

Abstract
The RecA and some related proteins possess a simple motif, called (KR)X(KR), that (in RecA) consists of two lysine residues at positions 248 and 250 at the subunit-subunit interface. This study and previous work implicate this RecA motif in the following: (a) catalyzing ATP hydrolysis in trans,(b) coordinating the ATP hydrolytic cycles of adjacent subunits, (c) governing the rate of ATP hydrolysis, and (d) coupling the ATP hydrolysis to work (in this case DNA strand exchange). The conservative K250R mutation leaves RecA nucleoprotein filament formation largely intact. However, ATP hydrolysis is slowed to less than 15% of the wild-type rate. DNA strand exchange is also slowed commensurate with the rate of ATP hydrolysis. The results reinforce the idea of a tight coupling between ATP hydrolysis and DNA strand exchange. When a plasmid-borne RecA K250R protein is expressed in a cell otherwise lacking RecA protein, the growth of the cells is severely curtailed. The slow growth defect is alleviated in cells lacking RecFOR function, suggesting that the defect reflects loading of RecA at stalled replication forks. Suppressors occur as recA gene alterations, and their properties indicate that limited dissociation by RecA K250R confers the slow growth phenotype. Overall, the results suggest that recombinational DNA repair is a common occurrence in cells. RecA protein plays a sufficiently intimate role in the bacterial cell cycle that its properties can limit the growth rate of a bacterial culture.
Homologous DNA recombination is an important part of DNA metabolism. The primary function of bacterial recombination systems is the recombinational DNA repair of stalled replication forks (1-4). Recombination is also responsible for the exchange of genetic material during meiosis in eukaryotes and conjugation in bacteria. RecA protein is the central recombinase in Escherichia coli. RecA homologues are present in nearly every organism, and closely related RecA proteins are found in virtually all bacterial species with the exception of a few endosymbionts (5). In E. coli, RecA participates not only in the restart of stalled replication forks but also the induction of the SOS response upon cellular DNA damage distress and translesion DNA synthesis via the error-prone DNA polymerase V (1, 6, 7).
Replication forks are thought to stall often in bacteria under normal growth conditions, but the actual rate is a matter of some dispute. Some estimates suggest that forks stall often, up to once per cell cycle in bacteria (1, 4, 8-11). At least some of these result in double strand breaks, with up to 50 double strand breaks per cell cycle occurring in humans (12). A direct measure of double strand break generation in bacteria suggests that fork stalling may be less frequent, or at least not always accompanied by double strand breaks (13). Notably, several pathways have been proposed for the reconstitution of replication forks that do not involve double strand breaks (1, 4, 11, 14). However, the frequency with which these pathways are utilized has not been established.
RecA protein is a DNA-dependent ATPase, and its active form is a DNA-bound (nucleoprotein) filament. RecA hydrolyzes ATP or dATP in a reaction that occurs uniformly throughout the filament (15). RecA promotes a series of DNA strand exchange reactions in vitro that are thought to account for the role of RecA in recombination (6, 16-18). When replication forks stall, single strand DNA gaps and double strand breaks are created. RecA filaments are loaded onto single-stranded DNA gaps and the processed ends of double strand breaks by the RecFOR mediator proteins and the RecBCD nuclease/helicase, respectively (17, 19-26).
RecA-mediated ATP hydrolysis is coupled to the end-dependent disassembly of the nucleoprotein filament (5, 18, 27). However, ATP hydrolysis at the interior subunits of a RecA filament does not result in dissociation and is associated with several additional RecA activities. These include bypass of heterologous insertions during DNA three-strand exchange, complete DNA strand exchange with DNA substrates longer than ∼3 kbp, strand exchange with four DNA strands, an indirect helicase function, and replication fork regression (5, 6, 18, 28-34). The ATP hydrolytic cycles between adjacent subunits in the RecA filament bound to double-stranded DNA (dsDNA)2 are coordinated such that waves of hydrolysis move sequentially through the filament with a separation of six subunits (27). RecA could potentially use the organized waves of ATP hydrolysis to act as a motor, driving completion of strand exchange beyond the barriers mentioned above, or to process replication forks (18). The detailed mechanism by which ATP hydrolysis is coordinated in the RecA-dsDNA nucleoprotein filament is currently unknown.
The central core domain of RecA protein defines a structural motif (now called the RecA fold) found in a variety of proteins that hydrolyze ATP, and many of these proteins are motors (5, 18, 35). The family includes helicases like PcrA, ABC transporters like MutS, and with somewhat less structural similarity, the AAA+ family of ATPases, including ClpA (36). Koonin and co-workers (35) have drawn attention to a new and simple motif, called the (KR)X(KR) motif, that is found in a subset of the proteins carrying the RecA fold. This motif is conserved among the DnaB, RecA, Sms, and KaiC protein families. Interestingly, the same motif is absent from the archaeal or eukaryotic homologues of RecA.
Where it occurs, the (KR)X(KR) motif is found at the subunit-subunit interface. For a number of these proteins, the motif has been implicated in the in trans catalysis of ATP hydrolysis. The gp4 of bacteriophage T7, a 5′ to 3′ hexameric ring helicase, possesses the motif and uses it to coordinate ATP hydrolysis between subunits (35, 37-39). It is proposed that Arg-522 of T7 gp4 senses hydrolysis of ATP, communicates the hydrolysis between subunits, and promotes conformational changes associated with ATP hydrolysis (37-39). In this manner, Arg-522 would have a role similar to an arginine finger found in Ras and its GTPase-activating protein. In the Ras-GAP system, it is postulated that Arg-789 of GAP similarly reaches across the GAP-Ras interface to stabilize the γ-phosphate of the Ras-bound ATP (40).
In E. coli RecA protein, the residues in the (KR)X(KR) motif are Lys-248 and Lys-250 (35). These two residues may be important for ATP hydrolysis and potentially for communicating the conformation changes associated with ATP hydrolysis between adjacent subunits in the RecA nucleoprotein filament, based on the similarities to homologous proteins described above. Lys-248 and Lys-250 are not near the ATP-binding site of the RecA filament as resolved in the first RecA crystal structure (41, 42). However, this early structure now appears to reflect an inactive conformation of RecA protein. A new examination of active filaments strongly suggests that the ATP hydrolytic site is at the subunit-subunit interface and that Lys-248 and Lys-250 are in very close proximity to the ATP hydrolytic site (43). Crystal structures of the archaeal RadA filament (44) and the eukaryotic Rad51 filament (45) support the revised location of the nucleotide between two RecA subunits.
In a recent study, we initiated an exploration of the function of (KR)X(KR) in RecA protein by examining residue Lys-248 (46). The K248A mutation was studied previously (47). Although the K248A mutant protein appears to fold normally, the K248A mutant does not form a filament. The new study focused on a more conservative mutation, K248R (46). The K248R mutant binds to DNA and forms a nucleoprotein filament, although it hydrolyzes neither ATP nor dATP at a readily measured rate. However, a combination of the K248R mutant and another ATP hydrolysis-deficient mutant, E96D (48, 49), partially restores dATP hydrolysis (46). In the new reconstructions of the RecA nucleoprotein filament, the Lys-248 and Glu-96 residues are located at the subunit-subunit interface near the bound nucleotide but are opposed across the interface. The capacity of Lys-248 to partially compensate for the E96D defect provided evidence for catalysis of ATP hydrolysis by Lys-248 in trans (46). The stability of the RecA K248R or RecA K248R/E96D filaments was insufficient for the promotion of DNA strand exchange.
In this study, we turn our attention to residue Lys-250. The K250N mutation was studied previously in vivo. The K250N mutant RecA protein was toxic to cells, catalyzed less recombination than wild-type RecA protein, and was a constitutive LexA co-protease (48). In contrast, we find that a conservative mutation, K250R, yields a highly active RecA nucleoprotein filament with interesting properties. Notably, RecA K250R is a slow RecA protein, promoting ATP hydrolysis and DNA strand exchange at rates that are 6-fold slower than the wild-type protein. When the protein is expressed in E. coli, a slow growth phenotype results.
EXPERIMENTAL PROCEDURES
Enzymes and Biochemicals—The E. coli SSB protein was purified as described previously (31). The E. coli wild-type RecA protein was purified as described previously (50) with the following exceptions: the polyethyleneimine pellet was washed with 100 ml of R Buffer (20 mm Tris-Cl buffer (80% cation, pH 7.5), 10% (w/v) glycerol, 1 mm dithiothreitol) plus 150 mm ammonium sulfate; protein was extracted from the polyethyleneimine pellet twice with 75 ml of R Buffer plus 300 mm ammonium sulfate; the ammonium sulfate pellet was resuspended in R Buffer plus 100 mm KCl; the wild-type RecA protein was then loaded onto the DEAE-Sepharose column and washed with two column volumes of R Buffer plus 100 mm KCl and then two column volumes of R Buffer plus 1 m KCl; and flow-through peak fractions and peak fractions eluted in R Buffer plus 1 m KCl were identified by SDS-PAGE and pooled together before dialyzing versus 20 mm phosphate buffer (10 mm KH2PO4, 10 mm K2HPO4, 10% (w/v) glycerol, 1 mm dithiothreitol) in preparation for the hydroxyapatite column.
The RecA K250R, K250Q, K250N, E96D, and K250R/A11V mutant proteins were purified by the same means as the wild-type RecA protein with the following exceptions: the plasmids encoding the mutant recA genes were transformed into the ΔrecA and nuclease-deficient strain STL2669 (pT7pol26; described in Ref. 50). Ammonium sulfate precipitation and the washing of the polyethyleneimine pellets varied slightly as determined by empirical tests. Proteins were purified using chromatography on some combination of DEAE-Sepharose, hydroxyapatite, PBE-94 (Amersham Biosciences), Source Q (Amersham Biosciences), and/or Ceramic HAP (Bio-Rad) columns. Details are available by e-mailing the corresponding author. Final pooled fractions were free of detectable nuclease activity.
The purified RecA proteins and SSB protein concentrations were determined by absorbance at 280 nm, using extinction coefficients of ε280 = 2.23 × 104 m-1 cm-1 (51) and ε280 = 2.38 × 104 m-1 cm-1 (52), respectively. RecA proteins and SSB preparations were free of detectable endo- and exonuclease activities on double-stranded DNA and single-stranded DNA. Unless otherwise noted, all reagents were purchased from Fisher. Ammonium sulfate was purchased from MP Biomedicals (Solon, OH), formerly ICN Biomedicals (Irvine, CA). Phosphoenolpyruvate (PEP), pyruvate kinase, lactate dehydrogenase, phosphocreatine, creatine kinase, ATP, dATP, and NADH were purchased from Sigma. ATPγS was purchased from Roche Applied Science. Restriction enzymes were purchased from Fermentas Life Sciences (Hanover, MD). Dithiothreitol was purchased from Research Organics (Cleveland, OH).
DNA Substrates—Poly(dT) linear single-stranded DNA (ssDNA)
was purchased from Amersham Biosciences, and the approximate average length
was 229 nucleotides (nt). Circular ssDNA (7229 nucleotides) from bacteriophage
M13mp8 was prepared as described
(53). The bacteriophage stocks
used in the preparation have a point mutation at position 2220 (adenosine to
guanosine), resulting in two BamHI restriction endonuclease recognition sites
instead of one. Bacteriophage ΦX174 circular ssDNA and replicative form I
circular duplex DNA were purchased from New England Biolabs (Ipswich, MA) and
Invitrogen, respectively. Linear duplex DNA (dsDNA) was generated by digesting
ΦX174 replicative form I DNA (5386 bp) with the XhoI endonuclease.
Concentrations of ssDNA and dsDNA were determined by absorbance at 260 nm
using 108 and 151 μm
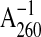 , respectively, as conversion
factors. All DNA concentrations are given in terms of total nucleotides.
, respectively, as conversion
factors. All DNA concentrations are given in terms of total nucleotides.
Reaction Conditions—All reactions were carried out at 37 °C in 25 mm Tris acetate buffer (80% cation; final pH after addition of all components was 7.56), unless otherwise stated. All reactions also included 10 mm magnesium acetate, 3 mm potassium glutamate, 5% (v/v) glycerol, 1 mm dithiothreitol, 3 mm ATP or dATP, an ATP-regenerating system (10 units/ml pyruvate kinase and 3 mm PEP), and concentrations of DNA and RecA protein as described below and in the figure legends. The coupled spectrophotometric assay to monitor (d)ATP hydrolysis catalyzed by the RecA proteins also contained 1.5 mm NADH and 10 units/ml lactate dehydrogenase. The reactions examined by electron microscopy included an alternative ATP-regenerating system composed of 10 units/ml creatine kinase and 12 mm phosphocreatine. Reactions were incubated for 10 min before (d)ATP (and single-stranded DNA-binding protein of E. coli (SSB) when included) was added to start the reaction.
ATP Hydrolysis Assays—A coupled spectrophotometric assay (54, 55) was used to measure the (d)ATP hydrolysis catalyzed by the RecA protein (50). The regeneration of ATP or dATP from ADP and PEP was coupled to the oxidation of NADH and monitored by the decrease in absorbance of NADH at 380 nm. The 380-nm wavelength was used instead of the absorption maximum at 340 nm so that the signal would remain within the linear range of the spectrophotometer for the duration of the experiment. The assays were executed using a Varian Cary 300 (Varian, Palo Alto, CA) dual beam spectrophotometer equipped with a temperature controller and 12-position cell changer. The cell path length and band pass were 1.0 cm and 2 nm, respectively. The NADH extinction coefficient at 380 nm of 1.21 mm-1 cm-1 was used to calculate rates of ATP or dATP hydrolysis.
DNA Three-strand Exchange Reactions—DNA three-strand exchange reactions were carried out at 37 °C in 25 mm Tris acetate buffer (80% cation, pH 7.5), 25 mm Tris acetate buffer (30% cation, pH 8.5), or 25 mm MES buffer (76% anion, pH 6.6). RecA protein was preincubated with ΦX174 circular ssDNA for 10 min. Then 3 mm (d)ATP and SSB were added to the reaction, followed by another incubation for 10 min. The reactions were initiated with the addition of ΦX174 linear dsDNA. Reactions were incubated for a total of 120 min with a 26-μl aliquot taken at 0, 15, 30, 60, 90, and 120 min. To stop the reaction, the 26-μl aliquot was added to 13 μl of a solution composed of 30% (w/v) glycerol, 72 mm EDTA, 0.12% (w/v) bromphenol blue, and 4% (w/v) SDS. Reactions were analyzed by agarose gel electrophoresis as described previously (56). Protein and DNA concentrations are provided in the figure legends.
For quantitation, gel images were captured with a Fotodyne FOTO/Analyst® CCD camera, PC Image acquisition software, and FOTO/Convertible dual transilluminator. The intensity of DNA bands was measured using the software package TotalLab version TL100 from Nonlinear Dynamics.
Electron Microscopy—A modified Alcian method was used to visualize RecA nucleoprotein filaments. Activated grids were prepared as described previously (50). Reaction mixtures included 25 mm Tris acetate buffer (80% cation, pH 7.5), 10 mm magnesium acetate, 5% (v/v) glycerol, 3 mm potassium glutamate, an ATP-regenerating system (12 mm phosphocreatine and 10 units/ml creatine kinase), 6 μm poly(dT) linear ssDNA or M13mp8 circular ssDNA, and 4 μm total RecA. The average length of the poly(dT) linear ssDNA was 229 nt, and the length of the M13mp8 circular ssDNA was 7229 nt. Reactions were incubated at 37 °C for 10 min before (d)ATP (to 3 mm) was added to start the filament formation. When included, SSB was added with the ATP to 0.6 μm. The reaction was again incubated at 37 °C for 10 min to allow RecA to form a filament on the DNA. ATPγS was then added to 3 mm, and the reaction was incubated at 37 °C for 3 min to stabilize the RecA-DNA filament. The reaction solution was then diluted 5- or 10-fold in buffer (200 mm ammonium acetate, 10 mm HEPES, pH 7.5, and 10% glycerol) and adsorbed onto Alcian grids for 3 min. The grid was then touched to a drop of the same buffer, followed by floating on a drop of the same buffer for 1 min. The sample was then stained by touching to a drop of 5% uranyl acetate followed by floating on a fresh drop of 5% uranyl acetate for 30 s. Finally, the grid was washed by touching it to a drop of double distilled water and one beaker of 100% ethyl alcohol. After the sample was dried, it was rotary-shadowed with platinum. This protocol is designed for visualization of complete reaction mixtures, and no attempt was made to remove unreacted material. Although this approach should yield results that provide insight into reaction components, it does lead to samples with a high background of unreacted proteins. Digital images of the nucleoprotein filaments were taken at ×15,000 magnification with a Phillips FEI Tecnai G212 twin transmission electron microscope equipped with a Gatan 890 CCD camera.
Filament lengths and pitches were measured using MetaMorph analysis software (Molecular Devices, Sunnyvale, CA). At least 8 filaments were measured for each RecA protein filament on M13mp8 circular ssDNA, and at least 52 filaments were measured for each RecA protein filament on poly(dT) linear ssDNA. Each filament was measured three times, and the average length was calculated. To measure the pitch of a RecA filament, the distances between individual striations were measured. At least 22 pitch distances from at least 6 different molecules were measured, and the average pitch distance was calculated.
A cytochrome c method was used to visualize poly(dT) linear ssDNA as a control and to check the purity of the M13mp8 circular ssDNA. Samples were prepared as described previously (57), except the spreading solution was assembled by combining 32 μl of 1 m Na2CO3, 40 μl of 0.126 m Na2EDTA, 400 μl of 37% HCHO, 292 μl of 100% formamide, and 10 μl of 5 m KOH. The final DNA concentration was 0.004 μg/μl.
Measurement of Sensitivity to Ultraviolet Light—Measurement of sensitivity to UV was carried out as described previously (50).
Growth Curves—LB media (25 ml) was inoculated with 0.25 ml of an overnight culture of a strain to be tested and grown for the indicated times at 37 °C with shaking at 200 rpm. Aliquots were removed every hour, and growth was monitored by absorption of the sample at a wavelength of 600 nm, using a Cary 100 spectrophotometer. Solutions were diluted as needed to bring the absorption into the linear range of the spectrophotometer.
RESULTS
Experimental Design—In this study, the phrase (d)ATP hydrolysis is used whenever a statement applies to RecA-mediated hydrolysis of either dATP or ATP. There are two parts to this study. In the first part (under “Characterization of RecA Proteins with Changes to Residue Lys-250”), we first examined the involvement of the subunit-subunit interface residue Lys-250 in (d)ATP hydrolysis catalyzed by the RecA protein by examining several purified proteins with single mutations at residue Lys-250 as well as a mutation at another subunit-subunit interface residue, Glu-96. The mutations we studied included K250R, K250N, K250Q, E96D, and the double mutant K250R/E96D. Our studies allowed us to investigate the contribution of subunit-subunit interface residues to an active RecA nucleoprotein filament. In the second part (under “Slow Growth Phenotype Associated with the Expression of RecA K250R in Vivo”), we next examined the effects of expression of the RecA K250R in vivo. A severe constraint on growth rate led to the isolation of suppressor mutations and to the examination of a RecA K250R/A11V double mutant protein that relieves the slow growth phenotype when expressed in the cell.
Characterization of RecA Proteins with Changes to Residue Lys-250
Changes at Lys-250 Affect (d)ATP Hydrolysis—As seen in Fig. 1, all mutant proteins catalyzed (d)ATP hydrolysis at a reduced rate relative to the wild-type protein when bound to poly(dT), linear ssDNA, or M13mp8 circular ssDNA. The K250R mutation resulted in a much less attenuated rate of (d)ATP hydrolysis. As displayed in Table 1, the K250R mutant protein catalyzed (d)ATP hydrolysis at a rate that was 13.1 to 15.3% of the wild-type rate, depending on the nucleoside triphosphate substrate and DNA bound. On the other hand, the E96D and K250N and K250Q single mutations of the RecA protein resulted in a reduced rate of (d)ATP hydrolysis that was 0.4 to 3.2% of the wild-type rate. The reduction in the rate of (d)ATP hydrolysis resulting from mutations at subunit-subunit interface residues Lys-250 and Glu-96 indicated the importance of these residues to (d)ATP hydrolysis.
FIGURE 1.

Mutations at residue Lys-250 affect (d)ATP hydrolysis catalyzed by the RecA protein. RecA protein-catalyzed (d)ATP hydrolysis was monitored. The RecA protein was preincubated with the DNA for 10 min before (d)ATP(+SSB) was added. Time 0 corresponds to the time of (d)ATP addition. The average observed rates of (d)ATP hydrolysis are reported in Table 1. A, reactions included 4 μm RecA protein and 6 μm poly(dT) linear ssDNA at pH 7.5. A solid line indicates reactions with dATP, and reactions with ATP are indicated with a dashed line. B, reactions included 4 μm RecA protein, 6 μm M13mp8 circular ssDNA, and 0.6 μm SSB protein at pH 7.5. A solid line indicates reactions with dATP, and reactions with ATP are indicated with a dashed line. WT, wild type.
TABLE 1.
Rates of (d)ATP hydrolysis catalyzed by wild-type and mutant RecA proteins
RecA protein-catalyzed (d)ATP hydrolysis was monitored as described in Fig. 1. Characteristic reactions for each solution condition are those presented in Fig. 1. Rates are in units of μm/min. WT is wild type; ND is not determined.
| RecA | Poly(dT)dATP/S.D. | n | % WT | Poly(dT)ATP/S.D. | n | % WT | M13mp8 dATP/S.D. | n | % WT | M13mp8 ATP/S.D. | n | % WT |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | 62.761 ± 2.583 | 29 | 100.0 | 54.804 ± 2.687 | 8 | 100.0 | 79.495 ± 2.192 | 8 | 100.0 | 63.283 ± 2.657 | 11 | 100.0 |
| K250R | 9.618 ± 0.268 | 8 | 15.3 | 7.193 ± 0.534 | 8 | 13.1 | 11.798 ± 0.195 | 4 | 14.8 | 9.126 ± 0.631 | 7 | 14.4 |
| K250N | 0.449 ± 0.044 | 7 | 0.7 | 0.289 ± 0.048 | 4 | 0.5 | 0.289 ± 0.048 | 4 | 0.4 | 0.248 ± 0.000 | 4 | 0.4 |
| K250Q | 0.434 ± 0.041 | 4 | 0.7 | 0.331 ± 0.138 | 6 | 0.6 | 0.351 ± 0.041 | 4 | 0.4 | 0.372 ± 0.045 | 6 | 0.6 |
| E96D | 1.405 ± 0.117 | 4 | 2.2 | 1.749 ± 0.062 | 6 | 3.2 | 1.384 ± 0.079 | 4 | 1.7 | 1.543 ± 0.179 | 6 | 2.1 |
| K250R/E96D | 3.515 ± 0.401 | 15 | 5.6 | 5.436 ± 0.421 | 9 | 9.9 | 2.572 ± 0.384 | 8 | 3.2 | 4.061 ± 1.175 | 7 | 6.4 |
| E96D + K250R | 20.397 ± 0.419 | 5 | 32.5 | ND | ND | ND | ||||||
| E96D + K250N | 10.165 ± 0.973 | 5 | 16.2 | ND | ND | ND | ||||||
| E96D + K250Q | 1.738 ± 0.165 | 7 | 2.8 | ND | ND | ND |
| RecA | No DNA dATP/S.D. | n | % WT | No DNA ATP/S.D. | n | % WT |
|---|---|---|---|---|---|---|
| WT | 0.744 ± 0.000 | 2 | 100.0 | 0.372 ± 0.058 | 2 | 100.0 |
| K250R | 0.661 ± 0.117 | 2 | 88.9 | 0.372 ± 0.058 | 2 | 100.0 |
| K250N | 0.331 ± 0.000 | 2 | 44.4 | 0.248 ± 0.000 | 2 | 66.7 |
| K250Q | 0.620 ± 0.175 | 2 | 83.3 | 0.413 ± 0.000 | 2 | 111.1 |
| E96D | 1.281 ± 0.292 | 2 | 172.2 | 1.818 ± 0.351 | 2 | 488.9 |
| K250R/E96D | 2.963 ± 0.263 | 7 | 398.4 | 4.339 ± 0.372 | 6 | 1166.7 |
The Mutant RecA Proteins Form Nucleoprotein Filaments—The ability of the mutant RecA proteins to bind DNA and form a nucleoprotein filament was examined by using electron microscopy to visualize the RecA proteins bound to poly(dT) linear ssDNA at pH 7.5 with dATP, as seen in Fig. 2. All reactions were diluted 10-fold before spreading, except the reaction with K250Q mutant protein that was diluted 5-fold. The reduced concentration of RecA K250Q nucleoprotein filaments indicated a reduced ability of this mutant protein to bind DNA and form a filament. No protein filaments were observed without DNA for any of the mutant proteins, which implied all nucleoprotein filaments were DNA-dependent.
FIGURE 2.

Wild-type and mutant RecA proteins form DNA-dependent nucleoprotein filaments. Electron micrographs show wild-type (WT) and mutant RecA proteins on linear ssDNA. Reactions included 4 μm RecA, 6 μm poly(dT) linear ssDNA, and 3 mm dATP at pH 7.5. The RecA protein was incubated with the DNA at 37 °C for 10 min before the addition of dATP. The reaction was incubated for another 10 min to allow the RecA protein to form a filament on the DNA before ATPγS was added (to 3 mm), and the reaction was incubated for 3 min to stabilize the filaments. Reactions were diluted 10-fold, except the K250Q reaction that was diluted 5-fold, before being adsorbed to the Alcian grid. Nucleoprotein filaments were viewed with the electron microscope. For each RecA protein, a total of at least 52 representative filaments was measured. Filaments shorter than 30 nm were difficult to distinguish from the background and were not included in the reported totals. A histogram of filament length for each protein is shown next to the electron microscopy image. Wild-type, K250R, and E96D proteins formed many filaments greater than 600 nm, implying end-to-end joining of filaments. The K250N and K250Q mutant RecA proteins formed shorter filaments, and K250Q filaments were reduced in concentration. A sample without DNA was examined for each RecA protein to test for DNA-independent filaments; all RecA proteins formed DNA-dependent filaments exclusively.
The poly(dT) linear ssDNA had an average length of 229 nt. The poly(dT) DNA was not observed when spread by itself with the cytochrome c method at the same magnification (46). Most of the filaments we observe with wild-type RecA protein reflect end-to-end joining of short filaments bound to the poly(dT). The average length of the filaments is thus a reflection of the interaction strength at the subunit-subunit interface in RecA filaments. The panels in Fig. 2 are typical for each of the mutant proteins. The longest filaments were seen with the wild-type protein and the K250R protein. The K250Q and K250N proteins produced filaments that were shorter and reduced in number, especially K250Q. The E96D mutant protein produced filaments nearly as long as the wild-type protein, as seen previously (46). The filaments generated for RecA E96D under these conditions were slightly shorter than those reported in our recent study (46), because the concentration of the mutant protein was decreased by 2-fold in this study.
The K250R Mutant RecA Protein Forms a Nucleoprotein Filament on M13mp8 Circular ssDNA—The wild-type RecA and E96D and K250R mutant proteins all bind to M13mp8 circular ssDNA and form complete circular filaments in the presence of SSB, as seen in Fig. 3. Reactions included 4 μm RecA, 6 μm M13mp8 circular ssDNA, 0.6 μm SSB, and 3 mm ATP. The K250R and E96D mutations resulted in circular filaments that appeared less smooth (most had one or more sharp bends) and somewhat shorter than the wild-type filaments. The average length of the wild-type RecA filament on M13mp8 was 3647.68 ± 56.15 nm. The average length of the RecA K250R filament on M13mp8 was 2987.09 ± 115.40 nm, and the average length of the RecA E96D filament on M13mp8 was 2466.65 ± 106.96 nm. We examined whether the shorter nucleoprotein filaments were because of the K250R and E96D mutations causing a reduction in the pitch of the nucleoprotein filament. The pitch of the nucleoprotein filament of each RecA protein bound to M13mp8 circular ssDNA was directly measured by examining linear segments of filaments where striations were clearly evident. The pitch varied for each RecA protein in a single nucleoprotein filament molecule. The pitch was measured directly in different areas of a single RecA-M13mp8 nucleoprotein filament molecule and among several molecules. The measured pitch of filaments on M13mp8 circular ssDNA for wild-type, K250R, and E96D RecA proteins were 10.972 ± 1.264 nm, 11.548 ± 1.484 nm, and 10.915 ± 1.136 nm, respectively. Within experimental error, our pitch measurements were the same as reported previously (43). Also within our error of measurement, the pitch of the RecA E96D and K250R nucleoprotein filaments was the same as the wild-type pitch and indicated the reduced length observed for E96D and K250R RecA filaments is not because of a reduced pitch of the nucleoprotein filament. In this type of sample preparation, the SSB protein binds to any long stretches of DNA not bound by RecA protein. No SSB-bound segments (easily characterized by their distinctive balls on a string appearance) were observed. It is possible that at the sharp bends of the E96D and K250R nucleoprotein filaments, unbound DNA was present that was too short to be bound by the SSB protein and thus could not be visualized. As discussed below, the reduced total filament lengths are not likely because of an impaired ability of K250R mutant RecA protein to form filaments in the presence of SSB. It is possible that the mutant RecA filaments are slightly less stable than the wild-type protein in these spreading conditions, and that some RecA protein is lost during spreading despite the ATPγS added to stabilize them.
FIGURE 3.

Wild-type, K250R, and E96D RecA mutant proteins form nucleoprotein filaments on M13mp8 circular ssDNA. Reactions were prepared as described for Fig. 2, except reactions included 4 μm RecA, 6 μm M13mp8 circular ssDNA, 0.6 μm SSB, and 3 mm ATP at pH 7.5. The RecA protein was incubated with the DNA at 37 °C for 10 min before the addition of ATP and SSB protein. The reaction was incubated for another 10 min to allow the RecA protein to form a filament on the DNA before ATPγS was added (to 3 mm), and the reaction was incubated for 3 min to stabilize the filaments. For each protein, the length of at least eight different molecules was measured three times and averaged. The pitch was directly examined by measuring the distances between individual striations in 8-11 areas of 6-7 different molecules for a total of 22-35 different pitch measurements for each protein. Wild-type (WT) RecA protein formed a nucleoprotein filament with an average length of 3647.68 ± 56.15 nm and an average pitch of 10.972 ± 1.264 nm. The E96D mutant protein formed a nucleoprotein filament with an average length of 2466.65 ± 106.96 nm and an average pitch of 10.915 ± 1.136 nm. The K250R mutant protein formed a nucleoprotein filament with an average length of 2987.09 ± 115.40 nm and an average pitch of 11.548 ± 1.484 nm. Within our error of measurement, the K250R mutant protein forms a nucleoprotein filament on M13mp8 circular ssDNA with the same pitch as wild-type RecA but with a length that is 81.9% of the wild-type length.
The K250R Mutant RecA Protein Catalyzes a Slow but Efficient (with Respect to Product Formation) DNA Three-strand Exchange Reaction—The ability of the mutant RecA proteins to catalyze the in vitro DNA three-strand exchange reaction was examined. As shown in Fig. 4, A and B, wild-type RecA protein catalyzed complete DNA three-strand exchange to form nicked circular product, with the first nicked circular product forming within 15 min with both ATP and dATP at pH 7.5. The RecA K250R mutant protein also catalyzed complete DNA three-strand exchange. However, the K250R mutant protein catalyzed the reaction at a rate much slower than wild-type RecA protein, with the first nicked circular product appearing within 60 min with either ATP or dATP at pH 7.5. The reduced rate of strand exchange catalyzed by the RecA K250R mutant protein was approximately proportional to the reduced rate of (d)ATP hydrolysis catalyzed by the mutant protein relative to wild-type RecA protein. The K250Q, K250N, and E96D mutant RecA proteins did not catalyze a complete DNA strand exchange reaction with either ATP or dATP at pH 7.5.
FIGURE 4.

DNA three-strand exchange catalyzed by wild-type and mutant RecA proteins. Reactions were carried out as described under “Experimental Procedures” and contained 10 μm ϕX174 circular ssDNA, 10 μm ϕX174 linear duplex DNA, 3 μm RecA protein or RecA protein mutant, 1 μm SSB protein, and 3 mm ATP or dATP. A and B, DNA three-strand exchange at pH 7.5 with ATP (A) or dATP (B); lds, linear dsDNA reactant; jm, joint molecule intermediate; nc, nicked circular product. Only wild-type (WT) and K250R mutant RecA proteins were able to catalyze complete DNA three-strand exchange at pH 7.5. C and D, DNA three-strand exchange with dATP at pH 6.6 (C) or pH 8.5 (D); lds, linear dsDNA reactant; jm, joint molecule intermediate; nc, nicked circular product. Reaction time points for each RecA protein were taken at 0 and 120 min. Only wild-type and K250R mutant RecA proteins were able to catalyze complete DNA three-strand exchange at pH 6.6 and pH 8.5.
The capacity of the mutant RecA proteins to catalyze DNA three-strand exchange at pH 6.6 and pH 8.5 with dATP was also examined. As seen in Fig. 4, C and D, both wild-type and K250R mutant RecA proteins catalyzed complete strand exchange to form nicked circular product at pH 6.6 and 8.5. A more efficient reaction was catalyzed at pH 6.6 than at pH 8.5, as indicated by the greater percentage of nicked circular product relative to linear dsDNA reactant. The K250Q, K250N, and E96D mutant RecA proteins were unable to catalyze DNA strand exchange at either pH 6.6 or pH 8.5, indicating that a pH change within this range does not improve their function.
Notably, although the final product yield of the reaction promoted by RecA K250R approached that of the reaction of the wild-type protein, the reaction proceeded much slower. Although products were clearly in evidence in the wild-type reaction at the 15-min time point, final products did not appear until 60-90 min after the reaction was initiated with RecA K250R. The slower generation of final products is roughly commensurate with the observed decrease in ATP hydrolysis, and is consistent with the coupling between ATP hydrolysis and DNA strand exchange reported elsewhere (18, 58, 59). The RecA K250R appears to be fully functional in DNA strand exchange; it is simply slow.
The K250R Mutant RecA Protein Can Displace SSB during Filament Extension—Nucleation of wild-type RecA protein onto ssDNA is inhibited by SSB pre-bound to the DNA. SSB stimulates extension of a RecA nucleoprotein filament if SSB is added to the reaction after allowing the RecA protein to nucleate on the ssDNA. SSB protein binds to and eliminates secondary structure than can form in ssDNA. After nucleation of RecA onto ssDNA, an extending RecA protein filament can displace SSB protein from the ssDNA and form a complete nucleoprotein filament to catalyze a maximal rate of (d)ATP hydrolysis. Poly(dT) linear ssDNA has a reduced propensity to form secondary structure and eliminates the requirement for the SSB protein. Wild-type RecA protein maintains the requirement for SSB protein to form a complete nucleoprotein filament on M13mp8 circular ssDNA. Without SSB, wild-type RecA protein is able to bind M13mp8 circular ssDNA and form a partial filament. We allowed wild-type and mutant RecA proteins to bind M13mp8 circular ssDNA without SSB. We then monitored the increase or decrease in dATP hydrolysis catalyzed by the RecA proteins upon addition of the SSB protein, reflecting the ability or inability, respectively, of the RecA protein to displace SSB during filament extension to form a full filament on the ssDNA. As seen in Fig. 5, both wild-type RecA and the K250R mutant RecA proteins were able to bind M13mp8 circular ssDNA and hydrolyze dATP. Both RecA proteins increased the rate of catalysis of dATP hydrolysis upon addition of SSB protein. This indicated the K250R mutant RecA protein is able to extend a nucleoprotein filament and displace SSB protein bound to the ssDNA. As mentioned earlier, the K250R RecA mutant protein bound to and coated 81.9% of M13mp8 circular ssDNA relative to wild-type RecA according to electron microscopic images of the nucleoprotein filaments. This was not likely because of the K250R RecA mutant protein being unable to compete with SSB to fully extend the nucleoprotein filament.
FIGURE 5.

K250R mutant RecA protein can extend a nucleoprotein filament against SSB protein bound to DNA. We monitored the hydrolysis of dATP catalyzed by wild-type (WT) and mutant RecA proteins when bound to M13mp8 circular ssDNA with and without SSB protein. Reactions included 4 μm RecA protein, 6 μm M13mp8 circular ssDNA, and 0.6 μm SSB at pH 7.5. The RecA protein was preincubated with the DNA for 10 min before dATP was added. Time 0 corresponds to the time of dATP addition. For all RecA proteins there are three conditions: SSB was added with dATP at time 0 min (continuous dashed line), SSB was added 12 or 30 min after dATP addition (solid line continued as dashed line after SSB addition indicated by an arrow), or SSB storage buffer was added 12 or 30 min after dATP addition (continuous solid line). SSB protein or storage buffer was added to the wild-type RecA protein reaction after 12 min, whereas SSB protein or storage buffer was added to the RecA mutant protein reactions after 30 min. The solid lines represent dATP hydrolysis before SSB protein was added to the reaction, and the dashed lines represent dATP hydrolysis after the addition of SSB protein. The decline or increase in the rate of dATP hydrolysis upon the addition of SSB protein was monitored. Both wild-type and K250R mutant RecA proteins were able to bind M13mp8 circular ssDNA without SSB protein. The addition of SSB protein stimulated the hydrolysis of dATP, presumably by binding to and melting secondary structure in the ssDNA to facilitate binding of the wild-type and K250R RecA proteins to the ssDNA and complete filament formation. This indicated the K250R mutant protein was able to extend a nucleoprotein filament against an SSB protein barrier.
A Combination of E96D and Lys-250 Mutant RecA Proteins Allows Limited Catalysis of dATP Hydrolysis—To study whether combining a Lys-250 mutant RecA protein with another subunit-subunit interface mutant protein could partially restore dATP hydrolysis activity to the nucleoprotein filament, we mixed each Lys-250 mutant RecA protein with the E96D mutant RecA protein in different ratios. Because the Glu-96 and Lys-250 residues are positioned on opposing sides of the subunit-subunit interface, the resulting mixture of single mutant proteins would generate a mixed filament featuring a combination of four different subunit-subunit interfaces (WT: WT, WT:K250X, E96D:WT, and E96D:K250X) as described previously (46). The E96D and Lys-250 mutant proteins were mixed in ratios varying from 1:3 to 1:2 to 1:1. As shown in Fig. 6, the maximal rate of dATP hydrolysis was observed at a 1:1 ratio of the E96D and Lys-250 mutant proteins. The E96D:K250Q, E96D:K250N, and E96D:K250R 1:1 mixtures restored dATP hydrolysis to 2.8, 16.2, and 32.5% of the wild-type rate, respectively (see Table 1).
FIGURE 6.

A mixture of the E96D and Lys-250 mutant proteins partially restores dATP hydrolysis activity to the RecA nucleoprotein filament. RecA-catalyzed dATP hydrolysis was monitored. Reactions included 4 μm total RecA protein and 6 μm poly(dT) linear ssDNA at pH 7.5. The maximal rate of dATP hydrolysis was observed at a 1:1 ratio of the E96D and Lys-250 mutant RecA proteins. This rate of dATP hydrolysis was 1.736 ± 0.165 μm/min for E96D:K250Q, 10.165 ± 0.973 μm/min for E96D:K250N, and 20.397 ± 0.419 μm/min for E96D:K250R mixed filaments compared with 62.761 ± 2.563 μm/min for the wild-type RecA protein under the same conditions.
The E96D:K250R mixed protein filament was examined further. One of the four possible subunit-subunit interfaces formed in the mixed filament would mimic that of the K250R/E96D double mutant protein. All subunit-subunit interfaces of the K250R/E96D double mutant protein have both mutations present together, and this mutant protein was purified and examined. As shown in Fig. 7A and Table 1, the K250R/E96D RecA mutant protein hydrolyzed (d)ATP at a relatively slow rate when bound to either poly(dT) linear ssDNA or M13mp8 circular ssDNA. We did observe some variability in the rate of dATP (but not ATP) hydrolysis, with some of the purified fractions (two of four separate preparations) increasing activity upon prolonged storage (4+ days) at 4 °C. The increased dATP hydrolysis did not signal a general activation of the protein, as it did not lead to the promotion of DNA three-strand exchange or even joint molecule formation (results not shown). An increase in NTP hydrolytic activity of this type has never before been observed with a RecA protein preparation in this laboratory, and we do not know to what to attribute it. The rates reported here, obtained with freshly thawed aliquots of the protein, were consistent from preparation to preparation.
FIGURE 7.

The K250R/E96D double mutant RecA protein hydrolyzes (d)ATP and binds DNA to form a nucleoprotein filament. A, RecA-catalyzed (d)ATP hydrolysis was monitored. Reactions included 4 μm RecA protein, 6 μm ssDNA, 0.6 μm SSB (only included in reactions with M13mp8), and 3 mm dATP or ATP at pH 7.5. The RecA protein was preincubated with the DNA for 10 min before (d)ATP(+SSB protein) was added. Time 0 corresponds to the time of (d)ATP addition. A solid line indicates reactions with dATP. A dashed line indicates reactions with ATP. Reactions for wild-type (WT) RecA protein are identical to those presented in Fig. 1 and Table 1. Rates of (d)ATP hydrolysis catalyzed by K250R/E96D mutant protein are 3.515 ± 0.401 μm/min with poly(dT) linear ssDNA and dATP, 5.436 ± 0.421 μm/min with poly(dT) linear ssDNA and ATP, 2.572 ± 0.384 μm/min with M13mp8 circular ssDNA and SSB protein and dATP, 4.061 ± 1.175 μm/min with M13mp8 circular ssDNA and SSB protein and ATP. B, electron micrograph showing K250R/E96D mutant RecA nucleoprotein filament with poly(dT) linear ssDNA and dATP at pH 7.5. Samples were prepared as those described in Fig. 2.
The DNA-independent rates of (d)ATP hydrolysis catalyzed by the K250R/E96D protein were slightly less than the DNA-dependent rates (Table 1). The DNA-independent rates for K250R/E96D were also faster than DNA-independent rates for the other RecA wild-type and mutant proteins. This suggests that the K250R and E96D mutations act together, perhaps synergistically, to stimulate a faster rate of (d)ATP hydrolysis in the absence of DNA.
The ability of K250R/E96D mutant protein to bind DNA and form a filament was examined using electron microscopy to visualize the protein bound to poly(dT) linear ssDNA with dATP. As shown in Fig. 7B, the K250R/E96D mutant protein does bind to and form nucleoprotein filaments on DNA, although the average length of the filaments did not approach that of the wild-type or K250R or E96D proteins. Thus, filament formation is compromised to a degree by the double mutant relative to either of the constituent single mutants. Despite the ability of the K250R/E96D mutant protein to bind DNA and catalyze (d)ATP hydrolysis, it cannot catalyze complete strand exchange at pH 7.5 with dATP (results not shown). The double mutant protein has a reduced capacity to displace SSB on ssDNA and did not form complete filaments on the M13mp8 ssDNA (data not shown). Individually, the E96D mutant protein binds DNA and forms a nucleoprotein filament, but it does not catalyze (d)ATP hydrolysis relative to wild-type RecA. A combination of the K250R and E96D mutations, whether in the E96D:K250R mixed filament or the K250R/E96D double mutant protein, restored limited (d)ATP hydrolysis activity to the nucleoprotein filament, to the rates were greater than those observed with E96D but reduced relative to K250R. The E96D mutant performs better in (d)ATP hydrolysis when an arginine is present across the subunit-subunit interface at position 250 than it does when a lysine residue is found at position 250, although the arrangement negatively affects filament formation.
Slow Growth Phenotype Associated with the Expression of RecA K250R in Vivo
Expression of RecA K250R Curtails Growth in E. coli—The RecA K250R mutant protein was expressed from its own promoter on plasmid pEAW451 and used to transform E. coli strain MG1655 ΔrecA. Unlike cells transformed with control plasmids expressing either the wild-type RecA protein or no protein, the cells expressing RecA K250R produced very small colonies. Individual colonies were picked and grown up in liquid media, and growth of the culture was monitored. Results are shown in Fig. 8. Transformation with the unmodified plasmid or with a plasmid expressing wild-type RecA protein had little effect on the growth curve. In contrast, transformation with a plasmid expressing RecA K250R led to a severe curtailment of growth in rich media. The culture did not approach stationary phase even after 9 h (Fig. 8A).
FIGURE 8.

Expression of RecA K250R confers a slow growth phenotype. The E. coli strain MG1655 ΔrecA was transformed with one of a series of plasmids expressing wild-type RecA protein, RecA K250R protein, another RecA variant, or no protein from the wild-type recA promoter as described under “Experimental Procedures.” The growth of cultures derived from selected transformed colonies is illustrated. All growth curves were repeated at least two times with consistent results. A, growth of cells expressing RecA K250R is compared with the growth of cells expressing wild-type RecA or no protein. B, growth of cells expressing RecA K250R is compared with the growth of cells expressing RecA variants containing both the K250R alteration and a second mutation that suppresses the slow growth phenotype.
Suppressor mutations appeared quite rapidly both on plates and in liquid culture. Thirty four separate suppressor mutants were analyzed by sequencing the plasmid-borne recA gene. All had an alteration of the recA K250R coding sequence (Table 2). In each case, the change in the plasmid sequence accounted for the suppression of the slow growth phenotype, because subsequent transformation resulted in recovery of culture growth to levels near that exhibited by cells transformed by plasmids expressing no gene or the wild-type recA gene (Fig. 8B).
TABLE 2.
Suppressor mutations isolated in cells expressing RecA K250R
Suppressor mutations were collected and identified as described under “Experimental Procedures.”
| Suppressor no. of colonies | Mutants of recA K250R plasmid pEAW451 mutation |
|---|---|
| 11 | IS10 insertion between Asp-100 and His-97 |
| 1 | IS10 insertion between His-97 and Asp-94 |
| 1 | IS10 insertion between Ser-117 and Leu-114 |
| 1 | Deletion between the recA promoter and Asn-236 |
| 1 | Deletion after Gly-59 to beyond the end of the gene |
| 1 | Gln-173 change to stop codon |
| 5 | Missing first base of amino acids 123 = frameshift after Gly-122 |
| 1 | A214E + K250R |
| 2 | E154A + K250R |
| 4 | A11V + K250R |
| 3 | L99R + K250R |
| 1 | A147V + K250R |
| 2 | I195N + K250R |
Sensitivity to UV light was measured for cells transformed with plasmids expressing each of the 13 different altered recA genes detected as slow growth suppressors, as well as the wild-type recA gene and recA K250R itself. Colonies appeared slowly in cells expressing RecA K250R, and counts indicated that survival of UV irradiation was reduced relative to cells expressing wild-type RecA protein. However, the reduction was less than that observed for a recA null mutant strain, indicating that some repair function was retained. For each of the suppressors, the UV resistance of the strains was reduced to levels comparable with cells lacking RecA function except in one case. Cells expressing the RecA double mutant K250R/A11V (Fig. 9) exhibited a partial retention of RecA function, comparable with the level of function seen in cells expressing RecA K250R.
FIGURE 9.

One RecA variant with a mutation that suppresses the slow growth phenotype of RecA K250R retains some RecA function. The UV sensitivity of cells expressing various RecA protein variants was measured as described under “Experimental Procedures.” The UV sensitivity of cells expressing RecA K250R falls between that of cells expressing wild-type (wt) RecA or those lacking RecA function. The A147V suppressor (see Table 2) is 1 of 12 different mutations that yield a recA null phenotype. The A11V suppressor mutation, in combination with K250R, retains the UV sensitivity seen with K250R alone.
We carried out additional experiments to determine whether the slow growth defect imparted by recA K250R expression could be suppressed by defects in other recombination functions. As shown in Fig. 10, the loss of recFOR function largely suppressed the slow growth phenotype. Of the three genes, loss of recF function consistently had the most beneficial effect. The same loss of recFOR function had no effect on cells transformed with the same plasmid expressing wild-type RecA. These results indicate that expression of the RecA K250R protein is less toxic if it cannot be loaded onto the DNA by the RecFOR mediator system, an event that is normally associated with recombinational DNA repair of replication forks. We attempted to examine the effects of expression of RecA K250R in cells lacking recB function. In four trials, we were unable to select any viable transformants when introducing plasmid pEAW451 into EAW81 (MG1655ΔrecAΔrecB) cells. Side-by-side in two of the same trials, transformants were obtained readily when the plasmids expressing either wild-type RecA protein or no gene at all were introduced. The results suggest that expression of RecA K250R is lethal in cells lacking RecB function.
FIGURE 10.

The slow growth phenotype of cells expressing RecA K250R is suppressed by recFOR mutations. An E. coli strain (MG1655 ΔrecA) and variants of the same strain lacking recF, recO, or recR function were transformed with plasmids expressing either wild-type (WT) RecA protein or RecA K250R protein from the wild-type recA promoter as described under “Experimental Procedures.” The growth of cultures derived from selected transformed colonies is illustrated. All growth curves were repeated at least two times with consistent results.
The A11V Suppressor Mutation Improves the Dynamics of RecA K250R Filaments—We purified the RecA K250R/A11V double mutant protein and characterized it. The protein promoted ATP hydrolysis and DNA strand exchange at rates that were quite comparable with the reactions promoted by RecA K250R (Fig. 11). Because the A11V mutation relieved the slow growth phenotype but did not significantly alter the rates of ATP hydrolysis or strand exchange, the “slow” nature of the reactions promoted by the RecA K250R mutant protein was not responsible for the slow growth of strains expressing it.
FIGURE 11.

The A11V mutation does not affect the rates of ATP hydrolysis and DNA strand exchange promoted by the RecA K250R protein. Reactions were carried out as described under “Experimental Procedures.” A, ATP hydrolysis promoted by RecA and RecA variants. Reactions contained 8 μm M13mp18 circular ssDNA, 5 μm RecA protein, or RecA protein mutant, 0.8 μm SSB protein, and 3 mm ATP. B, DNA strand exchange. Reactions were carried out and quantitated as described under “Experimental Procedures,” and the percent of duplex DNA present as the nicked circular product of strand exchange is plotted. Reactions contained 10 μm ϕX174 circular ssDNA, 10 μm ϕX174 linear duplex DNA, 4 μm RecA protein, or RecA protein mutant, 1 μm SSB protein, and 3 mm ATP. WT, wild type.
In a search for RecA filament properties that might be affected by the A11V alteration, we next examined filament dynamics. One convenient way to assess filament dynamics is to challenge reactions with a large addition of RecA K72R protein. RecA K72R can bind but not hydrolyze ATP (31, 60). If RecA protein dissociates from a filament and is replaced by RecA K72R, the net rate of ATP hydrolysis declines. RecA filament dissociation can thus be monitored as a decline in ATP hydrolysis over time. We carried out such experiments for the wild-type, K250R, and K250R/A11V variants of RecA protein (Fig. 12). As noted in previous work (61), filaments of wild-type RecA protein are quite dynamic when they are promoting DNA strand exchange. Addition of homologous dsDNA produces a slight decline in the rate of ATP hydrolysis, reflecting a filament change in state that accompanies the onset of DNA strand exchange (62, 63). In this state, there is measurable RecA dissociation and reassociation (61), as seen here when these filaments are challenged with RecA K72R. The rate of ATP hydrolysis promoted by the RecA K250R during DNA strand exchange was considerably reduced relative to that exhibited by wild-type RecA protein, as already noted. Addition of homologous dsDNA to initiate strand exchange had only a small effect on the rate of ATP hydrolysis. Notably, the addition of RecA K72R caused a small (33%) but abrupt decline in ATP hydrolysis. However, the new steady state was maintained in the presence of RecA K72R for over 7 h. The result was substantially different when the RecA K250R/A11V double mutant protein was examined. The decline in ATP hydrolysis upon challenge with K72R was more substantial and progressive over the first 2 h after challenge. The overall decline was just under 80%. The results suggest that the A11V mutation enhances the rate of dissociation of RecA from filaments promoting DNA strand exchange. This enhancement of filament dynamics provides a possible explanation for the relief of the slow growth phenotype in cells expressing the double mutant protein.
FIGURE 12.

The A11V mutation improves RecA filament dynamics relative to filaments made with RecA K250R. Reactions were carried out as described under “Experimental Procedures” and contained 4 μm M13mp18 circular ssDNA, 4 μm M13mp18 linear duplex DNA (where indicated), 2.5 μm RecA protein or RecA protein mutant, 0.4 μm SSB protein, and 3 mm ATP. A-C show reactions carried out by wild-type (WT) RecA protein, RecA K250R protein, and RecA K250R/A11V protein, respectively. ATP hydrolysis is monitored spectrophotometrically, as described under “Experimental Procedures.” Three reactions are shown for each protein. The linear duplex DNA was added to two reactions at the time indicated, illustrating the decline in ATP hydrolysis that generally accompanies the initiation of DNA strand exchange. One of these two reactions was further challenged by the addition of 5 μm RecA K72R protein at the time indicated. In each panel, the top line (labeled NA for no addition) represents a reaction that contains only ssDNA and the indicated RecA protein, with no dsDNA or RecAK72R additions. lds, linear dsDNA reactant.
DISCUSSION
This work allows us to draw two primary conclusions. First, the RecA K250R is a slow RecA protein variant. Its properties help to establish that the (KR)X(KR) motif has the following functions in RecA reactions. (a) These residues (at least Lys-248) are directly involved in the ATP hydrolytic activity of RecA, promoting ATP hydrolysis in trans across the subunit-subunit interface. (b) The (KR)X(KR) motif has a major role in coordinating the ATP hydrolytic cycles of adjacent subunits. (c) The Lys-250 residue has a role in governing the rate of RecA-mediated ATP hydrolysis. (d) The motif also has a role in coupling RecA-mediated ATP hydrolysis to work (in this case DNA strand exchange). A polar but uncharged residue is ineffective in substituting for the positive charge of Lys or Arg at position 250. Second, recombinational DNA repair occurs very commonly during the E. coli cell cycle. RecA protein plays a sufficiently intimate role in the recombinational DNA repair of stalled replication forks that the properties of RecA can limit the progression of the entire cell cycle.
The first conclusion arises from observations in this and a previous (46) study. Catalysis of ATP hydrolysis in trans is evident from the effects of changes at position 250 (K250R) relative to the E96D mutation present on the opposing side of the subunit-subunit interface. An even more dramatic compensation was observed in the RecA K248R mutant protein (46). The residues of the (KR)X(KR) motif are thus good candidates for mediators of subunit-subunit coordination of ATP hydrolytic cycles. This role is confirmed by the slow rate of ATP hydrolysis exhibited by the otherwise quite active K250R mutant.
RecA K250R is a bona fide slow RecA protein mutant. It forms filaments and promotes DNA strand exchange much like the wild-type protein does. However, it catalyzes both ATP hydrolysis and DNA strand exchange at much slower rates than the wild-type protein. Lys-250 clearly has a role in governing the rate of ATP hydrolysis in RecA. The mutant protein forms nucleoprotein filaments readily. The fact that the rates of both the ATP hydrolysis and DNA strand exchange are reduced by commensurate amounts reinforces the conclusion of a coupling between ATP hydrolysis and DNA strand exchange that has been evident in previous studies (18, 33, 58, 59, 64, 65), and it implies a role for Lys-250 in this coupling.
The K250N and K250Q mutant RecA proteins are much less robust than K250R. This appears to imply a requirement for not just a polar group at this position near the ATPase active site but a requirement for a group with a positive charge.
The (KR)X(KR) motif appears to be a feature of bacterial RecA proteins, but it is not present in eukaryotic RecA homologues. It is tempting to speculate that this motif represents one of the structural features that gives rise to the reactions or properties specific to bacterial RecA proteins.
Many attempts have been made to estimate the frequency with which replication forks stall or collapse during the bacterial replication cycle. Most of the data is based on the deleterious effects seen when a key repair function is removed (1, 4, 8-11). There are multiple types of DNA lesions that might affect fork progression, and multiple potential avenues for repair. Even for repair functions that have significant roles in replication fork repair and restart, the observed effects on cell survival may be small if redundant pathways for restart exist. The RecA K250R mutant protein has properties that uniquely implicate RecA as a surprisingly integrated component in the replication cycle. This is not a null mutant that does not bind to DNA and can be bypassed by other repair or replication restart mechanisms. It is also not binding randomly to dsDNA (RecFOR does not load RecA onto dsDNA, and recFOR mutants would not suppress the RecA K25R defect if random loading onto dsDNA was the mechanism of growth inhibition). RecFOR loads this mutant protein onto DNA, presumably at DNA gaps. When bound at those gaps, the protein functions, but it does so slowly. Perhaps more important, it does not readily dissociate when its function is completed. The progress of the cell cycle is curtailed dramatically. The associated slow growth phenotype is suppressed only by eliminating RecA function or by enhancing the dynamics of the RecA filament.
The results are potentially relevant to many recent proposals concerning the progress of replication forks that encounter DNA damage. Given the very slow rate of growth of cells expressing RecA K250R, it appears that most cells in the population undergo a repair and restart process involving RecA during every cell cycle. Recent estimates of low frequencies of double strand break occurrence (13) may indicate that many fork stalling events occur without generating such breaks. It is clear that replication restart mechanisms exist that permit the bypass of lesions without significant replication stalling (14). However, the current results and recent studies of UV-irradiated cells (66) indicate that there is a significant class of events requiring RecA function that are not readily bypassed. The apparent synthetic lethality of combining the expression of RecA K250R with a recB null allele suggests one pathway that might be operative. Based on the results seen with recFOR mutations, loss of RecA loading by RecB might be expected to be favorable rather than lethal. Thus, some other function of RecB must be involved in the lethality. If RecA K250R slowly generated regressed replication forks (33, 34) and then failed to dissociate, processing of the short arm of the resulting Holliday intermediate might require the action of the RecB nuclease (11, 67).
It is not the slow rate of ATP hydrolysis and DNA strand exchange mediated by RecA K250R that results in the slow growth phenotype of cells expressing this mutant protein. The A11V mutation in RecA effectively suppresses the slow growth phenotype of recA K250R but has only a minimal (if any) effect on the rates of ATP hydrolysis and strand exchange. The K250R mutation thus has the role delineated above in coordinating the ATP hydrolytic cycles of adjacent RecA subunits in a filament, and the amino-terminal domain (where A11V resides) appears to have a previously unappreciated role in subunit dissociation. Notably, although A11V suppresses slow growth, it does not improve the resistance of the cells to UV irradiation. The slow rates of reactions promoted by RecA K250R may not limit growth on their own, but they may well have a deleterious effect on a repair process that affects survival.
As this paper was being submitted, a new paper appeared reporting the first elucidation of a RecA filament bound to single-stranded DNA (68). This work confirms the concept of a different structure for active filaments, and it strongly reinforces the importance of Lys-248 and Lys-250 at the subunit-subunit interface of a RecA filament. Both Lys residues coordinate to the AlF4 stand-in for the γ-phosphate of ATP. Lys-250 also forms a hydrogen bond to Glu-96 (68). The authors suggest that Lys-248 and Lys-250 act as γ-phosphate sensors that help to establish the ATP dependence of DNA binding. The work highlights three mechanisms by which RecA stimulates ATP, two of which involve Lys-248 and Lys-250 (68).
Acknowledgments
We thank Elizabeth A. Wood for cloning of the RecA mutant proteins. We also thank the Martin Laboratory at the University of Wisconsin-Madison, Department of Biochemistry, for use of their MetaMorph analysis software.
This work was supported, in whole or in part, by National Institutes of Health Grant GM32335. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: dsDNA, double-stranded DNA; ATPγS, adenosine 5′-O-(3-thiotriphosphate); nt, nucleotide(s); PEP, phosphoenolpyruvate; SSB, single-stranded DNA-binding protein of E. coli; ssDNA, single-stranded DNA; MES, 2-(N-morpholino)ethanesulfonic acid.
References
- 1.Cox, M. M., Goodman, M. F., Kreuzer, K. N., Sherratt, D. J., Sandler, S. J., and Marians, K. J. (2000) Nature 40437 -41 [DOI] [PubMed] [Google Scholar]
- 2.Cox, M. M. (2001) Proc. Natl. Acad. Sci. U. S. A. 988173 -8180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kowalczykowski, S. C. (2000) Trends Biochem. Sci. 25156 -165 [DOI] [PubMed] [Google Scholar]
- 4.Kuzminov, A. (1999) Microbiol. Mol. Biol. Rev. 63751 -813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cox, M. M. (2007) in Topics in Current Genetics (Rothstein, R., and Aguilera, A., eds) pp.53 -94, Springer-Verlag, Heidelberg
- 6.Lusetti, S. L., and Cox, M. M. (2002) Annu. Rev. Biochem. 7171 -100 [DOI] [PubMed] [Google Scholar]
- 7.Schlacher, K., Cox, M. M., Woodgate, R., and Goodman, M. F. (2006) Nature 442883 -887 [DOI] [PubMed] [Google Scholar]
- 8.Rothstein, R., Michel, B., and Gangloff, S. (2000) Genes Dev. 141 -10 [PubMed] [Google Scholar]
- 9.Michel, B., Flores, M. J., Viguera, E., Grompone, G., Seigneur, M., and Bidnenko, V. (2001) Proc. Natl. Acad. Sci. U. S. A. 988181 -8188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michel, B., Grompone, G., Flores, M. J., and Bidnenko, V. (2004) Proc. Natl. Acad. Sci. U. S. A 10112783 -12788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michel, B., Boubakri, H., Baharoglu, Z., LeMasson, M., and Lestini, R. (2007) DNA Repair 6 967-980 [DOI] [PubMed] [Google Scholar]
- 12.Vilenchik, M. M., and Knudson, A. G. (2003) Proc. Natl. Acad. Sci. U. S. A 10012871 -12876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pennington, J. M., and Rosenberg, S. M. (2007) Nat. Genet. 39797 -802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heller, R. C., and Marians, K. J. (2006) Nature 439557 -562 [DOI] [PubMed] [Google Scholar]
- 15.Brenner, S. L., Mitchell, R. S., Morrical, S. W., Neuendorf, S. K., Schutte, B. C., and Cox, M. M. (1987) J. Biol. Chem. 2624011 -4016 [PubMed] [Google Scholar]
- 16.Cox, M. M. (2004) in The Bacterial Chromosome (Higgins, N. P., ed) pp.369 -388, American Society of Microbiology, Washington, DC
- 17.Cox, M. M. (2007) Crit. Rev Biochem. Mol. Biol. 4241 -63 [DOI] [PubMed] [Google Scholar]
- 18.Cox, M. M. (2007) Nat. Rev. Mol. Cell Biol. 8127 -138 [DOI] [PubMed] [Google Scholar]
- 19.Hobbs, M. D., Sakai, A., and Cox, M. M. (2007) J. Biol. Chem. 28211058 -11067 [DOI] [PubMed] [Google Scholar]
- 20.Shan, Q., Bork, J. M., Webb, B. L., Inman, R. B., and Cox, M. M. (1997) J. Mol. Biol. 265519 -540 [DOI] [PubMed] [Google Scholar]
- 21.Umezu, K., and Kolodner, R. D. (1994) J. Biol. Chem. 26930005 -30013 [PubMed] [Google Scholar]
- 22.Arnold, D. A., and Kowalczykowski, S. C. (2000) J. Biol. Chem. 27512261 -12265 [DOI] [PubMed] [Google Scholar]
- 23.Churchill, J. J., and Kowalczykowski, S. C. (2000) J. Mol. Biol. 297537 -542 [DOI] [PubMed] [Google Scholar]
- 24.Anderson, D. G., Churchill, J. J., and Kowalczykowski, S. C. (1999) J. Biol. Chem. 27427139 -27144 [DOI] [PubMed] [Google Scholar]
- 25.Morimatsu, K., and Kowalczykowski, S. C. (2003) Mol. Cell 111337 -1347 [DOI] [PubMed] [Google Scholar]
- 26.Spies, M., and Kowalczykowski, S. C. (2006) Mol. Cell 21573 -580 [DOI] [PubMed] [Google Scholar]
- 27.Cox, J. M., Tsodikov, O. V., and Cox, M. M. (2005) PLoS Biol. 3231 -243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jain, S. K., Cox, M. M., and Inman, R. B. (1994) J. Biol. Chem. 26920653 -20661 [PubMed] [Google Scholar]
- 29.Kim, J. I., Cox, M. M., and Inman, R. B. (1992) J. Biol. Chem. 26716444 -16449 [PubMed] [Google Scholar]
- 30.Kim, J. I., Cox, M. M., and Inman, R. B. (1992) J. Biol. Chem. 26716438 -16443 [PubMed] [Google Scholar]
- 31.Shan, Q., Cox, M. M., and Inman, R. B. (1996) J. Biol. Chem. 2715712 -5724 [DOI] [PubMed] [Google Scholar]
- 32.Cox, M. M. (2003) Annu. Rev. Microbiol. 57551 -577 [DOI] [PubMed] [Google Scholar]
- 33.Robu, M. E., Inman, R. B., and Cox, M. M. (2001) Proc. Natl. Acad. Sci. U. S. A 988211 -8218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robu, M. E., Inman, R. B., and Cox, M. M. (2004) J. Biol. Chem. 27910973 -10981 [DOI] [PubMed] [Google Scholar]
- 35.Leipe, D. D., Aravind, L., Grishin, N. V., and Koonin, E. V. (2000) Genome Res. 10 5-16 [PubMed] [Google Scholar]
- 36.Ye, J., Osborne, A. R., Groll, M., and Rapoport, T. A. (2004) Biochim. Biophys. Acta 1659 1-18 [DOI] [PubMed] [Google Scholar]
- 37.Sawaya, M. R., Guo, S., Tabor, S., Richardson, C. C., and Ellenberger, T. (1999) Cell 99 167-177 [DOI] [PubMed] [Google Scholar]
- 38.Singleton, M. R., Sawaya, M. R., Ellenberger, T., and Wigley, D. B. (2000) Cell 101589 -600 [DOI] [PubMed] [Google Scholar]
- 39.Crampton, D. J., Guo, S., Johnson, D. E., and Richardson, C. C. (2004) Proc. Natl. Acad. Sci. U. S. A. 1014373 -4378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scheffzek, K., Lautwein, A., Kabsch, W., Reza Ahmadian, M., and Wittinghofer, A. (1996) Nature 384591 -596 [DOI] [PubMed] [Google Scholar]
- 41.Story, R. M., Weber, I. T., and Steitz, T. A. (1992) Nature 355318 -325 [DOI] [PubMed] [Google Scholar]
- 42.Story, R. M., and Steitz, T. A. (1992) Nature 355374 -376 [DOI] [PubMed] [Google Scholar]
- 43.VanLoock, M. S., Yu, X., Yang, S., Lai, A. L., Low, C., Campbell, M. J., and Egelman, E. H. (2003) Structure (Camb.) 111 -20 [DOI] [PubMed] [Google Scholar]
- 44.Wu, Y., He, Y., Moya, I. A., Qian, X. G., and Luo, Y. (2004) Mol. Cell 15 423-435 [DOI] [PubMed] [Google Scholar]
- 45.Conway, A. B., Lynch, T. W., Zhang, Y., Fortin, G. S., Fung, C. W., Symington, L. S., and Rice, P. A. (2004) Nat. Struct. Mol. Biol. 11791 -796 [DOI] [PubMed] [Google Scholar]
- 46.Cox, J. M., Abbott, S. N., Chitteni-Pattu, S., Inman, R. B., and Cox, M. M. (2006) J. Biol. Chem. 28112968 -12975 [DOI] [PubMed] [Google Scholar]
- 47.Nguyen, T. T., Muench, K. A., and Bryant, F. R. (1993) J. Biol. Chem. 2683107 -3113 [PubMed] [Google Scholar]
- 48.Campbell, M. J., and Davis, R. W. (1999) J. Mol. Biol. 286417 -435 [DOI] [PubMed] [Google Scholar]
- 49.Campbell, M. J., and Davis, R. W. (1999) J. Mol. Biol. 286437 -445 [DOI] [PubMed] [Google Scholar]
- 50.Lusetti, S. L., Wood, E. A., Fleming, C. D., Modica, M. J., Korth, J., Abbott, L., Dwyer, D. W., Roca, A. I., Inman, R. B., and Cox, M. M. (2003) J. Biol. Chem. 27816372 -16380 [DOI] [PubMed] [Google Scholar]
- 51.Craig, N. L., and Roberts, J. W. (1981) J. Biol. Chem. 2568039 -8044 [PubMed] [Google Scholar]
- 52.Lohman, T. M., and Overman, L. B. (1985) J. Biol. Chem. 2603594 -3603 [PubMed] [Google Scholar]
- 53.Neuendorf, S. K., and Cox, M. M. (1986) J. Biol. Chem. 2618276 -8282 [PubMed] [Google Scholar]
- 54.Lindsley, J. E., and Cox, M. M. (1990) J. Biol. Chem. 2659043 -9054 [PubMed] [Google Scholar]
- 55.Morrical, S. W., Lee, J., and Cox, M. M. (1986) Biochemistry 251482 -1494 [DOI] [PubMed] [Google Scholar]
- 56.Drees, J. C., Lusetti, S. L., Chitteni-Pattu, S., Inman, R. B., and Cox, M. M. (2004) Mol. Cell 15 789-798 [DOI] [PubMed] [Google Scholar]
- 57.Schnos, M., and Inman, R. B. (2000) Mol. Biotechnol. 1677 -86 [DOI] [PubMed] [Google Scholar]
- 58.Bedale, W. A., and Cox, M. (1996) J. Biol. Chem. 2715725 -5732 [DOI] [PubMed] [Google Scholar]
- 59.Nayak, S., and Bryant, F. R. (1999) J. Biol. Chem. 27425979 -25982 [DOI] [PubMed] [Google Scholar]
- 60.Rehrauer, W. M., and Kowalczykowski, S. C. (1993) J. Biol. Chem. 2681292 -1297 [PubMed] [Google Scholar]
- 61.Shan, Q., and Cox, M. M. (1997) J. Biol. Chem. 27211063 -11073 [DOI] [PubMed] [Google Scholar]
- 62.Haruta, N., Yu, X. N., Yang, S. X., Egelman, E. H., and Cox, M. M. (2003) J. Biol. Chem. 27852710 -52723 [DOI] [PubMed] [Google Scholar]
- 63.Schutte, B. C., and Cox, M. M. (1987) Biochemistry 265616 -5625 [DOI] [PubMed] [Google Scholar]
- 64.MacFarland, K. J., Shan, Q., Inman, R. B., and Cox, M. M. (1997) J. Biol. Chem. 27217675 -17685 [DOI] [PubMed] [Google Scholar]
- 65.Shan, Q., and Cox, M. M. (1998) Mol. Cell 1309 -317 [DOI] [PubMed] [Google Scholar]
- 66.Rudolph, C. J., Upton, A. L., and Lloyd, R. G. (2007) Genes Dev. 21668 -681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Uzest, M., Ehrlich, S. D., and Michel, B. (1995) Mol. Microbiol. 171177 -1188 [DOI] [PubMed] [Google Scholar]
- 68.Chen, Z. C., Yang, H. J., and Pavletich, N. P. (2008) Nature 453489 -494 [DOI] [PubMed] [Google Scholar]