

Abstract
A convergent and enantioselective synthesis of the natural product amurensinine is described. The synthetic strategy takes advantage of mild and selective C–H and C–C bond insertion reactions, in addition to the palladium-catalyzed aerobic oxidative kinetic resolution recently developed in these laboratories.
The development of mild and selective C–H and C–C bond insertion reactions for complex molecule synthesis has the potential to enable strategic disconnections that would be inconceivable using traditional methods.1 Because of the sheer abundance of C–H and C–C bonds in most organic molecules and the general shortage of methods to perform selective reactions on these bonds, the application of C–H and C–C activation reactions in the context of natural product synthesis has been limited. Despite these challenges, the number of examples continues to rise.2 In this communication, we present a convergent and enantioselective synthesis of the natural product amurensinine (1) that employs mild and selective C–H and C–C bond insertion reactions as key strategic maneuvers.
Amurensinine (1) is a member of the isopavine family of alkaloids, which are exemplified by a characteristic tetracyclic tetrahydroisoquinoline core structure consisting of a doubly benzannulated azabicyclo[3.2.2]nonane (Figure 1).3,4 The isopavines exhibit important biological properties for the treatment of neurological disorders, such as Parkinson's and Alzheimer's disease.5 To date, there has been only one reported enantioselective synthesis of amurensinine (1), which was based on a chiral auxiliary approach.6
Figure 1.
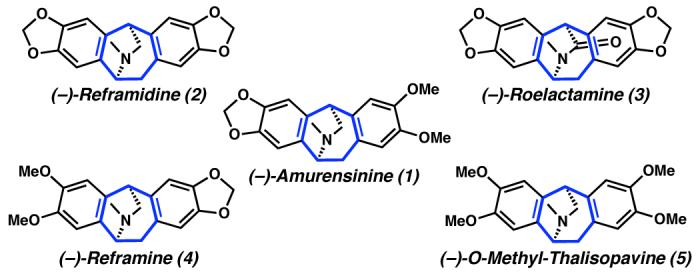
Representative isopavine natural products.
Our retrosynthetic strategy for the preparation of amurensinine ((+)-1) commences with the disconnection of the bridging amine functionality, exposing hydroxyester 6 as a synthetic intermediate (Scheme 1). We reasoned that this chiral benzylic alcohol could be produced enantioselectively by application of the palladium-catalyzed oxidative kinetic resolution methodology, recently developed in our group.7,8 Alcohol (±)-6 could be accessed from ketoester (±)-7, which contains the benzosuberane core carbocycle, an ideal retron for an efficient and mild C–C bond insertion reaction involving the acylalkylation of arynes, previously reported by our laboratories.9,10 Thus, aryne 8 and β-ketoester 9 were revealed as substrates for the C–C bond insertion reaction. The former may be generated in situ from o-trimethylsilyl triflate 10,11 and the latter by a position-selective Rh-catalyzed C–H bond insertion reaction of diazo compound 11.12
Scheme 1.
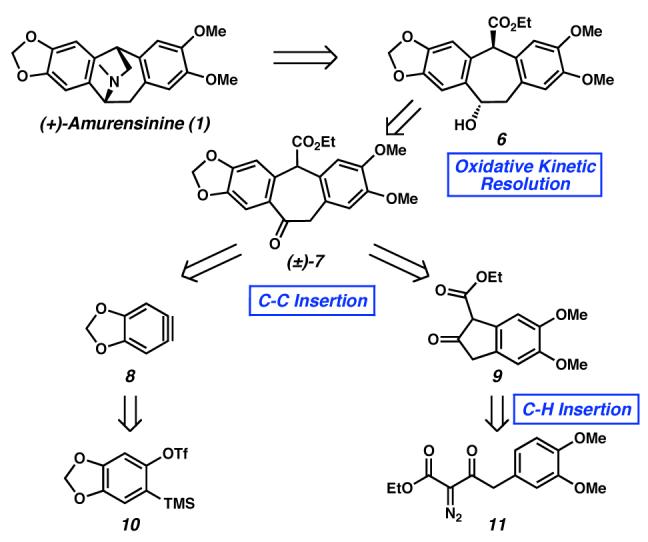
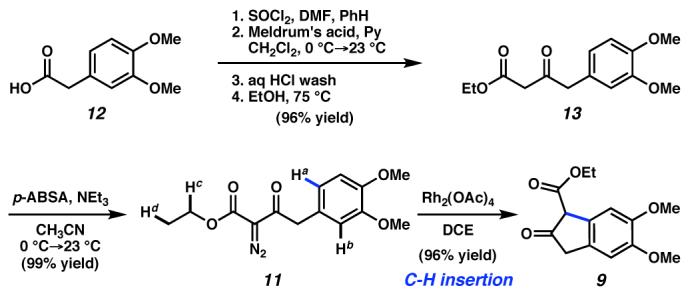
We began our efforts toward amurensinine ((+)-1) with the preparation of β-ketoester 9 (Scheme 2). Functionalization of (3,4-dimethoxyphenyl)acetic acid (12) by standard methods produced diazo compound 11, which was subjected to Rh2(OAc)4-catalyzed dediazotization.13 Despite the possibility for intramolecular insertion into a number of C–H bonds at sp3 and sp2 carbon centers (i.e., Ha-d), as well as intermolecular reactions, we observed the product of a single C–H insertion event into the aryl C–Ha bond producing the desired β-ketoester 9 in 96% yield.
Scheme 2.
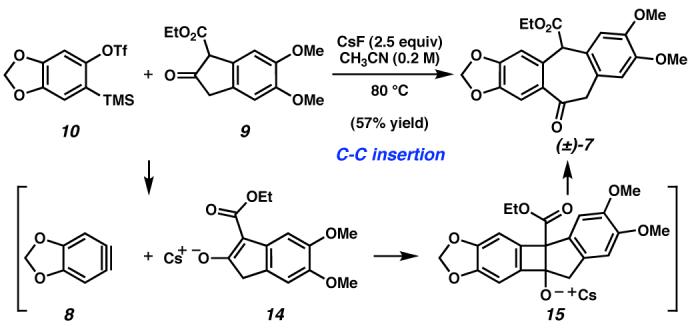
In the key bond-forming reaction of the synthesis, coupling of β-ketoester 9 and aryne precursor 109 in the presence of CsF produced ketoester (±)-7 in 57% isolated yield (Scheme 3). This single-step C–C insertion reaction generates the polycyclic carbon framework of amurensinine by direct acyl-alkylation of 8. From a strategic standpoint, the aryne/β-ketoester ringexpansive fragment coupling affords the concomitant production of the most synthetically challenging carbocycle of 1, as well as the convergent union of the two major synthetic subunits.
Scheme 3.
With the carbocyclic core structure of amurensinine (1) available (i.e., 7), we implemented our strategy for kinetic resolution and completion of the synthesis. To that end, ketoester (±)-7 was diastereoselectively converted to hydroxyester (±)-6 by chemoselective carbonyl reduction with L-Selectride. Gratifyingly, application of our oxidative kinetic resolution technology to (±)-6 using Pd(sparteine)Cl2 and O2 provided the enantioenriched intermediate (−)-6 in 90% ee (s = 19, Scheme 4). Treatment of hydroxyester (−)-6 with (PhO)2P(O)N3 in the presence of DBU,14 followed by reduction with Pd/C under an atmosphere of H2 spontaneously produced lactam (+)-16. Exhaustive reduction of the lactam with LiAlH4 and subsequent reductive methylation yielded the natural product amurensinine ((+)-1).
Scheme 4.
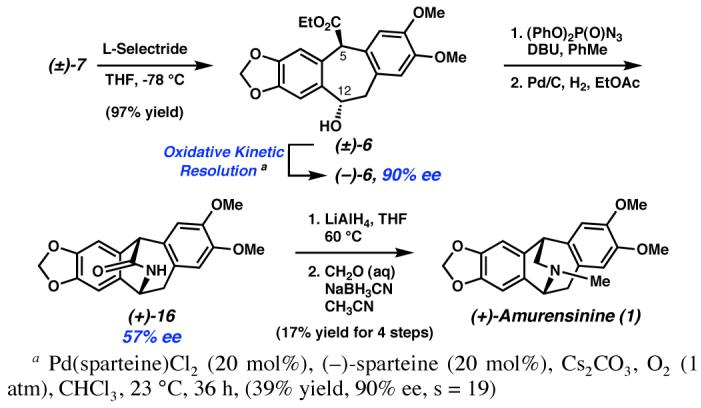
Although this reaction sequence converted ketoester (±)-7 to amurensinine ((+)-1) in a rapid fashion, the enantioenrichment produced by the oxidative kinetic resolution was degraded drastically. Most likely, this resulted from partial epimerization during the conversion of hydroxyester (−)-6 to the corresponding azide due to the acidic nature of the C(5) position15 and the propensity to produce achiral o-quinonedimethide intermediates following in situ activation of the C(12) hydroxyl.
In order to test this hypothesis and remedy the problem of racemization in our synthesis, we devised an alternate endgame strategy (Scheme 5). Hydroxyester (±)-6 was reduced to a diol, and the primary alcohol was silylated to furnish hydroxysilane (±)-17. This racemic alcohol was then subjected to the oxidative kinetic resolution conditions to provide alcohol (−)-17 in 47% yield and greater than 99% enantiomeric excess, corresponding to an associated s -factor of >47. Conversion of the enantioenriched alcohol (−)-17 to azide (−)-18 was straightforward and produced azidoalcohol of greater than 99% ee. Azide (−)-18 was then transformed to the desired secondary lactam (+)-16 in three simple steps. Importantly, the lactam produced by this new synthetic sequence suffered no loss in optical purity and was converted to amurensinine ((+)-1) as a single enantiomer.
Scheme 5.
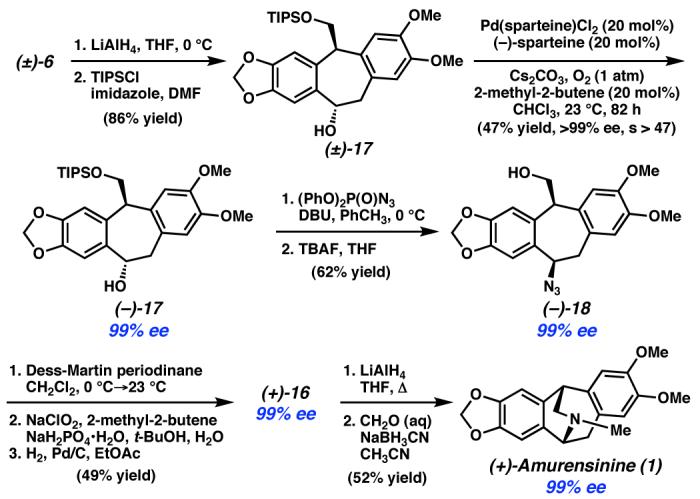
In summary, we have developed a convergent and enantioselective synthesis of amurensinine that takes advantage of sequential C–H and C–C bond insertion reactions to build the core structure of the isopavines in a rapid fashion. A palladiumcatalyzed enantioselective aerobic oxidation of hydroxysilane (±)-17 was utilized to generate enantioenriched amurensinine ((+)-1). Our work underscores the utility of selective C–H and C–C bond insertion reactions for strategic planning of multi-step syntheses and provides the first demonstration of the oxidative kinetic resolution in the context of natural product synthesis. Development and applications of these powerful reactions are ongoing.
Supplementary Material
Acknowledgment
The authors are grateful to the NIH-NIGMS (R01 GM65961-01), NDSEG (predoctoral fellowships to UKT and DCE), NSF (predoctoral fellowship to DCE), A. P. Sloan Foundation, Research Corporation, Bristol-Myers Squibb, Amgen, Merck, Pfizer, Novartis, Lilly, Roche, Abbott, AstraZeneca, and Caltech for financial support.
Footnotes
Supporting Information Available. Experimental details are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Kakiuchi F, Murai S. Topics in Organometallic Chemistry Series. In: Murai S, editor. Activation of Unreactive Bonds and Organic Synthesis. Vol. 3. Springer-Verlag; Berlin and New York: 1999. pp. 47–79. [Google Scholar]; (b) Murakami M, Ito Y. Topics in Organometallic Chemistry Series. In: Murai S, editor. Activation of Unreactive Bonds and Organic Synthesis. Vol. 3. Springer-Verlag; Berlin and New York: 1999. pp. 97–129. [Google Scholar]; (c) Crabtree RH. Chem. Rev. 1985;85:245–269. [Google Scholar]; (d) Labinger JA, Bercaw JE. Nature. 2002;417:507–514. doi: 10.1038/417507a. [DOI] [PubMed] [Google Scholar]; (e) Davies HML, Beckwith REJ. Chem. Rev. 2003;103:2861–2903. doi: 10.1021/cr0200217. [DOI] [PubMed] [Google Scholar]
- 2.For recent examples, see: Davies HML, Dai X, Long MS. J. Am. Chem. Soc. 2006;128:2485–2490. doi: 10.1021/ja056877l.O'Malley SJ, Tan KL, Watzke A, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2005;127:13496–13497. doi: 10.1021/ja052680h.Kurosawa W, Kobayashi H, Kan T, Fukuyama T. Tetrahedron. 2004;60:9615–9628.Wehn PM, Du Bois J. J. Am. Chem. Soc. 2002;124:12950–12951. doi: 10.1021/ja028139s.Johnson JA, Li N, Sames D. J. Am. Chem. Soc. 2002;124:6900–6903. doi: 10.1021/ja026130k.
- 3.(a) Boit HG, Flentje H. Naturwiss. 1960;47:180. [Google Scholar]; (b) Gözler B, Lantz MS, Shamma M. J. Nat. Prod. 1983;46:293–309. [Google Scholar]
- 4.For selected syntheses of isopavine alkaloids, see: Gözler B. Pavine and Isopavine Alkaloids. In: Brossi A, editor. The Alkaloids. Vol. 31. Academic Press; New York: 1987. pp. 343–356.Shinohara T, Takeda A, Toda J, Sano T. Heterocycles. 1998;48:981–992.Gottlieb L, Meyers AIJ. Org. Chem. 1990;55:5659–5662.Hanessian S, Mauduit M. Angew. Chem., Int. Ed. 2001;40:3810–3813. doi: 10.1002/1521-3773(20011015)40:20<3810::AID-ANIE3810>3.0.CO;2-8.Dragoli DR, Burdett MT, Ellman JA. J. Am. Chem. Soc. 2001;123:10127–10128. doi: 10.1021/ja016349j.
- 5.For discussions on the biological activity of the isopavine alkaloids, see: Weber E, Keana J, Barmettler P. PCT Int. Appl. WO 12575, 1990. Chem. Abstr. 1991;115:106019w.Childers WE, Abou-Gharbia MA. U.S. Patent 4940789, 1990 Chem. Abstr. 1990;113:191190w.
- 6.Carrillo L, Badía D, Domínguez E, Vicario JL, Tellitu I. J. Org. Chem. 1997;62:6716–6721. [Google Scholar]
- 7.(a) Ferreira EM, Stoltz BM. J. Am. Chem. Soc. 2001;123:7725–7726. doi: 10.1021/ja015791z. [DOI] [PubMed] [Google Scholar]; (b) Bagdanoff JT, Ferreira EM, Stoltz BM. Org. Lett. 2003;5:835–837. doi: 10.1021/ol027463p. [DOI] [PubMed] [Google Scholar]; (c) Bagdanoff JT, Stoltz BM. Angew. Chem., Int. Ed. 2004;43:353–357. doi: 10.1002/anie.200352444. [DOI] [PubMed] [Google Scholar]; (d) Trend RM, Stoltz BM. J. Am. Chem. Soc. 2004;126:4482–4483. doi: 10.1021/ja039551q. [DOI] [PubMed] [Google Scholar]
- 8.Similar methodology has been developed by Sigman, see: Jensen DR, Pugsley JS, Sigman MS. J. Am. Chem. Soc. 2001;123:7475–7476. doi: 10.1021/ja015827n.
- 9.Tambar UK, Stoltz BM. J. Am. Chem. Soc. 2005;127:5340–5341. doi: 10.1021/ja050859m. [DOI] [PubMed] [Google Scholar]
- 10.Subsequent to our report, similar aryne insertions into C-C bonds were disclosed: Yoshida H, Watanabe M, Ohshita J, Kunai A. Chem. Commun. 2005:3292–3294. doi: 10.1039/b505392g.Yoshida H, Watanabe M, Ohshita F, Kunai A. Tetrahedron Lett. 2005;46:6729–6731.
- 11.Himeshima Y, Sonoda T, Kobayashi H. Chem. Lett. 1983:1211–1214. [Google Scholar]
- 12.(a) Wenkert E, Davis LL, Mylari BL, Solomon MF, Da Silva RR, Shulman S, Warnet RJ, Ceccherelli P, Curini M, Pellicciari R. J. Org. Chem. 1982;47:3242–3247. [Google Scholar]; (b) Taber DF, Petty EH. J. Org. Chem. 1982;47:4808–4809. [Google Scholar]
- 13.Taber DF, Ruckle RE. J. Am. Chem. Soc. 1986;108:7686–7693. doi: 10.1021/ja00284a037. [DOI] [PubMed] [Google Scholar]
- 14.Thompson AS, Humphrey GR, DeMarco AM, Mathre DJ, Grabowski EJJ. J. Org. Chem. 1993;58:5886–5888. [Google Scholar]
- 15.Isopavine numbering convention (see ref 3b).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.