

Nitrogen–carbon bonds are ubiquitous in products ranging from chemical feedstock to pharmaceuticals. As ammonia is among the least expensive bulk chemicals produced in the largest volume, one of the greatest challenges of synthetic chemistry is to develop atom-efficient processes for the combination of NH3 with simple organic molecules to create nitrogen–carbon bonds. Transition-metal complexes can readily render a variety of N–H bonds reactive enough to undergo functionalization, including those of primary and secondary amines. However, with a few exceptions,[1,2] metals react with ammonia to afford supposedly inert Lewis acid–base complexes, as first recognized in the late 19th century by Werner.[3] Consequently, the homogeneous catalytic functionalization of NH3 remained elusive[4] until the recent discovery by Shen and Hartwig[5] and Surry and Buchwald[6] of the palladium-catalyzed coupling of aryl halides with ammonia in the presence of a stoichiometric amount of a base. An even more appealing process would be the addition of NH3 to carbon–carbon multiple bonds, a process that would occur ideally with 100% atom economy.[7] Although various homogeneous catalysts, including alkali metals,[8] early[9] and late transition metals,[10] and d-[11] and f-block elements,[12] have been used to effect the so-called hydroamination reaction, none of them were reported to be effective when NH3 is used as the amine partner.[13] Herein we report that cationic gold(I) complexes supported by a cyclic (alkyl)(amino)carbene (CAAC)[14] ligand readily catalyze the addition of ammonia to a variety of unactivated alkynes and allenes to provide a diverse array of linear and cyclic nitrogen-containing compounds.
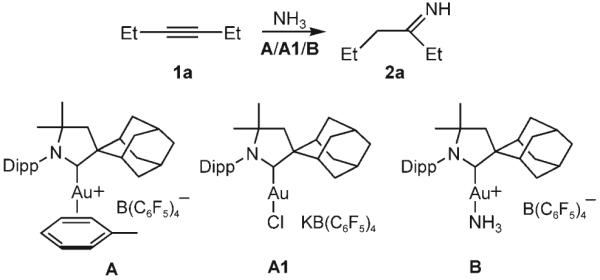
We showed recently that the cationic CAAC–gold complex A was very robust and exhibited unusual catalytic reactivity towards alkynes.[15] This discovery prompted us to investigate whether such a complex could activate alkynes sufficiently to enable the addition of NH3.[16] Thus, excess ammonia was condensed into a sealable NMR tube containing A (5 mol%), 3-hexyne, and deuterated benzene. Upon heating to 160 °C for 3.5 h, the clean addition of NH3 afforded the primary imine 2a, the expected tautomer of the corresponding enamine (Table 1).[17]
Table 1.
Catalytic hydroamination of 3-hexyne with ammonia.[a]
 | |||
|---|---|---|---|
| Catalyst [mol%] | t [h] | T [°C] | Conv.[%] |
| 5.0 | 3.5 | 160 | 95 |
| 1.0 | 14 | 160 | 95 |
| 0.1 | 17 | 160 | 58 |
| 0.1 | 20 | 200 | 93 |
Dipp = 2,6-diisopropylphenyl.
Complex A does not have to be isolated; when it was prepared in situ from an equimolar mixture of [(CAAC)AuCl]/KB(C6F5)4 (A1), identical results were obtained. When the related silver complex [(CAAC)AgCl]/KB(C6F5)4 or NH4B(C6F5)4 was used as the catalyst, no reaction was observed, which shows the importance of gold and rules out a Brønsted acid mediated reaction.[18] Finally, as AuCl, AuCl/ KB(C6F5)4, and even [(CAAC)AuCl] do not induce the hydroamination, it is clear that the gold center can only catalyze the addition of NH3 if it is coordinated by the CAAC ligand and rendered cationic by Cl abstraction.
To gain insight into the catalytic process, we performed a number of experiments: The addition of excess NH3 to complex A gave the Werner complex B instantaneously; the addition of 3-hexyne (1a; 1 equiv) to complex A gave the η2-bound alkyne complex C instantaneously (Scheme 1). Upon the exposure of a solution of C in benzene to excess NH3, 3-hexyne was immediately displaced from the gold center, and the Werner complex B was isolated in quantitative yield. This result suggests that NH3 does not add to the alkyne through an outer-sphere mechanism. Importantly, when a solution of complex B in benzene was treated at room temperature for 24 h with a large excess of 3-hexyne, the imine complex D was obtained quantitatively, even when the reaction vessel was open to a glovebox atmosphere. This experiment implies that NH3 does not dissociate from the metal by a simple ligand exchange with the alkyne. Therefore, an insertion mechanism, similar to that proposed by Tanaka and co-workers[19] and Nishina and Yamamoto[20] for gold-catalyzed hydroamination with aryl amines, is quite likely. Finally, the addition of excess NH3 to D liberated the imine 2a and regenerated complex B. From the results of these experiments, it can be concluded that the Werner complex B is the resting state of the catalyst. Indeed, B exhibits identical catalytic activity to that of A/A1. Consequently, the robust and readily available complex B (Figure 1)[21] was used in subsequent experiments.
Scheme 1.
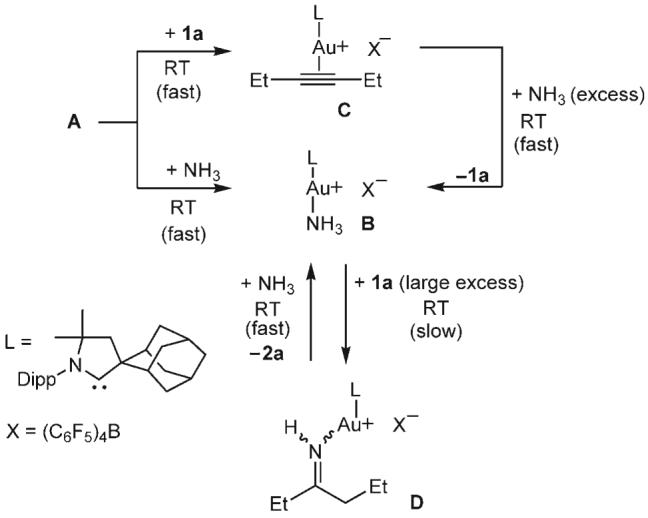
Experiments to probe the reaction mechanism. The results suggest that an insertion process is involved, and that B is the resting state of the catalyst.
Figure 1.
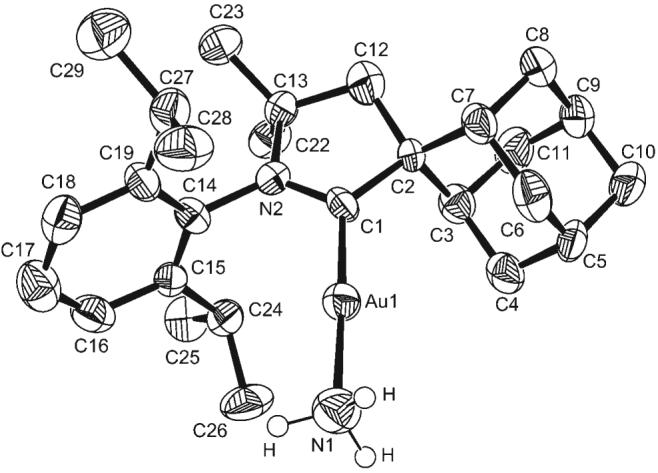
Molecular structure of complex B in the solid state. (Hydrogen atoms, except those at N1, and the (C6F5)4B anion are omitted for clarity; ellipsoids are drawn at 50% probability).
To test the scope of the reaction, the terminal alkyne 1b and the diaryl alkyne 1c were treated with NH3 in the presence of a catalytic amount of complex B (Scheme 2). With 1b, the reaction took place even at 110 °C to afford the Markovnikov imine 2b exclusively in 60% yield. When diphenyl acetylene (1c) was used, the 2-aza-1,3-diene 3c was formed cleanly in 95% yield. The different outcome of the reaction of 1c with respect to the results with substrates 1a,b can be rationalized by the presence of acidic benzylic hydrogen atoms in the imine. These acidic hydrogen atoms favor the formation of the enamine tautomer, which can then react further with a second molecule of the alkyne to afford 3c.
Scheme 2.
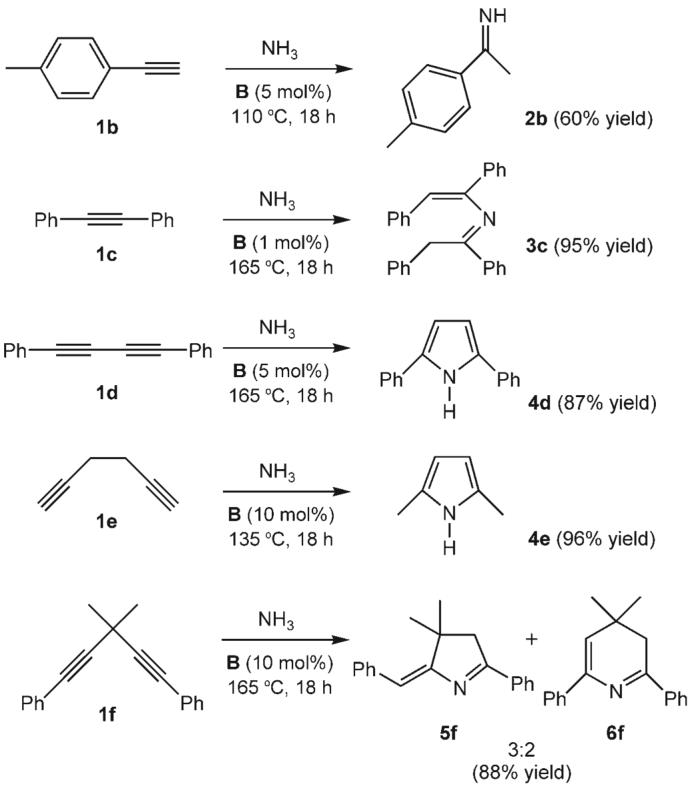
Catalytic hydroamination of various alkynes with ammonia.
Nitrogen heterocycles are an important class of compounds that occur widely in natural products and often display potent biological activity. On the basis of the results described above, we attempted the direct synthesis of hetero-cycles from diynes and NH3. When 1,4-diphenylbuta-1,3-diyne (1d) and hexa-1,5-diyne (1e) were used, the corresponding 2,5-disubstituted pyrroles 4d and 4e were produced in 87 and 96% yield, respectively. Both products result from the Markovnikov addition of NH3, followed by ring-closing hydroamination.[22,23] The treatment of the 1,4-diyne 1f with NH3 under similar conditions led to a 3:2 mixture of the five- and six-membered heterocycles 5f and 6f in 88% yield. The six-membered ring 6f arises from two consecutive Markovnikov hydroamination reactions, whereas the formation of 5f involves an anti-Markovnikov addition of NH3 or ring-closing step.
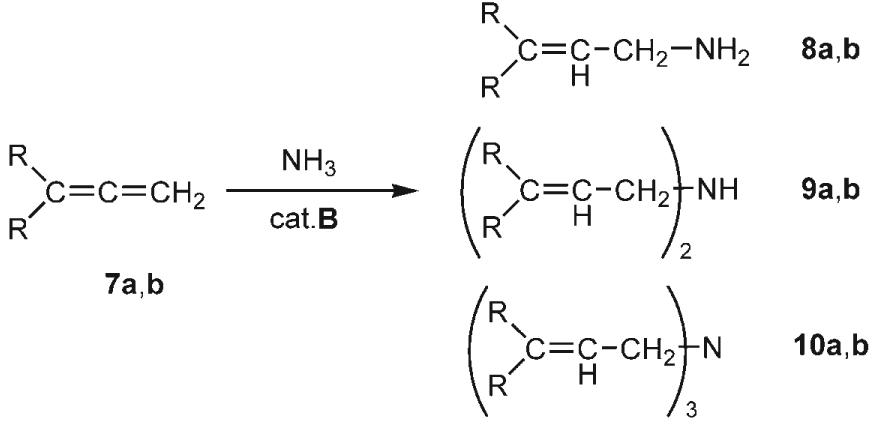
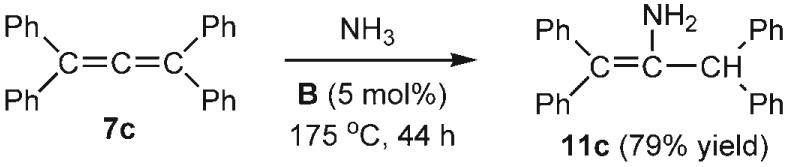
To expand the scope of the hydroamination reaction with NH3, we next tested allenes as substrates. When 1,2-propadiene (7a) was used, a mixture of mono- (8a), di- (9a) and triallylamine (10a) was obtained in excellent yield. Allyl amines are among the most versatile intermediates in synthesis and are of industrial importance. For example, the parent compound 8a, which is produced commercially from ammonia and allyl chloride, is used in antifungal preparations and the synthesis of polymers. By varying the NH3/allene ratio it is possible to control the selectivity of this reaction significantly (Table 2). In particular, the parent allylamine (8a) and triallylamine (10a) can be obtained with 86 and 91% selectivity, respectively, and further optimization of the conditions should be possible. The addition of NH3 to 1,2-dienes is not restricted to the parent allene 7a. The dialkyl-substituted derivative 7b was also converted into the corresponding allyl amines 8b–10b, with exclusive addition of the NH2 group at the less-hindered terminus; however the selectivity of this reaction for the mono-, di-, or trisubstituted amine product needs some improvement. Interestingly, even the tetrasubstituted allene 7c underwent hydroamination with ammonia. Probably because of steric factors, a different regioselectivity was observed, and only the monohydroamination product 11C was formed.[24]
Table 2.
Catalytic hydroamination of allenes with ammonia.
 | |||||||
|---|---|---|---|---|---|---|---|
| R | NH3/7 | B[mol%] | t [h] | T [°C] | Conv. [%] | 8/9/10 | |
| 1.4:1 | 1.2 | 22 | 175 | 98 | 1:4.3:3.7 | ||
| a | H | 40:1 | 4.3 | 16 | 175 | 96 | 6.2:1:– |
| 1:4.5 | 1.4 | 36 | 165 | 91 | 1:6:73 | ||
| b | Me | 0.8:1 | 1.3 | 22 | 155 | 87 | 1:18:47 |
 | |||||||
The results outlined herein demonstrate that (CAAC)-gold(I) cations readily catalyze the addition of NH3 to non-activated alkynes and allenes. This reaction leads to reactive nitrogen-containing compounds, such as imines, enamines, and allyl amines, and is therefore an ideal initial step for the preparation of simple bulk chemicals, as well as rather complex molecules, as illustrated by the preparation of heterocycles 4–6. This study paves the way for the discovery of catalysts that mediate the addition of ammonia to simple alkenes, a process considered to be one of the ten greatest challenges for catalytic chemistry.[25]
Experimental Section
All manipulations were performed under an inert atmosphere of argon by using standard Schlenk techniques. Water- and oxygen-free solvents were employed.
General procedure
The catalyst B (15 mg) and the appropriate amount (see Tables 1 and 2 and Scheme 2,) of an alkyne or allene were loaded into a Wilmad QPV thick-walled (1.4 mm) NMR tube. C6D6 (0.4 mL) and the internal standard benzyl methyl ether (5 mg) were added to the mixture. For experiments with a low catalyst loading (0.1 mol%), 3 mg of the catalyst and 0.1 mL of C6D6 were used. The NMR tube was connected to a high-vacuum manifold, and excess NH3 (typically 3–6 equivalents) was carefully condensed at −60 °C. For experiments with 1,2-propadiene, the allene was first condensed, with the subsequent addition of NH3. The tube was sealed, placed in an oil bath behind a blast shield, and heated at the specified temperature. (Caution: Sealed NMR tubes containing NH3 and/or 1,2-propadiene are under high pressure and pose an explosion hazard. Only new tubes with a wall thickness of at least 1.4 mm should be used).
B: Excess NH3 (approximately 1 mL) was condensed into a solution of A (1.00 g, 0.74 mmol) in toluene (3 mL) at −50 °C. The mixture was stirred for 1 min and then removed from the cold bath. The excess NH3 was removed, hexane (50 mL) was added, and the upper portion of the biphasic mixture was removed with a canula. The oily residue was dried under a high vacuum to afford complex B (0.90 g, 95%) as a colorless solid. M.p.: 112–114 °C; 1H NMR (300 MHz, C6D6): δ=1.19 (d, 3J=6.7 Hz, 6H, CH(CH3)2), 1.22 (d, 3J=6.7 Hz, 6H, CH(CH3)2), 1.30 (s, 6H, C(CH3)2), 1.70–1.97 (m, 12H), 2.31 (s, 2H, CH2), 2.53 (br s, 3H, NH3), 2.60 (sept, 3J=6.7 Hz, 2H, CH(CH3)2), 3.50 (d, 2J=12.0 Hz, 2H, CH2), 7.18 (d, J=7.7 Hz, 2H), 7.35 ppm (t, J=7.7 Hz, 1H); 13C NMR (75 MHz, C6D6): δ=22.8, 26.8, 27.4, 28.5, 28.8, 29.4, 34.4 (CH2), 35.9 (CH2), 37.3, 39.1 (CH2), 48.1 (CH2), 64.2 (Cq), 78.7 (NCq), 125.6 (CHm), 131.0 (CHp), 135.5 (ci), 135.6 (m, B–CAr), 137.4 (m, B–CAr), 138.9 (m, B–CAr), 140.8 (m, B–CAr), 145.1 (co), 147.9 (m, B–CAr), 151.1 (m, B–CAr), 236.7 ppm (Ccarbene); HRMS (ESI; CH3CN): m/z calcd for C29H42AuN2: 615.3008 [(M–NH3+CH3CN)]+; found: 615.3010; m/z calcd for C28H40AuN2: 601.2857 [(M–NH3+HCN)]+; found: 601.2856.
C: 3-Hexyne (1 equiv) was added to a solution of A (1.00 g, 0.74 mmol) in toluene (3 mL). The mixture was stirred for 1 min, and then hexane (50 mL) was added. The upper portion of the biphasic mixture was removed with a canula, and the oily residue was dried under a high vacuum to afford complex C (0.95 g, 96%) as a colorless solid. M.p.: 183–184 °C; 1H NMR (300 MHz, C6D6): δ=0.93 (t, 3J=7.5 Hz, 6H, CH2CH3), 1.24 (d, 3J=6.7 Hz, 6H, CH(CH3)2), 1.26 (d, 3J=6.7 Hz, 6H, CH(CH3)2), 1.37 (s, 6H, C(CH3)2), 1.73–1.98 (m, 12H), 2.12 (q, 3J=7.5 Hz, 4H, CH2CH3), 2.38 (s, 2H, CH2), 2.69 (sept, 3J=6.7 Hz, 2H, CH(CH3)2), 3.33 (d, 2J=12.7 Hz, 2H, CH2), 7.26 (d, J=7.7 Hz, 2H), 7.41 ppm (t, J=7.7 Hz, 1H); 13C NMR (75 MHz, C6D6): δ=13.7 (CH2CH3), 15.4 (CH2CH3), 23.2, 26.5, 27.3, 28.4, 28.9, 29.5, 34.2 (CH2), 36.5 (CH2), 37.3, 38.8 (CH2), 48.5 (CH2), 65.2 (Cq), 79.9 (NCq), 87.5 (C≡C), 126.2 (CHm), 131.3 (CHp), 135.9 (m, B–CAr), 135.8 (ci), 137.5 (m, B–CAr), 139.1 (m, B–CAr), 141.0 (m, B–CAr), 145.3 (co), 148.0 (m, B–CAr), 151.1 (m, B–CAr), 243.9 ppm (Ccarbene); HRMS (ESI; CH3CN): m/z calcd for C29H42AuN2: 615.3008 [(M–C6H10+CH3CN)]+; found: 615.3011.
D: 3-Hexyne (6.47 g, 78.7 mmol) was added to a solution of B (0.50 g, 0.39 mmol) in benzene (3 mL). The mixture was stirred for 24 h, and then hexane (100 mL) was added. The upper portion of the biphasic mixture was removed with a canula, and the oily residue was dried under a high vacuum to afford complex D (0.50 g, 99%; cis/trans 55:45) as a colorless solid. 1H NMR (300 MHz, CDCl3): δ=0.80 (t, J=7.1 Hz, 3H, CH3), 0.88 (t, J=7.8 Hz, 3H, CH3), 0.89 (t, J=7.8 Hz, 3H, CH3), 1.07 (t, J=7.1 Hz, 3H, CH3), 1.29 (d, 3J=6.7 Hz, 6H, CH(CH3)2), 1.30 (d, 3J=6.7 Hz, 6H, CH(CH3)2), 1.33 (br d, 3J=7.1 Hz, 12H, C(CH3)2), 1.41 (s, 6H, C(CH3)2), 1.42 (s, 6H, C(CH3)2), 1.45–1.55 (br m, J=7.2 Hz, 4H, CH2), 1.75–2.18 (m, 24H), 2.29 (t, J=8.2 Hz, 4H, HNC(CH2CH2CH3)), 2.40 (q, J=7.1 Hz, 4H, HNC-(CH2CH3), 2.42 (s, 4H, CH2), 2.76 (sept, 3J=6.7 Hz, 4H, CH(CH3)2), 3.50–3.75 (br m, 4H), 7.30 (d, J=7.7 Hz, 4H), 7.46 (t, J=7.7 Hz, 2H), 8.30–8.40 ppm (br s, 2H, NH); 13C NMR (75 MHz, CDCl3): δ=8.6 (CH2CH3), 11.1 (CH2CH3), 13.1 (CH2CH3), 13.8 (CH2CH3), 18.5 (CH2CH3), 20.2 (CH2CH3), 23.0, 23.1, 26.6, 26.7, 26.9, 27.9, 29.2, 29.4, 32.5 (CH2), 33.8 (CH2), 34.3 (CH2), 35.7 (CH2), 37.2, 38.8 (CH2), 40.7 (CH2), 42.4 (CH2), 48.5 (CH2), 64.3 (Cq), 64.4 (Cq), 78.6 (2 × NCq), 125.5 (CHm), 130.5 (CHp), 130.6 (CHp), 134.9 (m, B–CAr), 135.8 (br, ci), 136.8 (m, B–CAr), 138.0 (m, B–CAr), 140.0 (m, B–CAr), 145.0 (co), 145.1 (co), 146.6 (m, B–CAr), 150.1 (m, B–CAr), 199.6 (C=N), 200.2 (C=N), 238.1 (Ccarbene), 238.6 (Ccarbene).
Footnotes
We are grateful to the NIH (R01 GM 68825) and RHODIA Inc. for financial support of this research, the Alexander von Humboldt Foundation for a Postdoctoral Fellowship (G.D.F.), and the ACS for a Graduate Fellowship (V.L.).
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Bryan EG, Johnson BFG, Lewis K. J. Chem. Soc. Dalton Trans. 1977:1328–1330. [Google Scholar]; b) Hillhouse GL, Bercaw JE. J. Am. Chem. Soc. 1984;106:5472–5478. [Google Scholar]; c) Casalnuovo AL, Calabrese JC, Milstein D. Inorg. Chem. 1987;26:971–973. [Google Scholar]; d) Holl MMB, Wolczanski PT, Van Duyne GD. J. Am. Chem. Soc. 1990;112:7989–7994. [Google Scholar]; e) Zhao J, Goldman AS, Hartwig JF. Science. 2005;307:1080–1082. doi: 10.1126/science.1109389. [DOI] [PubMed] [Google Scholar]; f) Nakajima Y, Kameo H, Suzuki H. Angew. Chem. 2006;118:964–966. doi: 10.1002/anie.200503222. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2006;45:950–952. doi: 10.1002/anie.200503222. [DOI] [PubMed] [Google Scholar]
- 2.A nonmetallic system was reported recently to cleave NH3 under mild experimental conditions: Frey GD, Lavallo V, Donnadieu B, Schoeller WW, Bertrand G. Science. 2007;316:439–441. doi: 10.1126/science.1141474. see also:Kenward AL, Piers WE. Angew. Chem. 2008;120:38–42. doi: 10.1002/anie.200702816.Angew. Chem. Int. Ed. 2008;47:38–41.Lynam JM. Angew. Chem. 2008;120:843–845.Angew. Chem. Int. Ed. 2008;47:831–833. doi: 10.1002/anie.200704305.
- 3.Werner A. Z. Anorg. Chem. 1893;3:267. [Google Scholar]
- 4.A few examples of functionalization with NH3 in the presence of heterogeneous catalysts have been reported: Bodke AS, Olschki DA, Schmidt LD. Appl. Catal. A. 2000:13–22.Horn R, Mestl G, Thiede M, Jentoft FC, Schmidt PM, Bewersdorf M, Weber R, Schlögl R. Phys. Chem. Chem. Phys. 2004;6:4514–4521.Reppe W. Justus Liebigs Ann. Chem. 1956;601:81–84., andReppe W. Justus Liebigs Ann. Chem. 1956;601:128–138.Deeba M, Ford ME. J. Org. Chem. 1988;53:4594–4596.Mizuno N, Tabata M, Uematsu T, Iwamoto M. J. Catal. 1994;146:249–256.Hölderich WF. Catal. Today. 2000;62:115–130.Penzien J, Haessner C, Jentys A, Köhler K, Müller TE, Lercher JA. J. Catal. 2004;221:302–312.Pez GP, Galle JE. Pure Appl. Chem. 1985;57:1917–1926.Howk BW, Little EL, Scott SL, Whitman GM. J. Am. Chem. Soc. 1954;76:1899–1902.
- 5.Shen Q, Hartwig JF. J. Am. Chem. Soc. 2006;128:10028–10029. doi: 10.1021/ja064005t. [DOI] [PubMed] [Google Scholar]
- 6.Surry DS, Buchwald SL. J. Am. Chem. Soc. 2007;129:10354–10355. doi: 10.1021/ja074681a. [DOI] [PubMed] [Google Scholar]
- 7.For reviews, see: Severin R, Doye S. Chem. Soc. Rev. 2007;36:1407–1420. doi: 10.1039/b600981f.Matsunaga S. J. Synth. Org. Chem. Jpn. 2006;64:778–779.Hultzsch KC. Adv. Synth. Catal. 2005;347:367–391.Beller M, Seayad J, Tillack A, Jiao H. Angew. Chem. 2004;116:3448–3479. doi: 10.1002/anie.200300616.Angew. Chem. Int. Ed. 2004;43:3368–3398. doi: 10.1002/anie.200300616.Alonso F, Beletskaya IP, Yus M. Chem. Rev. 2004;104:3079–3159. doi: 10.1021/cr0201068.Roesky PW, Müller TE. Angew. Chem. 2003;115:2812–2814.Angew. Chem. Int. Ed. 2003;42:2708–2710. doi: 10.1002/anie.200301637.Pohlki F, Doye S. Chem. Soc. Rev. 2003;32:104–114. doi: 10.1039/b200386b.Müller TE, Beller M. Chem. Rev. 1998;98:675–703. doi: 10.1021/cr960433d.
- 8.a) Hartung CG, Breindl C, Tillack A, Beller M. Tetrahedron. 2000;56:5157–5162. [Google Scholar]; b) Horrillo-Martinez P, Hultzsch KC, Gil A, Branchadell V. Eur. J. Org. Chem. 2007:3311–3325. [Google Scholar]; c) Datta S, Gamer MT, Roesky PW. Organometallics. 2008;27:1207–1213. [Google Scholar]
- 9.For reviews, see: Roesky PW. Z. Anorg. Allg. Chem. 2006;632:1918–1926.Odom AL. Dalton Trans. 2005:225–233. doi: 10.1039/b415701j.Hazari N, Mountford P. Acc. Chem. Res. 2005;38:839–849. doi: 10.1021/ar030244z.Bytschkov I, Doye S. Eur. J. Org. Chem. 2003:935–946.Smolensky E, Kapon M, Eisen MS. Organometallics. 2007;26:4510–4527.Dochnahl M, Löhnwitz K, Pissarek J-W, Biyikal M, Schulz SR, Schön S, Meyer N, Roesky PW, Blechert S. Chem. Eur. J. 2007;13:6654–6666. doi: 10.1002/chem.200601765.
- 10.For recent examples, see: Zhang J, Yang C-G, He C. J. Am. Chem. Soc. 2006;128:1798–1799. doi: 10.1021/ja053864z.Komeyama K, Morimoto T, Takaki K. Angew. Chem. 2006;118:3004–3007. doi: 10.1002/anie.200503789.Angew. Chem. Int. Ed. 2006;45:2938–2941. doi: 10.1002/anie.200503789.Michael FE, Cochran BM. J. Am. Chem. Soc. 2006;128:4246–4247. doi: 10.1021/ja060126h.Johns AM, Sakai N, Ridder A, Hartwig JF. J. Am. Chem. Soc. 2006;128:9306–9307. doi: 10.1021/ja062773e.Takemiya A, Hartwig JF. J. Am. Chem. Soc. 2006;128:6042–6043. doi: 10.1021/ja058299e.Chianese AR, Lee SJ, Gagné MR. Angew. Chem. 2007;119:4118–4136.Angew. Chem. Int. Ed. 2007;46:4042–4059. doi: 10.1002/anie.200603954.Kovács G, Ujaque G, Lledós A. J. Am. Chem. Soc. 2008;130:853–864. doi: 10.1021/ja073578i.
- 11.For recent examples, see: Meyer N, Lohnwitz K, Zulys A, Roesky PW, Dochnahl M, Blechert S. Organometallics. 2006;25:3730–3734.Liu XY, Li CH, Che CM. Org. Lett. 2006;8:2707–2710. doi: 10.1021/ol060719x.Zulys A, Dochnahl M, Hollmann D, Lohnwitz K, Herrmann JS, Roesky PW, Blechert S. Angew. Chem. 2005;117:7972–7976.Angew. Chem. Int. Ed. 2005;44:7794–7798. doi: 10.1002/anie.200502006.
- 12.a) Gribkov DV, Hultzsch KC, Hampel F. J. Am. Chem. Soc. 2006;128:3748–3759. doi: 10.1021/ja058287t. [DOI] [PubMed] [Google Scholar]; b) Riegert D, Collin J, Meddour A, Schulz E, Trifonov A. J. Org. Chem. 2006;71:2514–2517. doi: 10.1021/jo052322x. [DOI] [PubMed] [Google Scholar]; c) Arnea E, Eisen MS. Coord. Chem. Rev. 2006;250:855–859. [Google Scholar]; d) Hong S, Marks TJ. Acc. Chem. Res. 2004;37:673–686. doi: 10.1021/ar040051r. [DOI] [PubMed] [Google Scholar]; e) Andrea T, Eisen MS. Chem. Soc. Rev. 2008;37:550–567. doi: 10.1039/b614969n. [DOI] [PubMed] [Google Scholar]; f) Aillaud I, Collin J, Hannedouche J, Schulz E. Dalton Trans. 2007:5105–5118. doi: 10.1039/b711126f. [DOI] [PubMed] [Google Scholar]; g) Rastätter M, Zulys A, Roesky PW. Chem. Eur. J. 2007;13:3606–3616. doi: 10.1002/chem.200601510. [DOI] [PubMed] [Google Scholar]
- 13.The hydroamination of short-chain alkenes with NH3 has been reported with zeolites[4d-g] and alkali metals as catalysts.[4h,i]
- 14.a) Lavallo V, Canac Y, Präsang C, Donnadieu B, Bertrand G. Angew. Chem. 2005;117:5851–5855. doi: 10.1002/anie.200501841. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2005;44:5705–5709. doi: 10.1002/anie.200501841. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lavallo V, Canac Y, DeHope A, Donnadieu B, Bertrand G. Angew. Chem. 2005;117:7402–7405. doi: 10.1002/anie.200502566. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2005;44:7236–7239. doi: 10.1002/anie.200502566. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Jazzar R, Dewhurst RD, Bourg JB, Donnadieu B, Canac Y, Bertrand G. Angew. Chem. 2007;119:2957–2960. doi: 10.1002/anie.200605083. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:2899–2902. doi: 10.1002/anie.200605083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Lavallo V, Frey GD, Kousar S, Donnadieu B, Bertrand G. Proc. Natl. Acad. Sci. USA. 2007;104:13569–13573. doi: 10.1073/pnas.0705809104. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Frey GD, Dewhurst RD, Kousar S, Donnadieu B, Bertrand G. J. Organomet. Chem. 2008;693:1674–1682. doi: 10.1016/j.jorganchem.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For recent reviews on gold chemistry, see: Widenhoefer RA, Han X. Eur. J. Org. Chem. 2006:4555–4563.Hashmi ASK. Chem. Rev. 2007;107:3180–3211. doi: 10.1021/cr000436x.Fürstner A, Davies PW. Angew. Chem. 2007;119:3478–3519.Angew. Chem. Int. Ed. 2007;46:3410–3449. doi: 10.1002/anie.200604335.Gorin DJ, Toste FD. Nature. 2007;446:395–403. doi: 10.1038/nature05592.
- 17.Primary imines are valuable intermediates for a variety of transformations, as exemplified by the dual-metal-catalyzed hydroaminomethylation with ammonia to give terminal primary amines via aldimines: Zimmermann B, Herwig J, Beller M. Angew. Chem. 1999;111:2515–2518. doi: 10.1002/(sici)1521-3773(19990816)38:16<2372::aid-anie2372>3.0.co;2-h.Angew. Chem. Int. Ed. 1999;38:2372–2375. doi: 10.1002/(sici)1521-3773(19990816)38:16<2372::aid-anie2372>3.0.co;2-h. Interestingly, the hydrogenation of ketimines 2a would lead to the complementary internal primary amine.
- 18.A few examples of acid-catalyzed hydroamination reactions (without NH3) have been reported: Schlummer B, Hartwig JF. Org. Lett. 2002;4:1471–1474. doi: 10.1021/ol025659j.Motokura K, Nakagiri N, Mori K, Mizugaki T, Ebitani K, Jitsukawa K, Kaneda K. Org. Lett. 2006;8:4617–4620. doi: 10.1021/ol0619821.Rosenfeld DC, Shekhar S, Takemiya A, Utsunomiya M, Hartwig JF. Org. Lett. 2006;8:4179–4182. doi: 10.1021/ol061174+.Anderson LL, Arnold J, Bergman RG. J. Am. Chem. Soc. 2005;127:14542–14543. doi: 10.1021/ja053700i.Lapis AM, Neto B. A. DaSilveira, Scholten JD, Nachtigall FA, Eberlin MN, Dupont J. Tetrahedron Lett. 2006;47:6775–6779.Li ZG, Zhang JL, Brouwer C, Yang CG, Reich NW, He C. Org. Lett. 2006;8:4175–4178. doi: 10.1021/ol0610035.
- 19.Mizushima E, Hayashi T, Tanaka M. Org. Lett. 2003;5:3349–3352. doi: 10.1021/ol0353159. [DOI] [PubMed] [Google Scholar]
- 20.Nishina N, Yamamoto Y. Angew. Chem. 2006;118:3392–3395. [Google Scholar]; Angew. Chem. Int. Ed. 2006;45:3314–3317. doi: 10.1002/anie.200600331. [DOI] [PubMed] [Google Scholar]
- 21.CCDC-677987 (B) and CCDC-677988 (5f) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 22.For examples of the synthesis of pyrrole derivatives by the catalytic hydroamination of diynes with primary amines, see: Ramanathan B, Keith AJ, Armstrong D, Odom AL. Org. Lett. 2004;6:2957–2960. doi: 10.1021/ol0489088.Chalk AJ. Tetrahedron. 1974;30:1387–1391.Schulte KE, Reisch J, Walker H. Chem. Ber. 1965;98:98–103.
- 23.Iminoalkynes are known to undergo cyclization readily, even in the absence of a catalyst: Sakamoto T, Kondo Y, Yamanaka H. Heterocycles. 1988;27:2225–2249.
- 24.A similar regioselectivity was observed in the intramolecular hydroamination of allenes: Ackermann L, Bergman RG. Org. Lett. 2002;4:1475–1478. doi: 10.1021/ol0256724.
- 25.Haggin J. Chem. Eng. News. 1993;71:23. [Google Scholar]