

Abstract
Complex I has reactive thiols on its surface that interact with the mitochondrial glutathione pool and are implicated in oxidative damage in many pathologies. However, the Cys residues and the thiol modifications involved are not known. Here we investigate complex I thiol modification within oxidatively stressed mammalian mitochondria, containing physiological levels of glutathione and glutaredoxin 2. In mitochondria incubated with the thiol oxidant diamide, complex I is only glutathionylated on the 75-kDa subunit. Of the 17 Cys residues on the 75-kDa subunit, 6 are not involved in iron-sulfur centers, making them plausible candidates for glutathionylation. Mass spectrometry of complex I from oxidatively stressed bovine heart mitochondria showed that only Cys-531 and Cys-704 were glutathionylated. The other four non-iron-sulfur center Cys residues remained as free thiols. Complex I glutathionylation also occurred in response to relatively mild oxidative stress caused by increased superoxide production from the respiratory chain. Although complex I glutathionylation within oxidatively stressed mitochondria correlated with loss of activity, it did not increase superoxide formation, and reversal of glutathionylation did not restore complex I activity. Comparison with the known structure of the 75-kDa ortholog Nqo3 from Thermus thermophilus complex I suggested that Cys-531 and Cys-704 are on the surface of mammalian complex I, exposed to the mitochondrial glutathione pool. These findings suggest that Cys-531 and Cys-704 may be important in preventing oxidative damage to complex I by reacting with free radicals and other damaging species, with subsequent glutathionylation recycling the thiyl radicals and sulfenic acids formed on the Cys residues back to free thiols.
Mammalian complex I is a large (∼1 MDa) protein comprising 45 subunits in the mitochondrial inner membrane that catalyzes NADH oxidation and ubiquinone reduction coupled to proton pumping across the inner membrane (1–5). Complex I is essential for oxidative phosphorylation, is a major source of reactive oxygen species (ROS),3 and damage to complex I contributes to a range of pathologies; consequently, there is considerable interest in understanding how it is affected by oxidative stress (1, 2). Glutathione (GSH) depletion, thiol reagents, and S-nitrosating agents all inhibit complex I through reaction with exposed thiols, suggesting that thiol modification is a major contributor to complex I oxidative damage (6–16). Previously, we have shown that isolated complex I contains reactive thiols on the 75- and 51-kDa subunits that become glutathionylated on exposure to glutathione disulfide (GSSG) (9–11), and this has been confirmed by others (17). The glutathione exchange enzyme glutaredoxin 2 (Grx2) (18, 19) catalyzes glutathionylation and deglutathionylation of both isolated complex I and the complex in mitochondrial membranes, even at relatively high GSH/GSSG ratios, and glutathionylation correlated with loss of complex I activity (10). As mitochondrial oxidative stress converts the mitochondrial GSH pool to GSSG, glutathionylation may contribute to the response of complex I to oxidative damage. Furthermore, there is growing interest in glutathionylation as a post-translational modification that protects proteins from oxidative damage or regulates their activity (20–24). However, it was unclear whether glutathionylation of complex I occurred within intact mitochondria; the Cys residues involved were not known, and the functional significance of the changes was uncertain (10). Here we show that complex I within intact, oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit and discuss the implications of these findings for the response of complex I to oxidative stress.
EXPERIMENTAL PROCEDURES
Materials—Rabbit antiserum against the bovine complex I 75-kDa subunit was from Prof. John E. Walker and cross-reacted with the rat 75-kDa subunit. The mouse monoclonal antibody against glutathionylated proteins (α-GSH) was from Viro-Gen Corp. Complete protease inhibitor was from Roche Applied Science. [35S]GSH (942.0 Ci/mmol) was from PerkinElmer Life Sciences, and dithiothreitol (DTT) was removed prior to experiments by extraction with ethyl acetate. His-tagged Grx2 was expressed and isolated as described (10). The biotinylated ethyl ester of glutathione was from Invitrogen. Rabbit antiserum against human sarcomeric mitochondrial creatine kinase was from AbCam and cross-reacted with the rat enzyme.
Preparation of Mitochondria, Membranes, and Complex I— Bovine and rat heart mitochondria were prepared as described (25). Heart mitochondrial preparations were stored on ice and used within 1–7 h of preparation over which time the mitochondria remained well coupled. Bovine heart mitochondrial membranes were prepared by disruption in a blender of mitochondria, prepared as described (26), followed by isolation of the membranes by centrifugation (27). Complex I was prepared by solubilization of membranes with dodecyl β-d-maltoside (DDM, Anatrace, OH) followed by ion-exchange chromatography (28). Pooled fractions were further purified by gel filtration, and the purified complex I was stored in buffer containing 0.1% DDM, 1 mm DTT, and 10% glycerol at -80 °C. Immediately prior to experiments, this buffer was replaced with one lacking DTT by centrifugation in a Micro Bio-Spin 6 chromatography column. Samples stored without DTT gave results indistinguishable from samples stored with DTT.
Glutathione Assays—GSH, GSSG, and protein-glutathione mixed disulfides (PrSSG) were assayed using the recycling assay adapted for a 96-well plate reader (29–31).
Two-dimensional Blue Native (BN)-PAGE—For BN-PAGE (32, 33), mitochondria (0.5 mg of protein) were treated with 50 mm NEM for 5 min, pelleted, and resuspended in 60 μl of extraction buffer: 1% DDM, 0.75 m ε-amino-n-caproic acid (ACA), 50 mm BisTris-HCl, pH 7.0 at 4 °C. After incubation on ice for 15 min, the suspension was clarified by centrifugation in an Airfuge™ (Beckman Coulter) at 17 p.s.i. (∼100 000 × g) for 15 min. Then 3.5 μl of sample buffer, comprising 5% (w/v) Coomassie Blue G 250 (Serva, Germany) and 500 mm ACA, was added, and samples were resolved on a 1-mm-thick 5–12% acrylamide gradient gel containing 0–20% (w/v) glycerol, 0.5 m ACA, 50 mm BisTris-HCl, pH 7.0 (4 °C), overlaid with a 3.9% acrylamide stacking gel in the same buffer. The anode buffer was 50 mm BisTris, pH 7.0, at 4 °C, and the cathode buffer was 0.02% (w/v) Coomassie Blue G-250, 50 mm Tricine, 15 mm BisTris, pH 7.0, at 4 °C. The gel was run at4 °C for 1 h at 100 V and then overnight at 40 V in cathode buffer without Coomassie Blue. Complex I bands were then excised and incubated for 5 min at room temperature in 125 mm Tris-HCl, pH 7.0, 1% SDS, and 50 mm NEM before insertion into the wells of a 3.7% acrylamide stacking gel in 0.13 m Tris-HCl, pH 6.8, overlaid on a 1-mm-thick SDS-polyacrylamide gel. SDS-PAGE was done using either a 12.5% acrylamide linear gel, or a 5–20% acrylamide gradient gel containing 0–15% (w/v) sucrose and 0.375 m Tris-HCl, pH 8.8. Stacking gel was then polymerized around the excised bands and proteins separated by electrophoresis at 100–120 V in a MiniProtean system (Bio-Rad) using 25 mm Tris, 0.192 m glycine, 0.1% SDS, pH 8.3, as running buffer.
Immunoblotting—Following SDS-PAGE, proteins were transferred to a 0.2-μm nitrocellulose membrane using a mini Trans-Blot system (Bio-Rad) in transfer buffer comprising 48 mm Tris, 39 mm glycine, 0.05% SDS, 20% (v/v) methanol, pH 8.3. The membrane was then blocked in PBST (PBS, 0.05% (v/v) Tween 20 with 1% (w/v) skimmed milk powder) and incubated with primary antiserum for 1–2 h at room temperature (αGSH, 1:1000; αCI75, 1:1000; α-creatine kinase (sarcomeric mitochondrial isoform), 1.25 μg/ml). Blots were incubated with 1:5000–1:10,000 dilutions of the appropriate secondary antiserum for 1 h at room temperature, treated with the chemiluminescent reagent (ECL or ECL Plus, Amersham Biosciences) according to the manufacturer's instructions, and visualized on Super RX photographic film (Fujifilm). To visualize biotinylated reagents, blots were incubated with ExtrAvidin-peroxidase conjugate (1:1000, Sigma) instead of a primary antibody. Blots were re-probed by stripping in 62.5 mm Tris-HCl (pH 6.8), 2% SDS, 100 mm β-mercaptoethanol for 30 min at 50 °C and then washed extensively in PBST.
Autoradiography—Radiolabeled proteins were transferred from gels to nitrocellulose as described above or the gel was dried onto filter paper. Gels or blots were then exposed to a phosphor screen (GE Healthcare), and the results were digitized with a Typhoon detector (GE Healthcare).
Protein Spot Excision and Mass Spectrometry—Following SDS-PAGE, protein bands were excised with a razor blade, transferred to a 0.5-ml tube that had been pre-washed with 50% methanol, and digested by “in-gel” cleavage (34). To do this, the gel slice was washed in high pressure liquid chromatography grade water (100 μl) for 30 min; 20 mm Tris-HCl, pH 7.0 (100 μl) for 30 min; 100 μl of 50% acetonitrile, 20 mm Tris-HCl, pH 7.0 (100 μl) for 30 min; and 100% acetonitrile (20 μl) for 10 min. The gel pieces were then dried completely in a SpeedVac at 37 °C for ∼ 20 min. The gels were rehydrated with 3–7 μl of 20 mm Tris-HCl, pH 7.0, 5 mm CaCl2 containing 12.5 ng/μl trypsin (Roche Applied Science) or with 25 ng/μl sequencing grade endoproteinase Asp-N (Roche Applied Science) in 20 mm Tris-HCl, pH 7.0, and digested overnight at 37 °C. Peptides were extracted from the gel with 4% formic acid (ARISTAR grade, Merck), 60% acetonitrile (Romil). All digests were examined in a MALDI-TOF-TOF mass spectrometer (model 4700 Proteomics Analyzer, Applied Biosystems) using α-cyano-hydroxy-trans-cinnamic acid as the matrix. For trypsin digests, the instrument was calibrated with bovine trypsin autolysis products (m/z 2163.057 and 2273.160) and a calcium related matrix ion (m/z 1060.048). For Asp-N digestions, the instrument was calibrated with Pseudomonas fragi Asp-N autolysis product (m/z 1723.743) and two complex I 75-kDa Asp-N lysis products (m/z 830.404 and 1329.753).
Peptide sequences were obtained by tandem MS in a MALDI-TOF-TOF mass spectrometer. Peptide masses and peptide fragmentation data were compared against the National Center for Biotechnology Information nr 20080103 data base against all entries using MASCOT (35). The parameters used were as follows: peptide tolerance ± 25 ppm, MS/MS tolerance ± 0.8 Da, allow for 1 missed cleavage and NEM as a variable modification. MS and tandem MS data were generally acquired automatically, and peptide fragmentation spectra were interpreted automatically with the Mascot MS-MS web-based tool (35). When glutathionylation on complex I was investigated, peaks from MALDI-TOF spectra were identified manually in Data Explorer (Applied Biosystems). Identifications were subsequently supported by MALDI-TOF-TOF data that were analyzed manually and also using the ion fragmentation calculator feature in Data Explorer software (Applied Biosystems). The digest containing the rat Cys-531 glutathionylated peptide was purified by capillary reverse phase chromatography, and a portion was analyzed by MALDI-TOF-TOF.
ROS Assays—To measure superoxide formation, bovine heart mitochondrial membranes (1 mg protein/ml) were first incubated with 1 mm DTT in KPi buffer (50 mm KPi, pH 8, 1 mm EGTA, 100 μm DTPA, 100 μm neocuproine) at 37 °C for 10 min, pelleted by centrifugation (14,000 × g for 7 min), and then washed twice in 0.5 ml of KPi buffer. Membranes (0.2 mg of protein/ml) were then incubated in KPi buffer at 30 °C with 2 μm coelenterazine (CLZ) in a luminometer (Berthold Auto-LumatPlus LB 953) with further additions as indicated in the figure legends, and the chemiluminescence was recorded for 5 s every 30 s over a 5-min incubation (25, 36). Chemiluminescence was stable for the duration of the incubation, and the average chemiluminescence was recorded.
Superoxide production by heart mitochondria was measured by two methods as follows: CLZ chemiluminescence (25, 36) and by dihydroethidine oxidation (37, 38). For the chemiluminescence measurements, mitochondria (100 μg of protein/ml) were incubated ±1 mm diamide in 120 mm KCl, 10 mm HEPES, 1 mm EDTA, 100 μm DTPA, pH 7.2, supplemented with 2 μm CLZ, and 5 mm succinate with 8 μg/ml rotenone or 10 mm glutamate/malate were used as substrates. CLZ chemiluminescence was measured as described above. For the dihydroethidine assay, mitochondria (0.5 mg of protein/ml) were incubated in 120 mm KCl, 10 mm HEPES, 1 mm EDTA, 100 μm DTPA, pH 7.2, and 10 μm dihydroethidine. Succinate (10 mm) and 4 μg/ml rotenone or 10 mm glutamate/malate were used as respiratory substrates. The mitochondria were incubated ±1 mm diamide or ±2.5 μm antimycin for 5 min, and then 0.1% Triton X-100 was added followed 1 min later by 50 μg/ml salmon sperm DNA. The fluorescence of the dihydroethidine oxidation products intercalated into DNA was measured in a 3-ml stirred cuvette at 30 °C in a Shimadzu RF-5301 fluorimeter (λex 520 nm, λem 590 nm). Control experiments showed that this amount of DNA was sufficient to maximize the fluorescence of 10 μm ethidium bromide and that the presence of diamide did not quench the fluorescence. This assay assesses the production of both 2-hydroxyethidium and ethidium, whereas production of 2-hydroethidium is specific for superoxide, there are other pathways to the formation of ethidium under conditions of oxidative stress (37, 38).
General Assays—To measure the effect of diamide on enzyme activities, rat heart mitochondria (0.5 mg of protein/ml) were incubated in KCl buffer (120 mm KCl, 10 mm HEPES, 1 mm EGTA, pH 7.2 or 7.5) at 37 °C for 5 min in the presence of 10 mm glutamate/malate and then pelleted by centrifugation. Complex I activity was measured using 20–40 μg of protein/ml at 30 °C as the rotenone-sensitive oxidation of NADH (ε340 = 6.22 mm-1·cm-1) in 50 mm KCl, 1 mm EDTA, 10 mm Tris-HCl, pH 7.4, supplemented with 300 nm antimycin, 100 μm NADH, and 50 μm decylubiquinone containing phosphatidylcholine (10 mg/ml) (39). The rotenone-sensitive rate was typically ∼90% of the uninhibited rate. Complex II/III activity was measured in 40 mm KPi, 0.5 mm EDTA, pH 7.4, 20 mm succinate, and 2 mm KCN using 20 μg of protein/ml at 30 °C. After 10 min pre-equilibration 30 μm cytochrome c was added and its rate of reduction measured (ε550 = 21 mm-1·cm-1). The background rate in the presence of 2 μm antimycin was negligible. Respiration rates were measured using a Clark-type oxygen electrode (Rank Brothers, Bottisham, Cambridge, UK). Rat heart mitochondria (1–2 mg protein/ml) were incubated in the stirred 3-ml electrode chamber at 30 °C in 120 mm KCl, 10 mm HEPES, 1 mm EGTA, pH 7.2, in the presence of 0, 0.5, or 1 mm diamide. After 2 min, 10 mm succinate in the presence of 4 μg/ml rotenone or 10 mm glutamate/malate was added, and the rate of coupled respiration was measured. After a further 2–3 min, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP; 1 μm) was added, and the rate of uncoupled respiration was measured. Protein concentration was determined by the bicinchoninic acid assay using bovine serum albumin as a standard (40).
Multiple Sequence Alignments and Comparative Modeling of the Complex I 75-kDa Subunit—Orthologous protein sequences of the subunits of complex I from Thermus thermophilus were identified in the UniProt resource using BLASTP (41). Sequences were aligned using ClustalW (42) with the T. thermophilus structure used to weight the gap penalties for the preferential placement of gaps in loop regions, rather than secondary structure elements. A set of nonredundant sequences was selected from diverse organisms to determine the extent of residue conservation across members of the protein family, including T. thermophilus, Deinococcus radiodurans, Reclinomonas americana, Bos taurus, Drosophila melanogaster, Caenorhabditis elegans, Neurospora crassa, Arabidopsis thaliana, Phytophthora infestans, Acanthamoeba castellani, and Dictyostelium discoideum. The extent of residue conservation in species more closely related to B. taurus was determined from a set, including protein sequences from mammals, Gallus gallus, Xenopus laevis, and Danio rerio. The alignment of the protein sequences for the bovine and T. thermophilus complex I 75-kDa subunits was used to identify the location in the bovine protein of the non-iron-sulfur center Cys residues in the structure. The comparative protein modeling program MODELLER (43) was used to calculate a model of subunits of bovine complex I that are orthologous to those of T. thermophilus, by using the pairwise sequence alignments. PyMOL was used to visualize and analyze the protein structures and the local environment of residues.
RESULTS AND DISCUSSION
Complex I 75-kDa Subunit Is Glutathionylated in Oxidatively Stressed Mitochondria—To induce oxidative stress, we exposed heart mitochondria to the thiol oxidant diamide, which converted GSH to GSSG and led to the formation of protein-mixed disulfides with glutathione (PrSSG) by thiol-disulfide exchange (Fig. 1A). To visualize glutathionylated mitochondrial proteins, we lysed mitochondria in the presence of the thiol alkylating reagent NEM to prevent transglutathionylation, separated the proteins by nonreducing SDS-PAGE, and probed with an antibody against glutathionylated proteins (Fig. 1B, αGSH). Diamide treatment led to the glutathionylation of a number of proteins that were reversed by the thiol reductant DTT (Fig. 1B, αGSH). Reprobing with antiserum against the complex I 75-kDa subunit showed that this subunit co-migrated with a glutathionylated protein (Fig. 1B, αCI75).
FIGURE 1.

Glutathionylation of the 75-kDa subunit complex I in oxidatively stressed mitochondria. A, rat heart mitochondria (1 mg of protein/ml) were incubated in KCl buffer with 10 mm glutamate/malate for 5 min at 37 °C ± 0.5 mm diamide. Then GSH, GSSG, and PrSSG were measured. Data are means ± S.E. of three independent experiments. Control mitochondria contained 1.0 ± 0.3 nmol of GSH equivalents (= GSH + 2GSSG + PrSSG)/mg of protein (n = 3 ± S.E.). Recovery of GSH equivalents after diamide treatment was ∼64% because of GSH loss from oxidatively stressed mitochondria and/or oxidation of GSH to higher oxidation states than GSSG. B, mitochondria were treated as in A and then resuspended in loading buffer containing 50 mm NEM (-DTT) or 100 mm DTT (+DTT), and 50 μg of protein was separated by SDS-PAGE and electrotransferred to nitrocellulose. Glutathionylated proteins were visualized using a specific antibody (αGSH), and the blot was then re-probed with an antibody against the complex I 75-kDa subunit antibody (αCI75). Samples were resolved on a single gel separated by empty lanes to prevent DTT leaching. These empty lanes have been removed, and a gray vertical line indicates where the two parts of the blots have been rejoined. The arrow indicates a minor glutathionylated protein that co-migrates with the 75-kDa subunit. C, mitochondria (1 mg of protein/ml) were incubated ± diamide as in A, and then treated ±10 mm DTT for 10 min at 37 °C followed by addition of 50 mm NEM and separation by BN-PAGE. The complex I band was then separated by nonreducing SDS-PAGE, electrotransferred to nitrocellulose, and probed with an antibody against glutathionylated proteins. D, mitochondria were incubated ± diamide as in A and then glutathionylation of complex I was assessed by BN-PAGE followed by nonreducing SDS-PAGE as in C (αGSH). The blot was then re-probed with an antibody against the 75-kDa complex I subunit (αCI75). E, mitochondria (∼20 mg of protein/ml) were preincubated with the biotinylated ethyl ester of glutathione (1.42 mm) for 30 min on ice, pelleted, and washed in 250 mm sucrose, 1 mm EGTA, 10 mm Tris-HCl, pH 7.4. The mitochondria were then incubated ± 0.5 mm diamide as in A, and biotinylation of complex I was assessed following BN-PAGE and nonreducing SDS-PAGE as in C by probing with ExtrAvidin. The blot was then re-probed with an antibody against the complex I 75-kDa subunit antibody (αCI75). F, mitochondria (∼20 mg of protein/ml) were first incubated with [35S]GSH (40 μCi/ml; 37 nm) in STE for 30 min on ice, pelleted, and washed. The [35S]GSH-loaded mitochondria were then incubated ± diamide, separated, and electrotransferred onto nitrocellulose as in C, and [35S]GSH-containing protein bands were detected using a PhosphorImager. The blot was then re-probed with an antibody against the complex I 75-kDa subunit (αCI75). G, [35S]GSH-loaded mitochondria were incubated ± diamide, and complex I was separated by BN-PAGE followed by nonreducing SDS-PAGE as in F. The gel was dried, and [35S]GSH-containing protein bands were detected using a PhosphorImager ([35S]GSH). Two bands labeled by [35S]GSH (gel, bands A and B) were excised, and identification was attempted by peptide mass fingerprinting. H, band A from G above was analyzed by peptide mass fingerprinting following in-gel digestion with trypsin. A Mascot score greater than 80 is significant (p < 0.05). Additional evidence for the identification of the 75-kDa subunit is provided by the analysis of three peptides by tandem MS (MALDI-TOF-TOF). The MASCOT ion scores for the three indicated peptides are listed. There was no significant identification for band B.
To analyze complex I, the intact protein was isolated from oxidatively stressed mitochondria by BN-PAGE and then dissociated into its subunits by nonreducing SDS-PAGE (Fig. 1C). An antibody against glutathionylated proteins revealed a single, DTT-sensitive band present in diamide-treated but not control samples (Fig. 1C). Reanalysis with antiserum against the bovine complex I 75-kDa subunit showed that the glutathionylated protein co-migrated with the 75-kDa subunit (Fig. 1D). Although SDS-PAGE used in the second dimension may not resolve all complex I polypeptides (44, 45), the full complement of complex I subunits should be present. Protein glutathionylation was analyzed by an alternative method using the membrane-permeant biotinylated ethyl ester of glutathione. After incubation with this glutathione derivative, the mitochondria were washed and treated with diamide, and then complex I was isolated as before by BN-PAGE followed by nonreducing SDS-PAGE (Fig. 1E). Diamide treatment led to the DTT-sensitive binding of biotinylated glutathione to a protein that co-migrated with the complex I 75-kDa subunit (Fig. 1E). To further investigate glutathionylation, we preincubated mitochondria with [35S]GSH (46) and after washing exposed them to diamide. Complex I was isolated by BN-PAGE, and following nonreducing SDS-PAGE, [35S]GSH binding was assessed (Fig. 1F). This showed a DTT-sensitive [35S]GSH band at about 75-kDa that co-migrated with the complex I 75-kDa subunit (Fig. 1F). Another [35S]GSH-labeled band at ∼45 kDa was not detected using the αGSH antibody or biotinylated glutathione ester. To identify the [35S]GSH-labeled proteins, the gel was dried, and [35S]GSH-containing bands were visualized, and the bands at ∼75 and ∼45 kDa were excised (Fig. 1G). Peptide mass fingerprinting and tandem mass spectrometry confirmed that one band was the complex I 75-kDa subunit (Fig. 1H). No identification was made of the [35S]GSH-containing band at ∼45 kDa; however, other experiments (supplemental Fig. 1) suggested that it was sarcomeric creatine kinase, a protein that is known to become glutathionylated (47).
Previous work showed glutathionylation of the 75- and 51-kDa subunits of purified complex I on incubation with GSSG (9–11, 17). However, when GSSG binding to purified complex I was investigated using low concentrations of radiolabeled or biotinylated GSH, the 75-kDa subunit predominated, along with a number of other minor bands, whereas higher GSSG concentrations were required for glutathionylation of the 51-kDa subunit (data not shown). As incubation of purified complex I with GSSG leads to artifactual glutathionylation because of denaturation of the complex (9), glutathionylation of the 51-kDa subunit may only occur in vitro. Our findings confirm that complex I is glutathionylated on the 75-kDa subunit within oxidatively stressed mitochondria and suggest that this is the only complex I subunit glutathionylated.
Identification of the Cysteine Residues Glutathionylated on the 75-kDa Subunit of Purified Complex I—The bovine (P15690) and rat (Q66HF1) complex I 75-kDa subunits contain a 23-amino acid N-terminal targeting sequence that is removed after mitochondrial import leaving 704 amino acid polypeptides that are 94.8% identical (48). The mature 75-kDa polypeptides contain 17 Cys residues, with 11 (Cys-41, -52, -54, -69, -105, -108, -114, -153, -156, -159, and -203) involved in the 75-kDa subunit three iron-sulfur centers (Fig. 2) (2, 4). The remaining six Cys residues (Cys-30, -344, -531, -541, -687, and -704) are potential candidates for glutathionylation and are shown in red in Fig. 2.
FIGURE 2.

Amino acid sequences of the mature bovine complex I 75-kDa subunit. The primary sequence of mature bovine complex I 75-kDa subunit is shown (P15690). The location of the Cys residues is the same in the corresponding rat sequence (Q66HF1). The cysteine-containing peptides identified by MALDI-TOF following proteolysis of complex I are marked by gray boxes. The 11 Cys residues involved in iron-sulfur centers are shown in yellow typeface. The six Cys residues that are not involved in iron-sulfur centers are in red and are surrounded by yellow boxes.
To determine whether mass spectrometry could be used to reliably assess glutathionylation of these Cys residues, purified bovine complex I was reduced and denatured in SDS, and protein thiols were alkylated with NEM, and the 75-kDa subunit was separated by nonreducing SDS-PAGE. Following in-gel proteolysis with Asp-N or trypsin, peptide masses were measured by MALDI-TOF-MS (Table 1), and their sequences were confirmed by tandem mass spectrometry in a MALDI-TOF-TOF (supplemental Table 1). We detected 12 of the 15 Cys-containing peptides in their NEM-modified forms, accounting for 13 of the 17 Cys residues (Table 1). The four Cys residues not detected were all involved in the coordination of iron-sulfur centers (Table 1), and consequently were unlikely candidates for glutathionylation.
TABLE 1.
MALDI-TOF analysis of NEM-alkylated, Cys-containing peptides from the bovine and rat complex I 75-kDa subunit Experimental masses were obtained from isolated and denatured complex I (2 mg/ml) treated with DTT (10 mm) and SDS (1%) for 10 min at 37°C; the DTT was removed; and the samples were treated with NEM. Bovine or rat mitochondria (1 mg of protein/ml) were incubated with diamide (0.5 mm) for 5 min at 37°C. After incubation all samples were alkylated with NEM (25–50 mm) and separated by electrophoresis (one-dimensional SDS-PAGE for isolated complex I samples and two-dimensional BN/SDS-PAGE for mitochondria) before in-gel digestion and MALDI-TOF.
|
Cys
no.a |
Sequenceb |
Enzymec |
FeSd |
Mass of NEM-alkylated peptide (MH+) |
|||
|---|---|---|---|---|---|---|---|
|
Calculatede |
Observed |
||||||
| Denatured complex I | Bovine mitochondria (b) | Rat mitochondria (r) | |||||
| 30 | TATAASNLIEVFVDGQSVMVEPGTTVLQACEK (b) | T | No | 3433.6865 | 3433.6977 | 3433.6301 | |
| TGTAASNLIEVFVDGQSVMVEPGTTVLQACEK (r) | 3419.6707 | 3419.7310 | |||||
| 41 | FCYHER | T | Yes | 979.4090 | 979.4006 | 979.4030 | 979.4123 |
| 52 | LSVAGNCR | T | Yes | 944.4618 | 944.4646 | 944.4555 | 944.4660 |
| 55 | MCLVEIEK | T | Yes | 1089.5319 | 1089.5171 | 1089.5236 | 1089.5527 |
| 69 | VVAACAMPVMK | T | Yes | 1244.6200 | 1244.6146 | 1244.6105 | 1244.6212 |
| 105,108 | DCPIC or DCPIC | D | Yes | 675.2475 | |||
| DCPIC | 800.2945 | ||||||
| 114 | DQGGEC | D | Yes | 733.2456 | |||
| 153,156 | CIQCTR or CIQCTR | T | Yes | 848.3753 | 848.3709 | 848.3825 | 848.3458 |
| CIQCTR | 973.4230 | 973.4194 | 973.4202 | 973.4334 | |||
| 159 | CIR | T | Yes | 516.2597 | |||
| 203 | DICPVGALTSKPYAFTARPW | D | Yes | 2318.1564 | 2318.1790 | 2318.2058 | 2318.2073 |
| 344 | VDSDTLCTEEVFPTAGAGTDLR (b) | T | No | 2422.1078 | 2422.0918 | 2422.0857 | |
| VDSDTLCTEEIFPNEGAGTDLR (r) | 2507.1241 | 2507.1228 | |||||
| 531 | MLFLLGADGGCITR (b) | T | No | 1591.7970 | 1591.7957 | 1591.7982 | |
| LLFLLGADGGCITR (r) | 1573.8405 | 1573.8358 | |||||
| 541 | DCFIVYQGHHG | D | No | 1400.5979 | 1400.5907 | 1400.6039 | 1400.6027 |
| 687 | DSISRASQTMAKCVKAVTEGAHAVE (b) | D | No | 2714.3163 | 2714.3508 | 2714.3940 | |
| DSISRASQTMAKCVKAVTEGAQAVE (r) | 2705.3231 | 2705.3904 | |||||
| 704 | AVTEGAHAVEEPSIC (b) | T | No | 1637.7475 | 1637.7476 | 1637.7484 | |
| AVTEGAQAVEEPSIC (r) (–NEM) | 1503.6995 | 1503.6367 | |||||
Cysteine residue numbers are shown, which are the same for bovine and rat sequences
Unless specified as bovine (b) or rat (r), peptide sequences are the same for both species
T indicates trypsin, and D indicates Asp-N
Data indicate whether the cysteine residue is part of an iron-sulfur cluster
Calculated masses of peptides were alkylated with NEM (+125.047 Da)
To see if glutathionylated peptides could be detected, we reduced complex I with DTT and then denatured it with SDS. After removing the DTT, the denatured complex was incubated with 20 mm GSSG for 30 min, followed by NEM to prevent subsequent transglutathionylation. After nonreducing SDS-PAGE and proteolytic digestion, the Cys-containing peptides from the 75-kDa subunit were analyzed by MALDI-TOF-MS for a +305 m/z shift consistent with glutathionylation (Table 2). Eight of the 15 peptides containing Cys residues were detected as glutathionylated peptides (Table 2), and in all cases glutathionylation was reversed by treatment with DTT (data not shown). These Cys residues included all six of the non-iron-sulfur center Cys residues. Glutathionylation of Cys-41 and Cys-203, which are components of iron-sulfur centers, was also detected, suggesting that these residues became available for modification on denaturation of purified complex I.
TABLE 2.
MALDI-TOF analysis of glutathionylated, Cys-containing peptides from the bovine and rat complex I 75 kDa subunit Experimental masses were obtained from isolated and denatured complex I (2 mg/ml) and treated with DTT (10 mm) and SDS (1%) for 10 min at 37°C; the DTT was removed; and the samples were treated with GSSG (20 mm) for 30 min at 37°C. For native complex I, isolated bovine complex I was incubated with GSSG (20 mm) for 30 min at 37°C; bovine or rat mitochondria (1 mg of protein/ml) were incubated with diamide (0.5 mm) for 5 min at 37°C. After incubation all samples were alkylated with NEM (25–50 mm) and separated by electrophoresis (one-dimensional SDS-PAGE for isolated CI samples and two-dimensional BN/SDS-PAGE for mitochondria) before in-gel digestion and MALDI-TOF.
|
Cys
no.a |
Sequenceb |
Enzymec |
FeSd |
Mass (MH+) + GSSG (305.068) |
||||
|---|---|---|---|---|---|---|---|---|
|
Calculatede |
Observed |
|||||||
| Denatured complex I | Native complex I | Bovine mitochondria (b) | Rat mitochondria (r) | |||||
| 30 | TATAASNLIEVFVDGQSVMVEPGTTVLQACEK (b) | T | No | 3613.7074 | 3613.7356 | |||
| TGTAASNLIEVFVDGQSVMVEPGTTVLQACEK (r) | 3559.6917 | |||||||
| 41 | FCYHER | T | Yes | 1159.4295 | 1159.4165 | |||
| 52 | LSVAGNCR | T | Yes | 1124.4822 | ||||
| 55 | MCLVEIEK | T | Yes | 1269.5529 | ||||
| 69 | VVAACAMPVMK | T | Yes | 1424.6405 | ||||
| 105,108 | DCPIC or DCPIC | D | Yes | 980.3164 | ||||
| DCPIC | 1160.3368 | |||||||
| 114 | DQGGEC | D | Yes | 913.2668 | ||||
| 153,156 | CIQCTR or CIQCTR | T | Yes | 1403.5430 | ||||
| CIQCTR | 1583.5630 | |||||||
| 159 | CIR | T | Yes | 696.2809 | ||||
| 203 | DICPVGALTSKPYAFTARPW | D | Yes | 2498.1842 | 2498.1648 | 2498.1743 | ||
| 344 | VDSDTLCTEEVFPTAGAGTDLR (b) | T | No | 2602.1135 | 2602.1125 | |||
| VDSDTLCTEEIFPNEGAGTDLR (r) | 2687.1451 | |||||||
| 531 | MLFLLGADGGCITR (b) | T | No | 1771.8175 | 1771.8016 | 1771.8175 | 1771.8191 | |
| LLFLLGADGGCITR (r) | 1753.8453 | 1753.8760 | ||||||
| 541 | DCFIVYQGHHG | D | No | 1580.6261 | 1580.6172 | 1580.6201 | ||
| 687 | DSISRASQTMAKCVKAVTEGAHAVE (b) | D | No | 2894.3440 | 2894.3035 | 2894.3816 | ||
| DSISRASQTMAKCVKAVTEGAQAVE (r) | 2885.3441 | |||||||
| 704 | AVTEGAHAVEEPSIC (b) | T | No | 1817.7680 | 1817.7848 | 1817.7869 | 1817.7720 | |
| AVTEGAQAVEEPSIC (r) | 1808.7681 | |||||||
Cysteine residue numbers are shown, which are the same for bovine and rat sequences
Unless specified as bovine (b) or rat (r), peptide sequences are the same for both species
T indicates trypsin, and D indicates Asp-N
Data indicate whether the cysteine residue is part of an iron-sulfur cluster
Calculated masses of peptides were glutathionylated on the Cys residues (+305.068 Da)
Next, glutathionylation of Cys residues in native complex I was investigated by incubating intact complex I with 20 mm GSSG followed by NEM, and then protease digests of the 75-kDa subunit were analyzed by mass spectrometry. Five Cys residues (Cys-203, -531, -541, -687, and -704) were glutathionylated in the native complex. Cys-203 is involved with iron-sulfur center ligation, suggesting some denaturation of the complex on incubation with GSSG. When intact complex I was exposed to a lower concentration of GSSG (5 mm) only three Cys residues (Cys–531, -687, and -704) were glutathionylated (data not shown). This identifies Cys residues 531, 687, and 704 of the 75-kDa subunit as plausible candidates for glutathionylation within intact mitochondria.
Recently Cys-344 (Cys-367 in the precursor sequence) of purified bovine complex I 75-kDa subunit was reported to be glutathionylated on exposure to GSSG (17). We found that this residue was only glutathionylated in denatured, purified complex I and that it was not glutathionylated in native, purified complex I or in diamide-treated mitochondria, nor was there decreased recovery of NEM-alkylated Cys-344. In Ref. 17, the sequence coverage of the 75-kDa subunit was limited so that Cys-344 was the only non-iron-sulfur center Cys residues investigated, so glutathionylation of the other five Cys residues would not have been detected (17). It is possible that the purified complex I preparations used were somewhat denatured, giving a glutathionylation pattern similar to that of our SDS-treated complex I preparations (Table 2).
Identification of Glutathionylated Peptides from the 75-kDa Subunit of Complex I within Oxidatively Stressed Mitochondria— To assess glutathionylation within mitochondria of the 75-kDa subunit, bovine and rat heart mitochondria were incubated ± diamide, then the remaining free Cys residues were blocked with NEM, and complex I was isolated by BN-PAGE followed by nonreducing SDS-PAGE. Peptides from the 75-kDa subunit were analyzed for NEM and glutathione modification by MALDI-TOF-MS (Tables 1 and 2), with confirmation of the peptide sequences by tandem mass spectrometry (supplemental Table 1).
In the absence of diamide, 12 of the 15 Cys-containing peptides in the 75-kDa subunit of complex I were detected with NEM modifications by MALDI-TOF. After treatment of bovine mitochondria with diamide, analysis of digests of the 75-kDa subunit revealed that peptides containing Cys-531 and Cys-704 were glutathionylated (Table 2). Similar experiments in rat heart mitochondria also showed that Cys-531 was glutathionylated. However, glutathionylation of Cys-704 in that rat 75-kDa subunit was not detected, although the amount of peptide containing unmodified Cys-704 was decreased by diamide.
Portions of typical MALDI-TOF mass spectra for peptides containing Cys-531 or Cys-704 from bovine heart mitochondria incubated ± diamide are shown in Fig. 3. In the presence of diamide there are significant peaks at m/z 1771.8 and at m/z 1817.8 (Fig. 3), corresponding to monoglutathionylation of the peptides containing Cys-531 and Cys-704, respectively. In mitochondria that had not been incubated with diamide, there was no detectable glutathionylation of peptides containing these two Cys residues, and subsequent DTT treatment removed these glutathionylated peaks (Fig. 3). These data are consistent with glutathionylation of Cys-531 and Cys-704 within oxidatively stressed bovine heart mitochondria.
FIGURE 3.

Analysis by mass spectrometry of glutathionylated peptides from the complex I 75-kDa subunit in oxidatively stressed mitochondria. Bovine heart mitochondria were incubated ± diamide; the 75-kDa subunit of complex I was then isolated by BN-PAGE followed by nonreducing SDS-PAGE as described in Fig. 1. After protease digestion, the peptides containing Cys-531 and Cys-704 were analyzed for glutathionylation by MALDI-TOF-MS as described in Table 2. An 8-Da portion of the mass spectra from the analysis of the modified peptides is shown. Data shown for + diamide and - diamide traces are typical MALDI-TOF traces, repeated at least three times with the same result.
Location of Modified Cysteine Residues within Glutathionylated Peptides—Glutathionylated peptides containing Cys-531 and Cys-704 from diamide-treated mitochondria were analyzed by tandem mass spectrometry (MALDI-TOF-TOF) to determine the sites of glutathione modification. In addition to the conventional b and y ion series, further fragmentations adjacent to the disulfide bond of the glutathionylated Cys residues were observed and are illustrated in Fig. 4A (49). Fragmentation of the 1771.8-Da peptide, corresponding to glutathionylation of the bovine Cys-531 peptide, gave a y*-type ion series for y4 and above, consistent with glutathionylation of Cys-531 (Fig. 4B). Additional y-type ions were present consistent with fragmentation at an amide bond within the glutathione adduct or on either side of the disulfide bond linking the adduct to Cys-531 (Fig. 4B, yΔ, yo, y†). Fragmentation of the 1753.8 peptide, corresponding to glutathionylation of the rat Cys-531 peptide, also showed a y* series consistent with glutathionylation of Cys-531 (data not shown). Fragmentation of the 1817.8-Da peptide, corresponding to glutathionylation of the bovine Cys-704 peptide, gave a y* series consistent with glutathionylation of Cys-704, along with other y series consistent with fragmentation within the glutathione adduct (Fig. 4C). The b series ions, notably b14, contained no glutathione demonstrating the glutathionylation of the terminal Cys-704 residue. The fragmentation patterns for the NEM-alkylated peptides containing Cys-531 and Cys-704 (1591.8 and 1637.7 Da, respectively) from purified complex I were similar to those seen for the glutathionylated peptides (data not shown), indicating that glutathionylation does not distort the peptide fragmentation pattern. Therefore, tandem mass spectrometry identifies the modification sites in these peptides and confirms glutathionylation of both Cys-531 and Cys-704 of the 75-kDa subunit in diamide-treated bovine heart mitochondria and glutathionylation of Cys-531 in diamide-treated rat heart mitochondria.
FIGURE 4.

Fragment ion spectra of glutathionylated peptides from complex I 75-kDa subunit containing Cys-531 and Cys-704. A, structure of a Cys-containing peptide modified by a disulfide link to glutathione. The sites of fragmentation observed in tandem mass spectrometry experiments are also shown. In subsequent fragment ion spectra, the asterisk indicates fragment ions containing an intact glutathione molecule, whereas the superscripts Δ, o, or † indicate ions resulting from fragmentation at the bond indicated. B and D, MALDI-TOF-TOF spectra of the glutathionylated peptides obtained from heart mitochondria incubated with diamide as in Fig. 1 with complex I 75-kDa subunit prepared for mass spectrometry as described in Table 2. B, fragment ion spectrum of a 1771.8 ion corresponding to a glutathionylated peptide containing Cys-531 from bovine complex I. C, fragment spectrum of a 1817.8 Da ion corresponding to a glutathionylated peptide containing Cys-704 from bovine complex I.
Estimation of the Extent of Cysteine Residue Modification in the Complex I 75-kDa Subunit within Oxidatively Stressed Mitochondria—The experiments so far have identified the glutathionylated Cys residues within the complex I 75-kDa subunit, but the extent of their modification was unclear. This was estimated by quantifying the loss of the Cys residue free thiol, assessed by measuring decreased NEM alkylation. Any unmodified Cys residues should be equally reactive with NEM in diamide-treated and control samples. In contrast, glutathionylation of a Cys residue should lead to decreased recovery of the NEM-alkylated peptide that is DTT-sensitive. However, one caveat is that a glutathionylated peptides that broke down during analysis to yield a free thiol that then reacted with NEM would not be detected.
To do this, mitochondria were incubated ± diamide and then treated ± DTT before addition of NEM. Then complex I was isolated by BN-PAGE; the 75-kDa subunit was separated by nonreducing SDS-PAGE, and the NEM-alkylated peptides containing the six non-iron-sulfur center Cys residues (30, 344, 531, 541, 687, and 704) were analyzed by MALDI-TOF-MS. To account for differential peptide recovery, all peak intensities were normalized relative to a non-Cys-containing peptide from the 75-kDa subunit (FEAPLFNAR, [MH+] 1064.6 Da for trypsin digests and DVVLLVGTNPRF [MH+] 1329.8 Da for Asp-N digests). Representative experiments for the NEM-alkylated peptide containing Cys-531 (1591.8 Da) and Cys-704 (1637.7 Da) from bovine heart mitochondria are shown in Fig. 5A. The peak intensity decreased on diamide treatment (+ diamide) and was recovered on subsequent incubation with DTT (+ diamide/DTT) (Fig. 5A). This approach was applied to all six candidate Cys residues of the 75-kDa subunit in bovine heart mitochondria, and the changes in peak intensity of the relevant NEM peptide were determined as a percentage of control mitochondrial incubations (Fig. 5B). Diamide led to a statistically significant decrease of ∼55% in both the Cys-531 and Cys-704 residues that was reversed by DTT, consistent with extensive glutathionylation of these residues. In contrast, no significant loss of NEM-alkylated peak intensity was observed on diamide treatment for peptides containing Cys residues 30, 344, 541, or 687 (Fig. 5B). Similar analysis of rat heart mitochondria showed that there was an ∼50% decrease in the intensity of alkylation of the Cys-531 residue that was largely reversed by DTT (data not shown). In rat mitochondria, Cys-704 reacted poorly with NEM, so instead the amount of the unalkylated peptide (1503.7 Da) was assessed, and its amount decreased by ∼63% on diamide treatment (data not shown). That these losses of the NEM-alkylated peptides were reversed by DTT treatment indicates that the modification is unlikely to be irreversible oxidation to a sulfinic or sulfonic acid. Furthermore, modification of peptides containing Cys-531 and Cys-704 to sulfenic, sulfinic, or sulfonic acids was searched for but none was found in mass spectra. Therefore, these data are consistent with extensive glutathionylation of Cys-531 and Cys-704 in oxidatively stressed bovine heart mitochondria occurring with minimal modification of other Cys residues in the 75-kDa subunit of complex I. However, the possibility remains that other DTT-reversible modifications may also contribute to the loss of the NEM-alkylated peptides seen in Fig. 5.
FIGURE 5.

Quantification of the extent of oxidation of complex I 75-kDa subunit Cys residues during diamide treatment of mitochondria. Heart mitochondria were incubated ± diamide; some diamide-treated samples were further incubated with DTT, and then all samples were treated with NEM. The peptides containing NEM-alkylated Cys residues were then analyzed by MALDI-TOF, and their peak intensities are expressed as a percentage of an appropriate non-Cys containing reference peptide peak from the 75-kDa subunit digest. A, representative MALDI-TOF spectra of the NEM-alkylated peptides containing Cys-531 (1591.8 Da) or Cys-704 (1637.7 Da) from control, diamide-treated, and diamide- and DTT-treated bovine heart mitochondria. B, effect of treatment of bovine heart mitochondria with diamide or with diamide and DTT on peak intensities of NEM-alkylated peptides. Peak intensities are plotted as a percentage of the control incubation (-diamide) for that experiment. Data are the means ± S.E. of three to four independent experiments, except for the +diamide/DTT experiments, which were done once. *, p ≤ 0.05 relative to control samples by a paired Student's t test.
Glutathionylation of Cys Residues on the 75-kDa of Complex I by Endogenous Oxidative Stress within Mitochondria—The proximal ROS produced by the respiratory chain is superoxide, which rapidly dismutates to H2O2 (50). Therefore, we next exposed bovine heart mitochondria to oxidative stress in ways designed to mimic the types and amounts of ROS encountered by complex I in pathological situations. This was done by adding H2O2 to mitochondria, or by inducing superoxide production from the respiratory chain at complex I by reverse electron transport or at complex III using antimycin (51). The glutathionylation of the complex I 75-kDa subunit at Cys-531 and Cys-704 was then assessed as before by MALDI-TOF-MS.
Induction of superoxide production by antimycin or by RET, and by direct addition of H2O2, all led to the glutathionylation of Cys-704 (Fig. 6A), with only minor changes in Cys-531 (data not shown). Measurement of these changes over multiple experiments confirmed that these forms of ROS led to statistically significant glutathionylation of Cys-704 (Fig. 6B). Therefore, glutathionylation of Cys-704 occurs in response to physiologically relevant types and amounts of mitochondrial ROS.
FIGURE 6.

Assessment by mass spectrometry of glutathionylation of complex I in mitochondria exposed to endogenous oxidative stress. Bovine heart mitochondria (1 mg of protein/ml) were incubated for 5 min at 37 °C. Reverse electron transport (RET) incubations were done in 250 mm sucrose, 10 mm Hepes, 1 mm EGTA, pH 7.4, supplemented with 0.01% bovine serum albumin and 10 mm succinate. Antimycin incubations also contained antimycin (2.5 μm) and FCCP (400 nm), whereas control incubations were supplemented with FCCP (400 nm) alone. Incubations with 25 μm H2O2 were done in KCl buffer (pH 7.2). Complex I was assessed for glutathionylation of the 75-kDa subunit Cys-531 and Cys-704 residues by mass spectrometry as described in Fig. 1 and Table 2. A, representative MALDI-TOF spectra of the glutathionylated peptide containing Cys-704 is shown with peak intensities expressed as a percentage of a non-Cys containing reference peptide peak from the 75-kDa subunit (FEAPLFNAR, [MH+] 1064.6 Da). B, measurement of the effects of treatment on the peak intensities of the glutathionylated peptides containing the Cys-531 and Cys-704 residues. Peak intensities are plotted as a percentage of the control incubation and are the means ± S.E. of three independent experiments. *, p ≤ 0.05 relative to control samples by paired Student's t test.
Effect of Complex I Glutathionylation on ROS Production—Earlier we reported that incubation of complex I with 20 mm GSSG increased superoxide formation during oxidation of NADH, as measured by SOD-sensitive reduction of acetylated cytochrome c (9). Recently, another group using both spin trapping and acetylated cytochrome c to measure superoxide showed that 1–3 mm GSSG did not increase superoxide production by isolated complex I (17). Therefore we reassessed how complex I glutathionylation affected superoxide production by measuring the SOD-sensitive CLZ chemiluminescence by mitochondrial membranes respiring on NADH (25, 36). Addition of the complex I inhibitor rotenone increased superoxide formation, indicating that superoxide production from complex I was easily detected by this approach (Fig. 7A). However, incubation with GSSG did not increase superoxide production, and further addition of Grx2 to accelerate glutathionylation (Fig. 7A) or varying [GSSG] from 100 μm to 5 mm (data not shown) were also ineffective. There is extensive glutathionylation of complex I under these conditions (Ref. 10 and data not shown) indicating that glutathionylation of complex I does not necessarily lead to increased superoxide formation. Possible causes of the discrepancy between these findings and our earlier work may include the high GSSG concentrations used, changes in the NADH/NAD ratio, and decomposition of complex I during incubations (data not shown).
FIGURE 7.

Effects of glutathionylation on complex I function. A, superoxide formation by bovine mitochondrial membranes was determined by measuring CLZ chemiluminescence. Mitochondrial membranes were incubated with 1 mm NADH, 500 μm GSSG, 4 μg/ml rotenone, 1 unit/ml Grx2, or 100 units/ml CuZn-SOD as indicated. Data are means ± S.E. of three to five independent experiments, each determined in triplicate. RLU = relative light units. B, oxidation of dihydroethidine by rat heart mitochondria incubated with diamide (1 mm) or antimycin (2.5 μm). Data are means ± range of two independent experiments. AFU = arbitrary fluorescence units. C, rat heart mitochondria (0.5 mg protein/ml) were incubated in 120 mm KCl, 10 mm HEPES, 1 mm EGTA, pH 7.5, at 37 °C for 5 min with 10 mm glutamate/malate with the indicated concentrations of diamide. The activity of complex I and of complexes II/III were measured. Data are the means ± S.D. of three independent measurements, each determined in triplicate. D, rat heart mitochondria were incubated with 0.5 mm diamide for 5 min as in C and pelleted by centrifugation, and then the mitochondria were broken by three cycles of freeze/thawing and incubated with DTT (1 mm) or β-mercaptoethanol (5 mm) for 2 min, when complex I activity was measured. Data are the means ± S.D. of three independent determinations, each in duplicate. BME = β-mercaptoethanol. E, irreversible inhibition of complex I after diamide treatment. Mitochondria were incubated with 0.5 mm diamide at 37 °C for 5 min, pelleted to remove the diamide, and incubated at 25 °C in KCl buffer (pH 7.5) with 10 mm glutamate/malate for 0–30 min, and then complex I activity was measured. Data are means ± S.D. of three measurements. F and G, effect of diamide on GSSG levels in rat heart mitochondria. Mitochondria were incubated with diamide as in E (Pre-wash). The diamide-treated mitochondria were then washed and incubated in KCl buffer (pH 7.5) with 10 mm glutamate/malate (Recovery) for 0–30 min and assayed for GSH and GSSG. F shows the GSH and GSSG contents, and G shows the GSH/GSSG ratio. Data are means ± S.D. of six incubations.
Treatment of isolated mitochondria with tert-butylhydroperoxide increased H2O2 efflux, consistent with increased superoxide formation by glutathionylated complex I (9). However, H2O2 efflux is an indirect measure of superoxide formation as changes in the activity of thiol-dependent mitochondrial H2O2 degradation pathways such as peroxiredoxin III may also contribute, and glutathione depletion by 1-chloro-2,4-dinitrobenzene has been shown to increase hydrogen peroxide efflux from heart mitochondria (52). Therefore, we measured mitochondrial ROS production more directly by dihydroethidine oxidation to 2-hydroxyethidium or ethidium, which we detected by their fluorescence in the presence of excess DNA (Fig. 7B) (37, 38). Addition of antimycin to mitochondria respiring on succinate increased dihydroethidine oxidation, consistent with increased superoxide formation by mitochondria. However, incubation of mitochondria respiring on succinate or on glutamate/malate with various concentrations of diamide for 5 min (1 mm, Fig. 7B, 10, 100, and 250 μm; data not shown) did not increase dihydroethidine oxidation. Mitochondria incubated with CLZ showed an increase in chemiluminescence on addition of antimycin (data not shown); however, addition of 0.5 mm diamide to mitochondria respiring on succinate or glutamate/malate did not increase CLZ chemiluminescence (data not shown). As diamide treatment of mitochondria under these conditions leads to extensive glutathionylation of complex I, these data, in contrast to our earlier suggestion (9), show that complex I glutathionylation within intact mitochondria does not correlate with increased superoxide formation.
Effect of Mitochondrial Exposure to Diamide on Complex I Activity—Exposure of mitochondrial membranes to GSSG, or to the disulfides cystine and β-hydroxyethylenedisulfide, leads to the loss of complex I activity (9, 10). Interestingly, this loss of activity is not ameliorated by treatment with thiol reductants, even if complex I glutathionylation is reversed (10). To see if there was a similar relationship between complex I activity and exposure to an oxidized glutathione pool within mitochondria, we exposed rat heart mitochondria to various concentrations of diamide for 5 min (Fig. 7C). This led to extensive loss of complex I and of complex II/III activity (Fig. 7C). Exposure of isolated mitochondria to diamide (0.5–2 mm) also decreased both coupled and phosphorylating respiration on succinate or glutamate/malate (data not shown). These data are consistent with diamide exposure leading to oxidation of the mitochondrial glutathione pool with effects on a range of mitochondrial processes.
To see if the loss of complex I activity on diamide treatment was readily reversible, we incubated mitochondria with diamide, lysed the mitochondria, and exposed them to the thiol reductants DTT, β-mercaptoethanol (Fig. 7D), or tricarboxyethylphosphine (data not shown). None of these treatments restored the activity of complex I (Fig. 7D) or of complex II/III (data not shown), even though these treatments readily removed glutathione residues from complex I (Figs. 1 and 3) (data not shown). To see if restoration of the glutathione pool within intact mitochondria could result in reactivation of complex I, we first exposed mitochondria to diamide and then washed them to remove the oxidant (Fig. 7E). Subsequent incubation of these mitochondria for up to 1 h did not lead to restoration of complex I activity, even though the mitochondrial glutathione pool was partially restored (Fig. 7, F and G).
Therefore within intact mitochondria, exposure to the thiol oxidant diamide leads to the rapid inactivation of complex I; however, subsequent exposure to thiol reductants does not restore complex I activity. This is similar to the situation for complex I exposed to GSSG in membranes and indicates that exposure to an oxidized glutathione pool rapidly leads to the inactivation of complex I, but there is not a simple correlation between complex I glutathionylation and its activity. Even so, complex I glutathionylation may affect its function by other mechanisms, such as by the recruitment of binding partners.
Location of Glutathionylated Cys Residues on Complex I—Of the non-iron-sulfur Cys residues, Cys-30 and Cys-344 were not glutathionylated in isolated complex I; Cys residues 531, 541, 687, and 704 were glutathionylated in isolated complex I, whereas only Cys-531 and Cys-704 were glutathionylated in intact mitochondria. The six non-iron-sulfur center Cys residues in the bovine and rat complex I 75-kDa subunit are conserved in mammals, G. gallus, and X. laevis, and all except Cys-541 are conserved in D. rerio. However only Cys-30 and Cys-687 are conserved more widely; Cys-30 is conserved in R. americana, C. elegans, N. crassa, A. thaliana, and D. discoideum; Cys-687 is conserved in D. melanogaster, N. crassa, A. thaliana, and A. castellanii, but not in C. elegans or D. discoideum. The location and environment of these Cys residues within complex I may indicate why there was residue-selective glutathionylation. The structure of mammalian complex I is not known, but that of the hydrophilic arm of complex I from T. thermophilus has been determined (4, 53) (Protein Data Bank 2FUG). Comparisons of the sequence alignments between orthologous subunits of bovine and Thermus complex I, and a model of the bovine complex I built by comparative modeling with the T. thermophilus structure, were used to identify the location of the six non-iron-sulfur center Cys residues (Fig. 8). The model of the structure of bovine complex I derived from the T. thermophilus structure is shown in Fig. 8A with the 75-kDa subunit shown in black. The six non-iron-sulfur center Cys residues shown in Fig. 8A are colored yellow. Cysteine residues 344, 531, 541, 687, and 704 are close to each other and are surrounded by a large red oval in Fig. 8A, whereas Cys-30 is surrounded by a small red circle. The iron-sulfur centers are indicated as yellow and gray spheres in Fig. 8A. The flavin mononucleotide where NADH is oxidized is indicated, and the probable locations of the mitochondrial inner membrane and the coenzyme Q-binding sites are indicated.
FIGURE 8.

Model of the bovine complex I 75-kDa subunit. A model comprising subunits of the bovine complex I that are orthologous to T. thermophilus subunits was built by using comparative modeling. A, structure of the bovine complex I. The 75-kDa subunit is colored dark gray, and the six noniron-sulfur Cys residues are shown with a yellow stick representation and outlined with a red oval (Cys-344, Cys-531, Cys-541, Cys-687, and Cys-704) and a red circle (Cys-30). The remaining subunits are shown in light gray, and the iron-sulfur centers are shown as yellow and gray spheres; the location of the flavin mononucleotide (FMN), which accepts electrons from NADH, is indicated by an arrow, the approximate locations of the membrane surface and the coenzyme Q are shown as a dashed blue line and a Q, respectively. B, close-up of the complex I 75-kDa subunit showing the location of five of the non-iron-sulfur Cys residues. Residues that are on the surface of the subunit are underlined. C, surface representation of the complex I 75-kDa subunit. The side chains of the four non-iron-sulfur Cys residues that are on the surface of the subunit are colored yellow. D, local environment of the Cys-531 residue in complex I 75-kDa subunit showing the close proximity of the two basic residues, Arg-534 and Lys-516.
In this structural model the bovine residue Cys-30 aligned to Val-23 in the Thermus Nqo3 subunit. However, Val-23 is in an α-helix and does not have a solvent-accessible surface because it is masked by a β-sheet. The bovine residue Cys-344 aligns with Asp-370 in the Thermus subunit. The residue aligns with the end strand of a parallel β-sheet. The side chain is very near the surface of the protein, but it is obstructed from the solvent by the loop region of the neighboring β-strand. The bovine residue Cys-531 corresponds to an insertion in a short α-helical region of the Thermus subunit (Pro-572 to Leu-577) which is conserved in mammals, G. gallus, X. laevis, and D. rerio. The α-helix is located on the surface of the subunit, and thus Cys-531 is likely to be solvent-accessible. The bovine residue Cys-541 is found in the middle of the parallel β-sheet containing Cys-344, and the side chain of Cys-541 is solvent-accessible. Bovine Cys-687 aligns with Arg-750 in the Thermus subunit, which is in a loop region following the last β-turn in the subunit. The side chain of the residue is partially exposed to the solvent; however, two loop regions lie on either side of it and may hinder access. The bovine residue Cys-704 is at the C terminus of the protein sequence and corresponds to the end of the sequence (Ala-767) in the Thermus structure. This region is on the surface of the protein and exposed to solvent. As bovine complex I contains ∼30 more subunits than that of T. thermophilus, it is possible that Cys residues that are apparently surface-accessible in our model of the bovine 75-kDa subunit may actually be occluded by a subunit not present in T. thermophilus. Allowing for this caveat, the modeling of the 75-kDa subunit suggests that Cys-30 and Cys-344 were not glutathionylated because they were not exposed to the glutathione pool in solution. In contrast, the four Cys residues glutathionylated in the purified bovine 75-kDa subunit (Cys-531, -541, -687, and -704) are solvent-accessible and thus potentially accessible to glutathionylation. In Fig. 8B, the 75-kDa subunit is enlarged, and underlining shows with the location of the four non-iron-sulfur center Cys residues that are solvent-exposed, as well as Cys-344 which is not solvent-exposed. This is extended in Fig. 8C, which shows the 75-kDa subunit as a space-filling model with the four solvent-exposed Cys residues colored yellow.
The modeling data are consistent with the glutathionylation of solvent-exposed Cys residues 531, 541, 687, and 704 in purified complex I. However, it was unclear why there was preferential glutathionylation of Cys-531 and Cys-704 in mitochondria. The local environment of Cys residues can alter their reactivity by decreasing the pKa values through the proximity of positively charged amino acid residues, increasing the proportion in the reactive thiolate form (54, 55). We used the model of the bovine 75-kDa complex I subunit to see if there were any positive amino acid residues in the vicinity of the four solvent accessible Cys residues. Cys-531 has a neighboring Arg-557 and Lys-539 within 5 Å of the residue (Fig. 8D). Cys-541 has Lys-562 in close proximity, although its side chain is on the opposite face of the β-sheet, whereas Arg-325 is ∼8 Å away. Cys-687 has Arg-655 within 5 Å, and although Lys-709 is close its side chain is probably inaccessible. There are no positively charged residues in the immediate vicinity of Cys-704, although Lys-692 is ∼8 Å away. Therefore, the proximity of positively charged amino acids close to Cys-531 may explain its greater propensity for glutathionylation, although it was unclear why Cys-704 was also particularly susceptible to glutathionylation. However, as bovine complex I contains ∼30 more subunits than that of T. thermophilus, it is possible that the local environment of Cys-704 may be affected by a subunit not present in T. thermophilus.
To conclude, modeling the location of the six non-iron-sulfur Cys residues in the complex I 75-kDa subunit suggests that only four are solvent-accessible and is consistent with their glutathionylation in purified complex I. Assessment of the local electrostatic environment of these four Cys residues suggests that Cys-531 may be the more reactive, hints that Cys-541 may be less reactive, but gave little indication of reasons for the different extents of glutathionylation between Cys-704 and Cys-687.
Conclusions—The glutathionylation of Cys-531 and Cys-704 is
of particular pathological interest because glutathione depletion leads to
complex I damage in Parkinson disease and other disorders
(6–8,
15,
56). Our finding of a
significant interaction between Cys-531 and Cys-704 and the mitochondrial
glutathione pool suggests that disruption of this interaction by glutathione
depletion may contribute to complex I oxidative damage and dysfunction in
pathologies. Reactive nitrogen and oxygen species such as
·NO2, ONOO-,
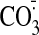 ,
and ·OH are thought to be particularly important in the
oxidative damage to complex I
(57,
58). These species react
rapidly with thiols as follows:
,
and ·OH are thought to be particularly important in the
oxidative damage to complex I
(57,
58). These species react
rapidly with thiols as follows:
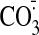 ,
k ∼5 × 106
m-1·s-1
(59); ONOO-,
k ∼700 m-1·s-1
(59);
·NO2 k ∼3–5 ×
107 m-1·s-1
(57); and
·OH, k = 2–4 × 1010
m-1·s-1
(59). (In contrast, the direct
reaction of thiols with H2O2 or superoxide are slow
compared with the reactions of these ROS with other antioxidant systems
(60,
61).) The fast reaction rates
of thiols mean that Cys residues on the surface of proteins are likely to
react with and inactivate these reactive species near the surface of complex
I, thereby preventing damage to other amino acids or prosthetic groups, such
as nitration of tyrosine residues by ·NO2
(57,
58). These reactions convert
the Cys thiol to a sulfenic acid or a thiyl radical, preventing further
antioxidant protection by the thiol, so the oxidized thiol must be recycled,
and this is done by reaction with the mitochondrial glutathione pool and Grx2
(10,
23). The protein sulfenic acid
reacts with GSH to generate a mixed disulfide, whereas the thiyl radical
reacts with GSH to generate a disulfide radical anion, which then reacts with
O2 to generate superoxide and a mixed disulfide by the Winterbourn
reaction (62), with the
superoxide then being detoxified by matrix superoxide dismutase. The mixed
disulfides are returned to a free thiol and GSSG by reaction with GSH
catalyzed by Grx2 (24).
Without the rapid recycling of the sulfenic or thiyl radicals by GSH and Grx2,
the Cys thiols would be irreversibly oxidized to sulfinic or sulfonic acids
and would be unable to protect complex I from oxidative damage
(20,
23,
24). This putative antioxidant
cycle in the prevention of oxidative damage to complex I is illustrated in
Fig. 9
(20,
23).
,
k ∼5 × 106
m-1·s-1
(59); ONOO-,
k ∼700 m-1·s-1
(59);
·NO2 k ∼3–5 ×
107 m-1·s-1
(57); and
·OH, k = 2–4 × 1010
m-1·s-1
(59). (In contrast, the direct
reaction of thiols with H2O2 or superoxide are slow
compared with the reactions of these ROS with other antioxidant systems
(60,
61).) The fast reaction rates
of thiols mean that Cys residues on the surface of proteins are likely to
react with and inactivate these reactive species near the surface of complex
I, thereby preventing damage to other amino acids or prosthetic groups, such
as nitration of tyrosine residues by ·NO2
(57,
58). These reactions convert
the Cys thiol to a sulfenic acid or a thiyl radical, preventing further
antioxidant protection by the thiol, so the oxidized thiol must be recycled,
and this is done by reaction with the mitochondrial glutathione pool and Grx2
(10,
23). The protein sulfenic acid
reacts with GSH to generate a mixed disulfide, whereas the thiyl radical
reacts with GSH to generate a disulfide radical anion, which then reacts with
O2 to generate superoxide and a mixed disulfide by the Winterbourn
reaction (62), with the
superoxide then being detoxified by matrix superoxide dismutase. The mixed
disulfides are returned to a free thiol and GSSG by reaction with GSH
catalyzed by Grx2 (24).
Without the rapid recycling of the sulfenic or thiyl radicals by GSH and Grx2,
the Cys thiols would be irreversibly oxidized to sulfinic or sulfonic acids
and would be unable to protect complex I from oxidative damage
(20,
23,
24). This putative antioxidant
cycle in the prevention of oxidative damage to complex I is illustrated in
Fig. 9
(20,
23).
FIGURE 9.

Possible mechanism of protection against oxidative damage to complex I
by exposed surface Cys residues. An exposed thiol on the surface of
complex I can react rapidly with a range of species such as
·OH and ·NO2 to generate a
thiyl radical, or with ONOO- or H2O2 to
generate a sulfenic acid. These oxidized thiols can then be oxidized further
by O2 to sulfinic or sulfonic acids (-SOn),
thereby preventing the thiols from protecting complex I from oxidative damage
to essential amino acid residues (X), leading to an irreversibly
damaged complex I. Alternatively, the sulfenic acid can react with GSH to form
a mixed disulfide with GSH, or the thiyl radical can react with GSH to form a
disulfide radical anion that loses an electron to O2 to form
superoxide
(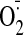 ).
The mixed disulfides then react rapidly with GSH catalyzed by Grx2 to give
GSSG, and the protein thiol is regenerated.
).
The mixed disulfides then react rapidly with GSH catalyzed by Grx2 to give
GSSG, and the protein thiol is regenerated.
Deglutathionylation of Cys residues by Grx2 is an important part of this antioxidant cycle. There is considerable evidence for a critical antioxidant role for Grx2 in mitochondria: decreasing Grx2 expression in cells increases oxidative damage and cell death (63, 64); overexpressing Grx2 protects cells against apoptosis induced by oxidative stress (65), and Grx2 is maintained in an inactive, iron-sulfur form that is activated by oxidative stress (66). Grx2, but not Grx1, is recycled directly by thioredoxin reductase as well as by GSH, implying that it can remain functional even when the mitochondrial GSH pool is oxidized (67). Finally, down-regulation of the cytosolic isoform Grx1 renders complex I in mouse brain more susceptible to damage by the parkinsonian neurotoxin 1-methyl-4-ophenyl-1,2,3,6-tetrahydropyridine, although how this isoform interacts with complex I is unclear (16). The extensive glutathionylation of Cys-531 and Cys-704 shows that these Cys residues readily interact with the mitochondrial glutathione pool by thiol-disulfide exchange. Glutathionylation can also occur in vivo in the presence of a reduced GSH pool (24). When mitochondria were exposed to H2O2 or to superoxide produced by the respiratory chain, this also led to glutathionylation of complex I, under conditions where there is negligible oxidation of the glutathione pool (51). Under these conditions glutathionylation probably occurs following ferrous iron released from FeS centers by superoxide reacting with H2O2 to form ·OH by the Fenton reaction, leading to oxidation of complex I thiols to thiyl radicals that are then converted to a glutathionylated Cys residue and then to a free thiol by glutathione. Therefore, in these experiments we have probably trapped a small amount of glutathionylated Cys residues as it is being recycled by the GSH pool after having detoxified a ·OH radical (Fig. 9). This contrasts with diamide treatment where the oxidized glutathione pool renders the Cys residues predominantly glutathionylated and no longer protective, perhaps contributing to the nonreversible damage to complex I that accumulates, whereas under milder oxidative stress damage to complex I was reversible by thiol reductants (6–8, 15, 56). It may be that the other two exposed Cys residues, Cys-541 and Cys-687, also contribute to this antioxidant cycle but that their glutathionylation is too transient to be measured under these conditions.
A further consideration for the protection of complex I by the antioxidant reactions of surface Cys residues is their concentration relative to that of GSH close to the surface of complex I. The solvent-exposed surface area of the T. thermophilus Nqo3 subunit, the ortholog of the bovine 75-kDa subunit, is ∼20,500 Å2 (53). The GSH concentration within heart mitochondria is 2–5 mm, so there are ∼1–3 GSH molecules within 50 Å of the 75-kDa subunit surface. Consequently, the four exposed surface thiols make a significant contribution to local thiol reactivity near the surface of complex I, consistent with a protective role for protein thiols.
Reactive thiols are also involved in modulating the complex I active/deactive transition (68), and the activity of complex I can be modified by S-nitrosation (13, 14, 69, 70). Our analysis suggests that Cys-531 may be particularly reactive; therefore it will be interesting to see whether complex I is S-nitrosated on this residue. As S-nitrosation often leads to glutathionylation (71), S-nitrosation and glutathionylation may turn out to be aspects of a related system to modulate and protect complex I activity (72).
Although more work is required to assess the importance of surface Cys residues in protecting complex I against oxidative damage, our results suggest that the interplay of thiols on the surface of complex I with the glutathione pool and Grx2 within mitochondria may be an important mode of preventing oxidative damage to complex I.
Supplementary Material
Acknowledgments
We thank Steven Sherwood, Richard Shannon, and Judy Hirst for supplying complex I; Michael Harbour for assistance with mass spectrometry; and Andrew James and Helena Cochemé for helpful comments on the manuscript.
This work was supported by the Medical Research Council (UK) and by a postgraduate research scholarship (to T. R. H.) from the Natural Sciences and Engineering Council of Canada. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1 and Figs. S1 and S2.
Author's Choice—Final version full access.
Footnotes
The abbreviations used are: ROS, reactive oxygen species; ACA, ε-amino-n-caproic acid; BN, Blue Native; CLZ, coelenterazine; DDM, dodecyl-β-d-maltoside; DTPA, N,N-bis(2 bis[carboxymethyl]aminoethyl) glycine; DTT, dithiothreitol; FCCP, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone; Grx2, glutaredoxin 2; MALDI-TOF, matrix-assisted laser desorption/ionization time-of-flight; TOF-TOF, tandem time-of-flight; MS, mass spectrometry; NEM,N-ethylmaleimide; PrSSG, glutathione-protein mixed disulfide; SOD, superoxide dismutase; BisTris, 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol; Tricine, N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
References
- 1.Brandt, U. (2006) Annu. Rev. Biochem. 75 69-92 [DOI] [PubMed] [Google Scholar]
- 2.Hirst, J., Carroll, J., Fearnley, I. M., Shannon, R. J., and Walker, J. E. (2003) Biochim. Biophys. Acta 1604 135-150 [DOI] [PubMed] [Google Scholar]
- 3.Walker, J. E. (1992) Q. Rev. Biophys. 25 253-321 [DOI] [PubMed] [Google Scholar]
- 4.Sazanov, L. A. (2007) Biochemistry 46 2275-2288 [DOI] [PubMed] [Google Scholar]
- 5.Carroll, J., Fearnley, I. M., Skehel, J. M., Shannon, R. J., Hirst, J., and Walker, J. E. (2006) J. Biol. Chem. 281 32724-32727 [DOI] [PubMed] [Google Scholar]
- 6.Jha, N., Jurma, O., Lalli, G., Liu, Y., Pettus, E. H., Greenamyre, J. T., Liu, R. M., Forman, H. J., and Andersen, J. K. (2000) J. Biol. Chem. 275 26096-26101 [DOI] [PubMed] [Google Scholar]
- 7.Hsu, M., Srinivas, B., Kumar, J., Subramanian, R., and Andersen, J. (2005) J. Neurochem. 92 1091-1103 [DOI] [PubMed] [Google Scholar]
- 8.Chinta, S. J., and Andersen, J. K. (2006) Free Radic. Biol. Med. 41 1442-1448 [DOI] [PubMed] [Google Scholar]
- 9.Taylor, E. R., Hurrell, F., Shannon, R. J., Lin, T. K., Hirst, J., and Murphy, M. P. (2003) J. Biol. Chem. 278 19603-19610 [DOI] [PubMed] [Google Scholar]
- 10.Beer, S. M., Taylor, E. R., Brown, S. E., Dahm, C. C., Costa, N. J., Runswick, M. J., and Murphy, M. P. (2004) J. Biol. Chem. 279 47939-47951 [DOI] [PubMed] [Google Scholar]
- 11.Lin, T. K., Hughes, G., Muratovska, A., Blaikie, F. H., Brookes, P. S., Darley-Usmar, V., Smith, R. A. J., and Murphy, M. P. (2002) J. Biol. Chem. 277 17048-17056 [DOI] [PubMed] [Google Scholar]
- 12.Brown, G. C., and Borutaite, V. (2004) Biochim. Biophys. Acta 1658 44-49 [DOI] [PubMed] [Google Scholar]
- 13.Dahm, C. C., Moore, K., and Murphy, M. P. (2006) J. Biol. Chem. 281 10056-10065 [DOI] [PubMed] [Google Scholar]
- 14.Burwell, L. S., Nadtochiy, S. M., Tompkins, A. J., Young, S., and Brookes, P. S. (2006) Biochem. J. 394 627-634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sriram, K., Shankar, S. K., Boyd, M. R., and Ravindranath, V. (1998) J. Neurosci. 18 10287-10296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kenchappa, R. S., and Ravindranath, V. (2003) FASEB J. 17 717-719 [DOI] [PubMed] [Google Scholar]
- 17.Chen, C. L., Zhang, L., Yeh, A., Chen, C. A., Green-Church, K. B., Zweier, J. L., and Chen, Y. R. (2007) Biochemistry 46 5754-5765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gladyshev, V. N., Liu, A., Novoselov, S. V., Krysan, K., Sun, Q. A., Kryukov, V. M., Kryukov, G. V., and Lou, M. F. (2001) J. Biol. Chem. 276 30374-30380 [DOI] [PubMed] [Google Scholar]
- 19.Lundberg, M., Johansson, C., Chandra, J., Enoksson, M., Jacobsson, G., Ljung, J., Johansson, M., and Holmgren, A. (2001) J. Biol. Chem. 276 26269-26275 [DOI] [PubMed] [Google Scholar]
- 20.Hurd, T. R., Costa, N. J., Dahm, C. C., Beer, S. M., Brown, S. E., Filipovska, A., and Murphy, M. P. (2005) Antioxid. Redox. Signal. 7 999-1010 [DOI] [PubMed] [Google Scholar]
- 21.Fratelli, M., Demol, H., Puype, M., Casagrande, S., Eberini, I., Salmona, M., Bonetto, V., Mengozzi, M., Duffieux, F., Miclet, E., Bachi, A., Vandekerckhove, J., Gianazza, E., and Ghezzi, P. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 3505-3510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giustarini, D., Rossi, R., Milzani, A., Colombo, R., and Dalle-Donne, I. (2004) J. Cell. Mol. Med. 8 201-212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas, J. A., Poland, B., and Honzatko, R. (1995) Arch. Biochem. Biophys. 319 1-9 [DOI] [PubMed] [Google Scholar]
- 24.Dalle-Donne, I., Milzani, A., Gagliano, N., Colombo, R., Giustarini, D., and Rossi, R. (2008) Antioxid. Redox. Signal. 10 445-473 [DOI] [PubMed] [Google Scholar]
- 25.Cocheme, H. M., and Murphy, M. P. (2008) J. Biol. Chem. 283 1786-1798 [DOI] [PubMed] [Google Scholar]
- 26.Smith, A. L. (1967) Methods Enzymol. 10 81-86 [Google Scholar]
- 27.Walker, J. E., Skehel, J. M., and Buchanan, S. K. (1995) Methods Enzymol. 260 14-34 [DOI] [PubMed] [Google Scholar]
- 28.Sharpley, M. S., Shannon, R. J., Draghi, F., and Hirst, J. (2006) Biochemistry 45 241-248 [DOI] [PubMed] [Google Scholar]
- 29.Anderson, M. (1985) Methods Enzymol. 113 548-555 [DOI] [PubMed] [Google Scholar]
- 30.Scarlett, J. L., Packer, M. A., Porteous, C. M., and Murphy, M. P. (1996) Biochem. Pharmacol. 52 1047-1055 [DOI] [PubMed] [Google Scholar]
- 31.Akerboom, T. P. M., and Sies, H. (1981) Methods Enzymol. 113 373-382 [DOI] [PubMed] [Google Scholar]
- 32.Schagger, H. (1995) Methods Enzymol. 260 190-202 [DOI] [PubMed] [Google Scholar]
- 33.Schagger, H., and von Jagow, G. (1991) Anal. Biochem. 199 223-231 [DOI] [PubMed] [Google Scholar]
- 34.Wilm, M., Shevchenko, A., Houthaeve, T., Breit, S., Schweigerer, L., Fotsis, T., and Mann, M. (1996) Nature 379 466-469 [DOI] [PubMed] [Google Scholar]
- 35.Perkins, D. N., Pappin, D. J., Creasy, D. M., and Cottrell, J. S. (1999) Electrophoresis 20 3551-3567 [DOI] [PubMed] [Google Scholar]
- 36.Lucas, M., and Solano, F. (1992) Anal. Biochem. 206 273-277 [DOI] [PubMed] [Google Scholar]
- 37.Zhao, H., Joseph, J., Fales, H. M., Sokoloski, E. A., Levine, R. L., Vasquez-Vivar, J., and Kalyanaraman, B. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 5727-5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao, H., Kalivendi, S., Zhang, H., Joseph, J., Nithipatikom, K., Vasquez-Vivar, J., and Kalyanaraman, B. (2003) Free Radic. Biol. Med. 34 1359-1368 [DOI] [PubMed] [Google Scholar]
- 39.James, A. M., Wei, Y.-H., Pang, C.-Y., and Murphy, M. P. (1996) Biochem. J. 318 401-407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith, P. K., Krohn, R. I., Hermanson, G. T., Mallia, A. K., Gartner, F. H., Provenzano, M. D., Fujimoto, E. K., Goeke, N. M., Olson, B. J., and Klenk, D. C. (1985) Anal. Biochem. 150 76-85 [DOI] [PubMed] [Google Scholar]
- 41.Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D. J. (1997) Nucleic Acids Res. 25 3389-3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994) Nucleic Acids Res. 22 4673-4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sali, A., and Blundell, T. L. (1993) J. Mol. Biol. 234 779-815 [DOI] [PubMed] [Google Scholar]
- 44.Carroll, J., Fearnley, I. M., Shannon, R. J., Hirst, J., and Walker, J. E. (2003) Mol. Cell. Proteomics 2 117-126 [DOI] [PubMed] [Google Scholar]
- 45.Carroll, J., Shannon, R. J., Fearnley, I. M., Walker, J. E., and Hirst, J. (2002) J. Biol. Chem. 277 50311-50317 [DOI] [PubMed] [Google Scholar]
- 46.McKernan, T. B., Woods, E. B., and Lash, L. H. (1991) Arch. Biochem. Biophys. 288 653-663 [DOI] [PubMed] [Google Scholar]
- 47.Reddy, S., Jones, A. D., Cross, C. E., Wong, P. S. Y., and Van Der Vliet, A. (2000) Biochem. J. 347 821-827 [PMC free article] [PubMed] [Google Scholar]
- 48.Runswick, M. J., Gennis, R. B., Fearnley, I. M., and Walker, J. E. (1989) Biochemistry 28 9452-9459 [DOI] [PubMed] [Google Scholar]
- 49.Rubino, F. M., Pitton, M., Brambilla, G., and Colombi, A. (2006) J. Mass Spectrom. 41 1578-1593 [DOI] [PubMed] [Google Scholar]
- 50.Raha, S., and Robinson, B. H. (2000) Trends Biochem. Sci. 25 502-508 [DOI] [PubMed] [Google Scholar]
- 51.Hurd, T. R., Prime, T. A., Harbour, M. E., Lilley, K. S., and Murphy, M. P. (2007) J. Biol. Chem. 282 22040-22051 [DOI] [PubMed] [Google Scholar]
- 52.Han, D., Canali, R., Rettori, D., and Kaplowitz, N. (2003) Mol. Pharmacol. 64 1136-1144 [DOI] [PubMed] [Google Scholar]
- 53.Sazanov, L. A., and Hinchliffe, P. (2006) Science 311 1430-1436 [DOI] [PubMed] [Google Scholar]
- 54.Jao, S. C., English Ospina, S. M., Berdis, A. J., Starke, D. W., Post, C. B., and Mieyal, J. J. (2006) Biochemistry 45 4785-4796 [DOI] [PubMed] [Google Scholar]
- 55.Hansen, R. E., Ostergaard, H., and Winther, J. R. (2005) Biochemistry 44 5899-5906 [DOI] [PubMed] [Google Scholar]
- 56.Chinta, S. J., Kumar, M. J., Hsu, M., Rajagopalan, S., Kaur, D., Rane, A., Nicholls, D. G., Choi, J., and Andersen, J. K. (2007) J. Neurosci. 27 13997-14006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ford, E., Hughes, M. N., and Wardman, P. (2002) Free Radic. Biol. Med. 32 1314-1323 [DOI] [PubMed] [Google Scholar]
- 58.Bharath, S., and Andersen, J. K. (2005) Antioxid. Redox. Signal. 7 900-910 [DOI] [PubMed] [Google Scholar]
- 59.Bonini, M. G., and Augusto, O. (2001) J. Biol. Chem. 276 9749-9754 [DOI] [PubMed] [Google Scholar]
- 60.Winterbourn, C. C., and Metodiewa, D. (1999) Free Radic. Biol. Med. 27 322-328 [DOI] [PubMed] [Google Scholar]
- 61.Winterbourn, C. C. (2008) Nat. Chem. Biol. 4 278-286 [DOI] [PubMed] [Google Scholar]
- 62.Winterbourn, C. C. (1993) Free Radic. Biol. Med. 14 85-90 [DOI] [PubMed] [Google Scholar]
- 63.Starke, D. W., Chock, P. B., and Mieyal, J. J. (2003) J. Biol. Chem. 278 14607-14613 [DOI] [PubMed] [Google Scholar]
- 64.Lillig, C. H., Lonn, M. E., Enoksson, M., Fernandes, A. P., and Holmgren, A. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 13227-13232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Enoksson, M., Fernandes, A. P., Prast, S., Lillig, C. H., Holmgren, A., and Orrenius, S. (2005) Biochem. Biophys. Res. Commun. 327 774-779 [DOI] [PubMed] [Google Scholar]
- 66.Lillig, C. H., Berndt, C., Vergnolle, O., Lonn, M. E., Hudemann, C., Bill, E., and Holmgren, A. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 8168-8173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johansson, C., Lillig, C. H., and Holmgren, A. (2004) J. Biol. Chem. 279 7537-7543 [DOI] [PubMed] [Google Scholar]
- 68.Gavrikova, E. V., and Vinogradov, A. D. (1999) FEBS Lett. 455 36-40 [DOI] [PubMed] [Google Scholar]
- 69.Clementi, E., Brown, G. C., Feelisch, M., and Moncada, S. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 7631-7636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Galkin, A., and Moncada, S. (2007) J. Biol. Chem. 282 37448-37453 [DOI] [PubMed] [Google Scholar]
- 71.Costa, N. J., Dahm, C. C., Hurrell, F., Taylor, E. R., and Murphy, M. P. (2003) Antioxid. Redox. Signal. 5 291-305 [DOI] [PubMed] [Google Scholar]
- 72.Burwell, L. S., and Brookes, P. S. (2008) Antioxid. Redox. Signal. 10 579-599 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.