakash A, Piening B, Whiteaker J, Zhang H, Shaffer SA, Martin D, Hohmann L, Cooke K, Olson JM, Hansen S, Flory MR, Lee H, Watts J, Goodlett DR, Aebersold R, Paulovich A, Schwikowski B. Assessing bias in experiment design for large scale mass spectrometry-based quantitative proteomics. Mol Cell Proteomics. 2007;6(10):1741–8. doi: 10.1074/mcp.M600470-MCP200. [DOI] [PubMed] [Google Scholar]
- 9.Biosystems A. Using Pro Group Reports. 2004 [Google Scholar]
- 10.Fisher RA. The Design of Experiments. 1937 [Google Scholar]
- 11.Kerr MK, Martin M, Churchill GA. Analysis of variance for gene expression microarray data. J Comput Biol. 2000;7(6):819–37. doi: 10.1089/10665270050514954. [DOI] [PubMed] [Google Scholar]
- 12.Wolfinger RD, Gibson G, Wolfinger ED, Bennett L, Hamadeh H, Bushel P, Afshari C, Paules RS. Assessing gene significance from cDNA microarray expression data via mixed models. J Comput Biol. 2001;8(6):625–37. doi: 10.1089/106652701753307520. [DOI] [PubMed] [Google Scholar]
- 13.Hill EG, Schwacke JH, Comte-Walters S, Slate EH, Oberg AL, Eckel-Passow JE, Therneau TM, Schey KL. A statistical model for iTRAQ data analysis. Journal of Proteome Research. 2007 doi: 10.1021/pr070520u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu H, Sadygov RG, Yates JR., 3rd A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76(14):4193–201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 15.Wang P, Tang H, Zhang H, Whiteaker J, Paulovich AG, Mcintosh M. Normalization regarding non-random missing values in high-throughput mass spectrometry data. Pacific Symposium of Biocomputing. 2006;11:315–326. [PubMed] [Google Scholar]
- 16.Choi DS, Lee JM, Park GW, Lim HW, Bang JY, Kim YK, Kwon KH, Kwon HJ, Kim KP, Gho YS. Proteomic Analysis of Microvesicles Derived from Human Colorectal Cancer Cells. J Proteome Res. 2007 doi: 10.1021/pr070192y. [DOI] [PubMed] [Google Scholar]
- 17.Keshamouni VG, Michailidis G, Grasso CS, Anthwal S, Strahler JR, Walker A, Arenberg DA, Reddy RC, Akulapalli S, Thannickal VJ, Standiford TJ, Andrews PC, Omenn GS. Differential Protein Expression Profiling by iTRAQ-2DLC-MS/MS of Lung Cancer Cells Undergoing Epithelial-Mesenchymal Transition Reveals a Migratory/Invasive Phenotype. Journal of Proteome Research. 2006;5:1143–1154. doi: 10.1021/pr050455t. [DOI] [PubMed] [Google Scholar]
- 18.Tang WH, Halpern BR, Shilov IV, Seymour SL, Keating SP, Loboda A, Patel AA, Schaeffer DA, Nuwaysir LM. Discovering known and unanticipated protein modifications using MS/MS database searching. Anal Chem. 2005;77(13):3931–3946. doi: 10.1021/ac0481046. [DOI] [PubMed] [Google Scholar]
- 19.Ballman KV, Grill DE, Oberg AL, Therneau TM. Faster cyclic loess: normalizing RNA arrays via linear models. Bioinformatics. 2004;20(16):2778–86. doi: 10.1093/bioinformatics/bth327. [DOI] [PubMed] [Google Scholar]
- 20.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 21.Wu Z, et al. A model-based background adjustment for oligonucleotide expression arrays. J Am Stat Assoc. 2004;99:909–917. [Google Scholar]
- 22.Jain N, Thatte J, Braciale T, Ley K, O’Connell M, Lee JK. Local-pooled-error test for identifying differentially expressed genes with a small number of replicated microarrays. Bioinformatics. 2003;19(15):1945–51. doi: 10.1093/bioinformatics/btg264. [DOI] [PubMed] [Google Scholar]
- 23.Hastie TJ, Tibshirani RJ. Some Theory for Additive Models. In: Cox DR, Rubin DVHD, Silverman BW, editors. Generalized Additive Models. Chapman and Hall; 1990. pp. 105–132. [Google Scholar]
- 24.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. 1995;57:289–300. [Google Scholar]
- 25.Jin W, Riley RM, Wolfinger RD, White KP, Passador-Gurgel G, Gibson G. The contributions of sex, genotype and age to transcriptional variance in Drosophila melanogaster. Nat Genet. 2001;29(4):389–95. doi: 10.1038/ng766. [DOI] [PubMed] [Google Scholar]
- 26.Eckel-Passow JE, Hoering A, Therneau TM, Ghobrial I. Experimental design and analysis of antibody microarrays: applying methods from cDNA arrays. Cancer Res. 2005;65(8):2985–9. doi: 10.1158/0008-5472.CAN-04-3213. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SAS programs that perform the backfitting algorithm and mixed effects models described in the text are provided. This material is available free at http://pubs.acs.org.