

Abstract

The enantioselective syntheses of naturally occurring kaempferol glycoside SL0101 (1a) and its analogues (1b-e), as well as their enantiomers have been achieved in 7 to 10 steps. The routes rely upon a diastereoselective palladium-catalyzed glycosylation, ketone reduction and dihydroxylation to introduce the rhamno-stereochemistry. The asymmetry of the sugar moiety of these flavone glycosides was derived from Noyori reduction of an acylfuran. An acetyl group shift from an axial (C-2) to equatorial position (C-3) under basic conditions was also described.
In an effort to find specific inhibitors of p90 Ribosomal S6 Kinase (RSK), Smith and Hecht screened an extensive collection of botanical extracts derived from rare plants.1 Using a dual high-throughput screen they found only one extract, which inhibited the RSK2 isoform (RSK2) without inhibiting the tyrosine kinase (FAK). The active extract was from a South American dogbane plant named Forsteronia refracta . More detailed bioactive fractionization revealed the active constituent to be a kaempferol glycoside, which was given the name SL0101 (Figure 1).1 SL0101 (1a) is a member of a class of acylated kaempferol L-rhamnosides which occur naturally with various degrees of acylation (e.g., 1a-e).2 SL0101 sans acetyl groups (1b) is also known as afzelin.2aThe kaempferol glycosides, like most flavonoids, have received a great deal of attention because they are believed to induce many positive biological effects.3
Figure 1.
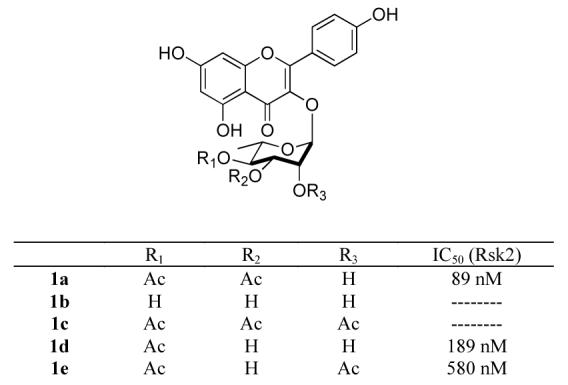
Kaempferol glycoside SL0101 (1a) and its analogues (1b-e), and their Rsk2 inhibitory activities.
In addition to this unique activity, our interest in SL0101 (1a) was peaked by the report that it displayed some 150 times greater activity than the simple aglycon, kaempferol. Similarly, we were intrigued by the importance of the specific placement of acetyl groups on the L-rhamnose and its effect on the SAR of SL0101 (Figure 1).4As part of an effort to elucidate the role of the sugar and acetyl portion of SL0101 to its activity, we decided to prepare both enantiomers of SL0101 (1a) and its analogues 1b-e (Figure 1).
Not long after the isolation and structure elucidation of SL0101 (1a), its first synthesis was reported by Professor Hecht.4 The Hecht synthesis derived the absolute and relative stereochemistry from rhamnose. In contrast, we were interested in the possibility of preparing all five members of this class of kaempferol glycosides (1a-e) via asymmetric catalysis. This de novo approach would have the additional advantage of preparing both the D-and the L-enantiomers for biological testing.
Recently we reported a diastereoselective palladium-catalyzed glycosylation reaction that used alcohols as nucleophiles and pyranones such as 4 as glycosyl donors.5We have also found several post glycosylation transforms, which subsequently install the desired sugar stereochemistry.6 This methodology also works well for other N -, O-nucleophiles, such as 6-chloropurine/benzimidizole and phenol.7 In order to produce this class of interesting compounds for activity studies, we decided to apply this methodology toward the syntheses of the flavone glycosides, SL0101 (1a) and its analogues (1b-e). In addition to providing material for biological study, this effort should also allow us study flavon-3-ol as a nucleophile in the palladium-catalyzed glycosylation.
Retrosynthetically, we envisioned that pyranone 2 could be derived from a Pd(0)-catalyzed glycosylation between flavonol 3 and pyranone 4 (Scheme 1). Subsequent application of NaBH4 reduction and an Upjohn dihydroxylation (OsO4/NMO)8 would install the manno-stereochemistry.9 The selective introduction of the C-4 acetyl group should occur by introducing an acylation reaction between the NaBH4 reduction and dihydroxylation. All that would remain would be to differentiate the C-2 hydroxyl group from the C-3 hydroxyl group. For this, we planned to use a combination of selective orthoester hydrolysis10 and acyl migration reactions.11 Since pyranone 4 has been prepared in either enantiomeric form,12 this procedure should be amenable to the preparation of both enantiomers of 1a-e. Herein we describe our successful efforts at the implementation of this strategy to this class of kaempferol glycosides (1a-e), which is noteworthy in that the various acetyl groups in 1a-e are installed without any hydroxyl protecting groups on the sugar.
Scheme 1.
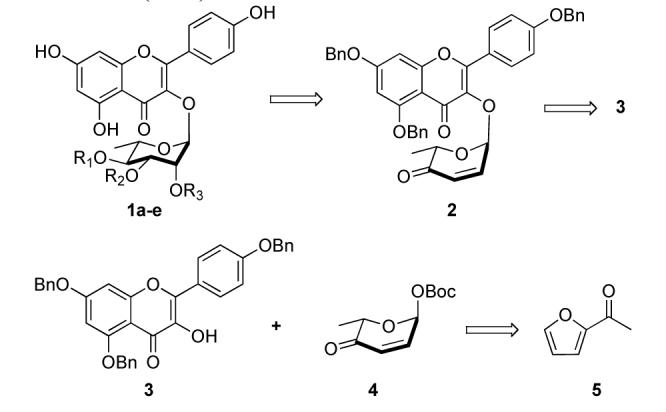
Retrosynthetic analysis of kaempferol rhamnosides (1a-e)
Our synthesis started with the known perbenzylated kaempferol 3, which was synthesized from narringin 6 in three steps (Scheme 2).4 The glycosylation was carried out with flavonol 3 and L-pyranone 4 under catalysis of 2.5 mol% Pd2(dba)3•CHCl3 and 10 mol% of PPh3 in CH2Cl2 at 0 °C, which afforded pyranone 2 in 85% yield with complete α-selectivity (Scheme 2). Reduction of the enone 2 by NaBH4 at —78 °C in CH2Cl2/MeOH resulted in allylic alcohol 7 in 73% yield with excellent diastereoselectivity (dr > 20:1). The rhamno-stereochemistry in 8 was diastereoselectively introduced upon exposure of 7 to the Upjohn conditions (OsO4/NMO, 96%). Debenzylation of 8 using Pearlman’s catalyst (10% Pd/C) in the presence of hydrogen gave kaempferol-3-α-L-rhamnoside (1b) in 80% yield.
Scheme 2.
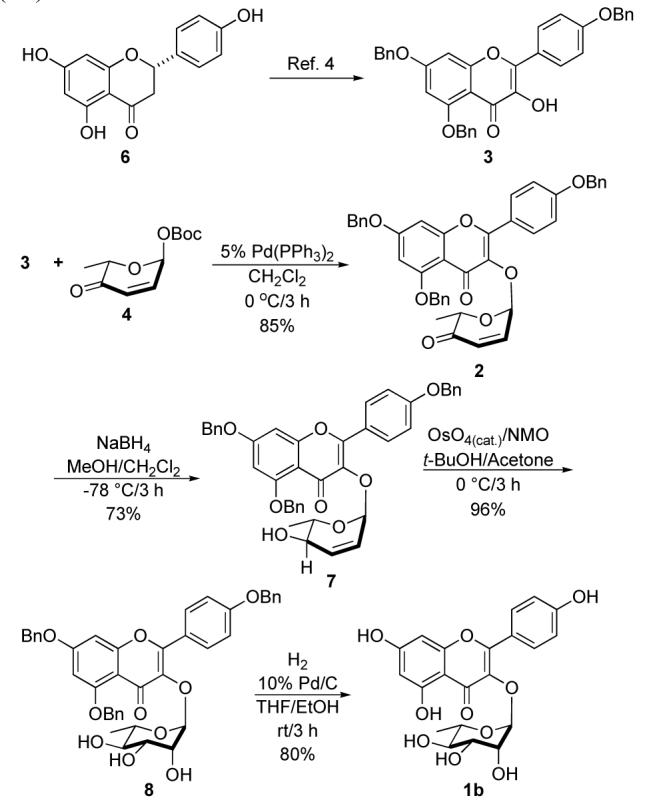
Synthesis of kaempferol-3-α-L-rhamnoside (1b).
In addition to the unacylated rhamno-sugar 1b, the peracylated sugar 1c could also be easily prepared in two steps from triol 8. Exhaustive acylation of the triol 8 with the excess acetic anhydride in presence of pyridine and 10% DMAP gave triacetate 9 in 86% yield (Scheme 3). Debenzylation of triacetate 9 by hydrogen using Pearlman’s catalyst (10% Pd/C) again produced kaempferol-3-α-L-2″,3″,4″-O-triacetylrhamnoside (1c) in 86% yield.
Scheme 3.
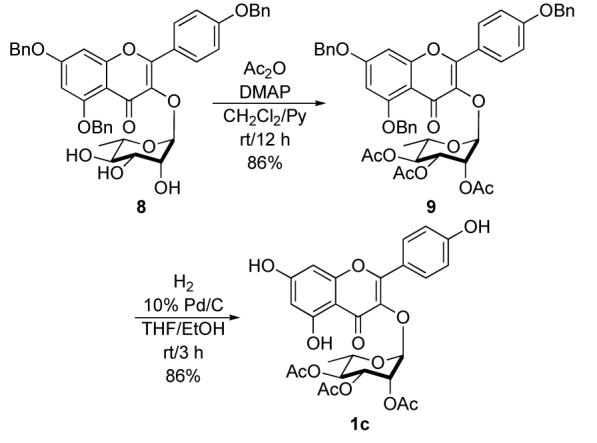
Synthesis of kaempferol-3-α-L-2″,3″,4″-O-triacetylrhamnoside (1c).
The selective installation of the C-4 acetyl group in 1d was easily achieved without the need of additional protecting groups from the previously described allylic alcohol 7 (Scheme 2). In this route (Scheme 4), the NaBH4reduction of enone 6 was followed by an acylation of the resulting allylic alcohol with acetic anhydride in the presence of pyridine and DMAP. This two-step procedure afforded acetate 10 in 70% overall yield. Once again, dihydroxylation using the Upjohn condition stereoselectively converted allylic acetate 10 into the rhamno-diol 11 in 77% yield. Global deprotection was accomplished under hydrogenolysis conditions by exposure of diol 11 to 1 atm of H2 in the presence of Pearlman’s catalyst (Pd/C), which furnished kaempferol-3-α-L-4″-O-acetylrhamnoside (1d) in 87% yield.
Scheme 4.
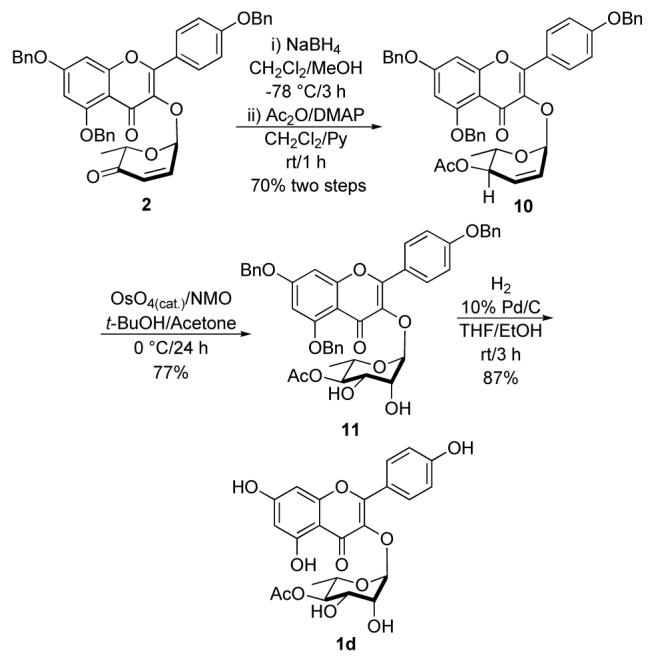
Synthesis of kaempferol-3-α-L -4″-O-acetylrhamnosid(1d).
Acylation of the C-2 axial hydroxyl group of diol 11 was selectively introduced using orthoester chemistry in excellent yield (99%).6 Exposure of diol 11 to trimethyl orthoacetate in the presence of 10% p-TsOH in CH2Cl2followed by hydrolysis with excess 90% HOAc/H2O(Scheme 5). Once again, reductive debenzylation of 12 with hydrogen and 10% Pd/C produced kaempferol-3-α-L-2″,4″-O-diacetylrhamnoside (1e) in 88% yield.
Scheme 5.
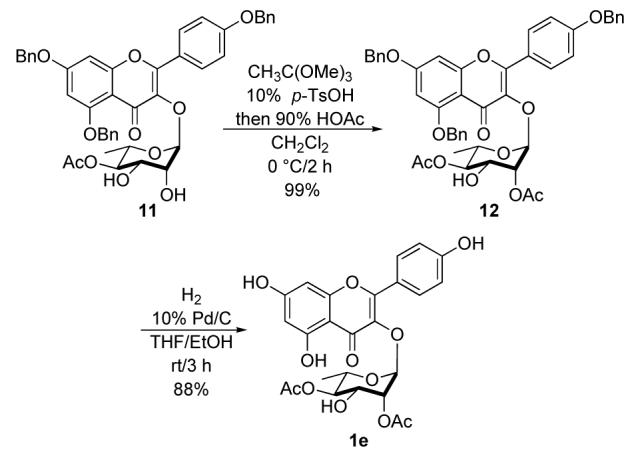
Synthesis of kaempferol-3-α-L-2″,4″-O-diacetylrhamnoside (1e).
In contrast to the selective C-2 acylation of the axial alcohol in 11 (Scheme 5), our synthesis of SL0101 required the selective acylation of the C-3 equatorial alcohol (Scheme 6). Unfortunately all of our attempts with Ac2O/Py at various temperature only furnished mixtures of diacetate 13 and 12, as well as triacetate 9 (∼1:1:1). We next turned to the isomerization of the less stable axial C-2 acetate in 12 to the more stable equatorial C-3 acetate in 13.
Scheme 6.
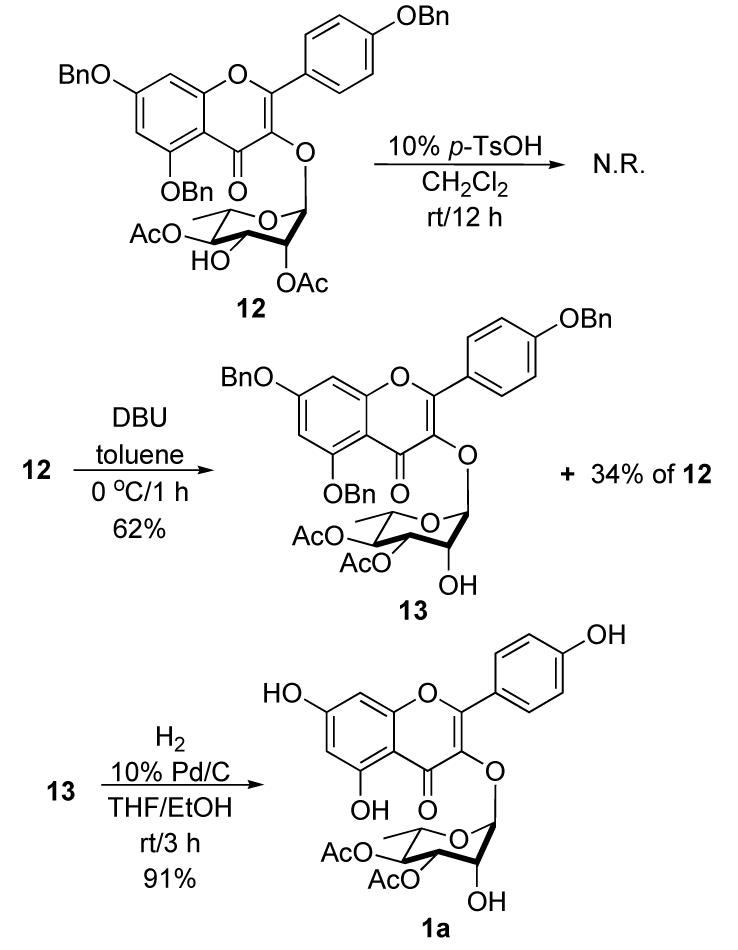
Synthesis of SL0101 (1a)
All attempts to shift the axial acetyl group of diacetate 12 to the equatorial position by using 10 mol % p-TsOH in CH2Cl2 at room temperature failed to give desired diacetate 13 (Scheme 6). In contrast, more promising results were seen with basic conditions. For instance, analysis of crude1H NMR of dilute toluene solutions of 12 with one equiv of DBU showed clean conversion to a 2:1 mixture of 13 and 12. In practice good yields of 13 (62%) could be obtained along with recovered starting material (34%) after SiO2chromatography. Finally, debenzylation of 13 under the similar condition as before produced SL0101 (1a) in 91% yield. Our synthetic products (1a-e) were physically and spectroscopically identical to the isolated natural materials in terms of melting point, optical rotation, Rf, 1H NMR, 13C NMR and MS.2,4
In conclusion, a divergent and highly enantio- and diastereoselective procedures for the preparation of naturally occurring SL0101 (1a) as well as four kaempferol rhamnoside analogues (1b-e) has been developed. This approach provides either enantiopode of kaempferol glycosides 1a-e without protecting any of the sugar hydroxyl groups. Our approach relied upon a diastereoselective palladium(0)-catalyzed glycosylation followed by a sequence of reduction/dihydroxylation reaction, as well as acylation. An acetyl group shift from an axial position to an equatorial position provided an alternative way for selective acylation of the equatorial hydroxyl group of a cis-diol in carbohydrate chemistry. The evaluation of the biological activities of these flavone glycosides is in progress.
Supplementary Material
Acknowledgment
We thank the NIH (GM63150) and NSF (CHE-0415469) for their generous support of our research program. Funding for a 600 MHz NMR and an LTQ-FT Mass Spectrometer by the NSF-EPSCoR (#0314742) is also gratefully acknowledged.
Footnotes
Supporting Information Available: Experimental procedures and spectral data for all new compounds can be found in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1(a).Smith JA, Poteet-Smith CE, Xu Y, Errington TM, Hecht SM, Lannigan DA. Cancer Res. 2005;65:1027–1034. [PubMed] [Google Scholar]; (b) Xu Y, Smith JA, Lannigan DA, Hecht SM. Bioorg. Med. Chem. 2006;14:3974–3977. doi: 10.1016/j.bmc.2006.01.066. [DOI] [PubMed] [Google Scholar]
- 2(a).For the isolation of 1a-b see ref. 1b and:Matthes HWD, Luu B, Ourisson G. Phytochem. 1980;19:2643–2650.For 1b, see:Kaouadji M. Phytochem. 1990;29:2295–2297. doi: 10.1016/0031-9422(90)85465-r. For 1b and 1d, see:Masuda T, Jitoe A, Kato S, Nakatani N, Phytochem. 1991;30:2391–2392. For 1a and 1e, see:Nakatani N, Jitoe A, Masuda T. Agric. Biol. Chem. 1991;55:455–460. For 1c see:Usia T, Iwata H, Hiratsuka A, Watabe T, Kadota S, Tezuka Y. J. Nat. Prod. 2004;67:1079–1083. doi: 10.1021/np030556a. For 1b, see: Deng J-Z, Marshall R, Jones SH, Johnson RK, Hecht SM. J. Nat. Prod. 2002;65:1930–1932. doi: 10.1021/np020285o.
- 3(a).Muir SR, Collins GJ, Robinson S, Hughes S, Bovy A, Ric De Vos CH, van Tunen AJ, Verhoeyen ME. Nat. Biotechnol. 2001;19:470–474. doi: 10.1038/88150. [DOI] [PubMed] [Google Scholar]; (b) Hertog MGL, Feskens EJM, Hollman PCH, Katan MB, Kromhout D. Lancet. 1993;342:1007–1011. doi: 10.1016/0140-6736(93)92876-u. [DOI] [PubMed] [Google Scholar]
- 4.Maloney DJ, Hecht SM. Org. Lett. 2005;7:1097–1099. doi: 10.1021/ol0500463. [DOI] [PubMed] [Google Scholar]
- 5(a).Babu RS, O’Doherty GA. J. Am. Chem. Soc. 2003;125:12406–12407. doi: 10.1021/ja037097k. [DOI] [PubMed] [Google Scholar]; (b) Babu RS, Zhou M, O’Doherty GA. J. Am. Chem. Soc. 2004;126:3428–3429. doi: 10.1021/ja039400n. [DOI] [PubMed] [Google Scholar]
- 6(a).Harris JM, Keranen MD, O’Doherty GA. J. Org. Chem. 1999;64:2982–2983. doi: 10.1021/jo990410+. [DOI] [PubMed] [Google Scholar]; (b) Harris JM, Keranen MD, Nguyen H, Young VG, O’Doherty GA. Carbohydr. Res. 2000;328:17–36. doi: 10.1016/s0008-6215(00)00031-8. [DOI] [PubMed] [Google Scholar]
- 7(a).Guo H, O’Doherty GA. Org. Lett. 2005;7:3921–3924. doi: 10.1021/ol051383e. [DOI] [PubMed] [Google Scholar]; (b) Guppi SR, Zhou M, O’Doherty GA. Org. Lett. 2006;8:293–296. doi: 10.1021/ol052664p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Babu RS, Guppi SR, O’Doherty GA. Org. Lett. 2006;8:1605–1608. doi: 10.1021/ol060254a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.VanRheenen V, Kelly RC, Cha DY. Tetrahedron Lett. 1976;17:1973–1976. [Google Scholar]
- 9.According to carbohydrate nomenclature, rhamnose is 6-deoxymannose
- 10(a).For the selective acylation of an axial alcohol see: ref. 6 and King JF, Allbutt AD. Can. J. Chem. 1970;48:1754–1769.Lowary TL, Hindsgaul O. Carbohydr. Res. 1994;251:33–67. doi: 10.1016/0008-6215(94)84275-2.
- 11(a).Doerschuk AP. J. Am. Chem. Soc. 1952;74:4202–4203. [Google Scholar]; (b) Pettit GR, Cragg GM, Suffness M. J. Org. Chem. 1985;50:5060–5063. [Google Scholar]
-
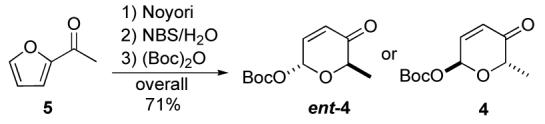
12.Pyranones such as 4 and its enantiomer ent-4 can be prepared in three steps from achiral acylfurans such as 5 in either enantiomeric form (D/L). The pyranone asymmetry is derived from a Noyori reduction; see ref 5b, 7a and: Li M, Scott JG, O’Doherty GA. Tetrahedron Lett. 2004;45:1005–1009.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.