

Abstract
The reaction of silylaryl triflates, CsF and ortho heteroatom-substituted benzoates affords a general and efficient way to prepare biologically-interesting xanthones, thioxanthones and acridones. This chemistry presumably proceeds by a tandem intermolecular nucleophilic coupling of the benzoate with an aryne and subsequent intramolecular electrophilic cyclization.
Introduction
Xanthones are secondary metabolites found in higher plant families, fungi and lichens.1 This class of compounds exhibits interesting pharmaceutical properties; specifically, anti-bacterial, anti-inflammatory, anti-cancer, and anti-viral activities have been observed.2 Some xanthone-containing plants, for example, cratoxylum cochinchinense (Lour.) Blume, have been used as traditional medicines to treat fever, coughing, diarrhea, itching, ulcers and abdominal complaints.3 Thioxanthone derivatives also exhibit interesting anti-cancer activities.4 Xanthones are usually synthesized through the intermediacy of benzophenones or diaryl ethers under harsh reaction conditions and/or in the presence of strong acids or toxic metals.5 Acridones are naturally-occurring compounds exhibiting a variety of biological activities. They are important anti-leishmanial, anti-fungal, anti-tumor and DNA-intercalating anti-cancer drugs.6 Acridones are usually prepared by the acid-induced ring closure of N-phenyl anthranilic acids, which are usually obtained from Ullmann condensation of anilines with ortho halogen-substituted benzoic acids. However, harsh reaction conditions and tedious workup procedures are generally required.7 An efficient and general synthesis of each of these heterocycles is thus highly desirable.

Benzyne, a highly reactive intermediate, was first proposed by Wittig in 1942,8 and the structure was confirmed by Roberts in 1956 using 14C isotope labeling.9 Since then, many methods have been developed to generate benzynes, for example, the base-promoted elimination of hydrogen halide from aryl halides,10 the elimination of o-dihaloaromatics with lithium amalgam or magnesium,11 or the recently reported decomposition of 2-magnesiated aryl sulfonates.12 In 1983, Kobayashi first reported a novel way to generate arynes from silylaryl triflate precursors in the presence of CsF.13 Later, nucleophiles bearing neighboring electrophiles, such as ureas,14 trifluoroacetanilides and sulfinamides,15 and β-keto esters16 have been shown to react with these aryne precursors to afford the C-N bond or C-C bond insertion products (Scheme 1). These nucleophiles first undergo intermolecular nucleophilic attack on the aryne. Subsequent intramolecular electrophilic cyclization, followed by fragmentation, affords the final insertion product.
Scheme 1.

Aryne Insertion Reactions.
Recently, we communicated a novel annulation reaction utilizing readily accessible salicylates and silylaryl triflates plus CsF, which affords an efficient one-step synthesis of biologically-interesting xanthones and thioxanthones (eq. 1).17 This chemistry presumably proceeds by a tandem intermolecular nucleophilic coupling of the benzoate and aryne, and subsequent intramolecular electrophilic cyclization. A fragmentation step, which is inevitable in the insertion examples, is not involved in this annulation process, because the intermediate obtained from the cyclization is a stable 6-membered ring system. Herein, we provide a full account of this efficient synthesis of xanthones and thioxanthones, plus, we also wish to report an extension of this coupling-cyclization strategy to the synthesis of biologically-interesting acridones.
(1).
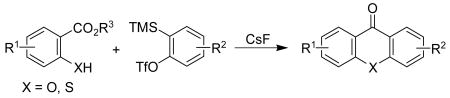
Results and Discussion
Optimization Studies
The reaction of methyl salicylate (1a) and the commercially available aryne precursor o-(trimethylsilyl)phenyl triflate (2a) was first conducted in the presence of 4 equiv of CsF in 5 mL of MeCN. After 12 h reaction at room temperature, an 80% combined yield of methyl 2-phenoxybenzoate (3a) and xanthone (4a) was obtained in a 40:60 ratio (Table 1, entry 1). Presumably, this reaction proceeds through the key intermediate B generated by nucleophilic coupling of the aryne and the aryloxide A (Scheme 2). The carbanion B can either undergo H abstraction to afford benzoate 3a or intramolecular electrophilic cyclization to generate the xanthone (4a). The major problem here is the proton abstraction process, which could be suppressed by adjusting the reaction conditions, for example, using different solvents and concentrations. Thus, we next conducted the coupling-cyclization reaction in acetone, and a 75% yield of diaryl ether 3a and xanthone (4a) was obtained in a 38:62 ratio (entry 2), which suggests that a less polar solvent should be examined. We then performed the reaction in CH2Cl2. However, only a trace of the xanthone (4a) was observed by GC-MS analysis after 24 h (entry 3). When THF was used as the solvent at room temperature, after 24 h reaction, a 20% yield of products 3a and 4a was obtained, and a lot of the starting materials 1a and 2a was observed by GC-MS analysis. However, the ratio of 3a to 4a was 5:95, suggesting that proton abstraction has been almost completely suppressed (entry 4). When this reaction was conducted in toluene, only a trace of the product was evident by GC-MS analysis (entry 5). MeNO2 also turned out to be an unsuitable solvent for this reaction (entry 6). At this point, THF seemed to be the best solvent, at least as far as the reaction selectivity was concerned. The same reaction was then carried out at 65 °C in THF for 24 h. A 75% yield of xanthone (4a) was isolated by flash chromatography and GC-MS analysis indicated only a trace of the diaryl ether 3a was obtained (entry 7). Further investigation indicated that a reaction temperature of 90 °C or 50 °C reduces the amount of xanthone product (entries 8 and 9). When DME was used as the solvent, the reaction afforded only a 70% yield of two isomers formed in a 25:75 ratio (entry 10).
Table 1.
Optimization Studies.
| entry | fluoride source | solvent | temp (°C) | time (h) | % yield (3a:4a)a |
|---|---|---|---|---|---|
| 1 | 4 CsF | MeCN | rt | 12 | 80 (40:60) |
| 2 | 4 CsF | acetone | rt | 4 | 75 (38:62) |
| 3 | 4 CsF | CH2Cl2 | rt | 24 | trace |
| 4 | 4 CsF | THF | rt | 24 | 20 (5:95) |
| 5 | 4 CsF | toluene | 100 | 48 | trace |
| 6 | 4 CsF | MeNO2 | rt | 24 | - |
| 7 | 4 CsF | THF | 65 | 24 | 82 (8:92) |
| 8 | 4 CsF | THF | 90 | 12 | 80 (14:86) |
| 9 | 4 CsF | THF | 50 | 24 | 30 (6:94) |
| 10 | 4 CsF | DME | 65 | 24 | 70 (25:75) |
| 11 | 4 TBAF | THF | 65 | 3 | 60 (83:17) |
| 12 | 2 CsF | THF | 65 | 24 | 65 (8:92) |
All reactions were conducted on a 0.25 mmol scale in 5 mL of solvent (sealed vial). The ratio of methyl salicylate to aryne precursor is 1 to 1.1. The ratio of 3a to 4a in parentheses has been determined by GC-MS analysis.
Scheme 2.

The effect of the fluoride source has also been examined in this process. When tetrabutylammonium fluoride (TBAF) was used as the fluoride source, the reaction proceeded much faster. After 3 h, all of the starting materials were consumed and the proton abstraction product 3a predominated (entry 11). A 65% yield of an 8:92 ratio of 3a and 4a was obtained when this reaction was carried out in the presence of 2 equiv of CsF (entry 12). In conclusion, the “optimal” reaction conditions for this one-step synthesis of xanthone utilize 4 equiv of CsF in THF solvent at 65 °C for 24 h (entry 7).
Synthesis of Xanthones
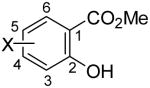
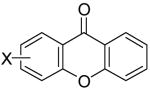
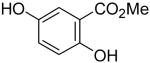
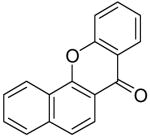
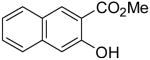
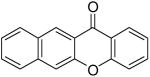
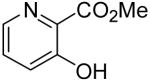
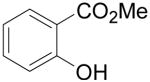
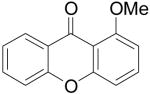
Employing our “optimal” reaction conditions, we have investigated the reaction scope and limitations of this process. These results are summarized in Table 2. We first examined the effect of a methoxy substituent on the salicylate ring to determine which position on the salicylate ring affords the best yield of xanthone. Thus, salicylates 1b, 1c, 1d and 1e were employed, and 35-69% yields of substituted xanthones 4b-4e were obtained (entries 2-5). Having an electron-donating methoxy group in the 5 position of the salicylate ring gave the highest yield (entry 4). The yields of xanthones from the 3- and 4-methoxy starting materials were only slightly lower, but the 6-methoxy isomer gave a much lower yield. When methyl 5-acetylsalicylate (1f) with an electron-withdrawing group in the 5 position was used as the starting material, a 58% yield of xanthone 4f was isolated by flash chromatography (entry 6). On the other hand, the reaction of methyl 5-fluorosalicylate (1g) and aryne precursor 2a affords an 83% yield of xanthone 4g (entry 7). The reaction of methyl 5-bromosalicylate (1h) with aryne 2a affords a 75% yield of the product 4h (entry 8). The phenyl- and methyl-substituted salicylates 1i and 1j afforded 64% and 71% yields of the corresponding xanthone products respectively (entries 9 and 10). When methyl 5-hydroxysalicylate (1k) is allowed to react with 2.5 equiv of aryne precursor 2a, the O-arylated xanthone product 4k was isolated in a 52% yield (entry 11). We have previously reported the facile arylation of phenols by these same aryne precursors.18 From these results with 5-substituted salicylates, there is no obvious correlation between the electronic properties of the substituent and the yield. Phenyl salicylate (1l) has been employed in this reaction and an 81% yield of xanthone 4a was obtained (entry 12). Assuming that 2-hydroxybenzoic acid would first form the corresponding phenyl ester 1l,18 which should then afford the corresponding xanthone (4a), we treated 2 equiv of benzyne precursor 2a and 2-hydroxybenzoic acid (1m) in the usual fashion. Unfortunately, none of the desired product was observed for reasons we do not really understand at this time (entry 13). Interestingly, the cross coupling of methyl 1-hydroxy-2-naphthoate (1n) with silylaryl triflate 2a affords a 73% yield of xanthone 4l, but the reaction using methyl 2-hydroxy-3-naphthoate (1o) only generates a 48% yield of the product 4m (entries 14 and 15). This latter reaction produced several side products which have not been identified. The annulation of 1p, which contains a pyridine ring, afforded none of the xanthone product under our “optimal” conditions (entry 16). Again, we are uncertain why this latter reaction failed.
Table 2.
Synthesis of Xanthones.a
| entry | salicylate | aryne | product(s) | % yield | entry | salicylate | aryne | product(s) | % yield | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
11 |
|
1k | 2a |

|
4k | 52 | ||||
| X | ||||||||||||
| 1 | H | 1a | 2a | 4a | 75 | 12 |
|
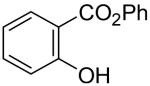
1l | 2a |
|
4a | 81 |
| 2 | 3-OMe | 1b | 2a | 4b | 59 | 13 |
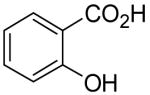
|
1m | 2a | 4a | 0 | |
| 3 | 4-OMe | 1c | 2a | 4c | 62b | 14 |
|
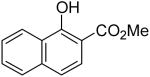
1n | 2a |
|
4l | 73 |
| 4 | 5-OMe | 1d | 2a | 4d | 69 | 15 |
|
1o | 2a |
|
4m | 46c |
| 5 | 6-OMe | 1e | 2a | 4e | 35 | 16 |
|
1p | 2a |
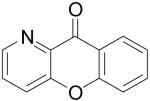
|
4n | 0 |
| 6 | 5-COMe | 1f | 2a | 4f | 58 | 17 |
|
1a | 2b |
|
4e | 62b |
| 7 | 5-F | 1g | 2a | 4g | 83 | 18 | 1a | 2c |
|
4o | 57b | |
| 8 | 5-Br | 1h | 2a | 4h | 75 | 19 | 1a | 2d |
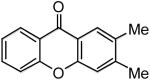
|
4p | 51b | |
| 20 | 1a | 2e |
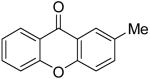
|
4j | ||||||||
| 9 | 5-Ph | 1i | 2a | 4i | 64 | 59b (1:1) | ||||||
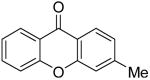
|
4q | |||||||||||
| 10 | 5-Me | 1j | 2a | 4j | 71 | 21 | 1a | 2f |
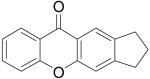
|
4r | 45b |
All reactions were conducted on a 0.25 mmol scale in the presence of 4 equiv of CsF and 1.1 equiv of silylaryl triflate in 5 mL of THF at 65 °C for 24 h (sealed vial) unless otherwise stated.
The reaction was conducted in 5 mL of THF at 90 °C.
The yield has been determined by GC analysis due to the impurities in the product.
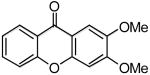
After investigating the effect of varying the salicylate structure, we examined the reaction efficiency using different aryne precursors (Scheme 3). A 62% yield of a single isomeric methoxyxanthone 4e was obtained from the reaction of methyl salicylate 1a with aryne precursor 2b after 24 h at 90 °C (entry 17). Note that a somewhat higher temperature was required to get a good yield. The regioselectivity of this reaction is due to the steric and electronic effects in the step involving nucleophilic attack on the aryne, which has been seen in several previous reactions involving this aryne.18 When the dimethoxysilylaryl triflate 2c was employed, a 57% yield of xanthone 4o was isolated (entry 18). Again a higher temperature was required. The reaction of aryne precursor 2d with methyl salicylate (1a) afforded a 51% yield of xanthone 4p (entry 19). When aryne precursor 2e was employed, a 59% yield of two isomeric xanthones 4j and 4q was obtained in a 1:1 ratio (entry 20). This is consistent with the intermediacy of an unsymmetrical methyl-substituted benzyne. Aryne precursor 2f afforded a 45% yield of a single xanthone product 4r (entry 21).
Scheme 3.

Aryne Precursors.
One of the major advantages of this methodology is that halogen atoms can be tolerated, which provides access to more structurally diverse xanthone skeletons via metal-catalyzed cross-coupling reactions. As illustrated in Scheme 4, the halogen-substituted xanthone product 4h can be further modified by Heck19 and Suzuki20 reactions, affording interesting xanthone derivatives 5a and 5b for further biological examination.
Scheme 4.

Diversification of Halogen-substituted Xanthones.
Synthesis of Thioxanthones
Biologically-interesting thioxanthone derivatives have also been prepared by this same methodology. All of the results are summarized in Table 3. Methyl thiosalicylate (6a) and 1.1 equiv of benzyne precursor 2a were treated with 4 equiv of CsF in 5 mL of THF at 65 °C, after 24 h, a 35% yield of thioxanthone (7a) was isolated. The lower yield in this example is presumably due to oxidative homocoupling of the thiols, since thiols are known to afford disulfides in the presence of CsF on a celite solid support in air.21 In order to suppress this undesired homocoupling process, the same reaction was repeated under an Ar atmosphere, and a 55% yield of thioxanthone (7a)was obtained. Further optimization indicated that a 64% yield of product 7a could be obtained under more dilute conditions, although a small amount of disulfide product and S-arylation product were present (Table 3, entry 1). The reaction of this thiol with benzyne precursor 2b afforded a 45% yield of the desired thioxanthone 7b (entry 2). When 2d was employed as the aryne precursor in this reaction, a 40% yield of the product 7c was isolated (entry 3). The reaction of 2e afforded a 56% yield of two regioisomers 7d and 7e in a 1:1 ratio (entry 4). Finally, aryne precursor 2f was allowed to react with thiosalicylate 6a, and a 62% yield of thioxanthone 7f was isolated by flash chromatography (entry 5).
Table 3.
Synthesis of Thioxanthones.a
| entry | thiosalicylate | aryne | product(s) | % yield | ||
|---|---|---|---|---|---|---|
| 1 |
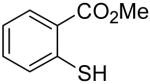
|
6a | 2a |
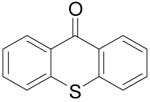
|
7a | 64 |
| 2 | 6a | 2b |
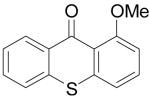
|
7b | 45b | |
| 3 | 6a | 2d |
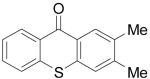
|
7c | 40b | |
| 4 | 6a | 2e |
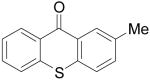
|
7d | 56b,c (1:1) | |
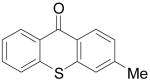
|
7e | |||||
| 5 | 6a | 2f |
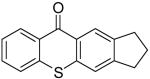
|
7f | 62b |
All reactions were conducted on a 0.25 mmol scale in the presence of 4 equiv of CsF and 1.1 equiv of silylaryl triflate in 10 mL of THF at 65 °C under Ar.
The reaction was conducted in 10 mL of THF at 90 °C under Ar (sealed tube).
The ratio in parentheses has been determined by 1H NMR spectroscopy.
Synthesis of Acridones
After we had a general and efficient synthesis of xanthones and thioxanthones in hand, we attempted to expand this methodology to the synthesis of acridones, a well-known class of anti-fungal, anti-tumor and anti-cancer compounds.6 Acridones have been prepared by the coupling of 3-halogeno-4-methoxybenzynes generated from 5-(3-halogeno-4-methoxyphenyl)thianthrenium perchlorates and LDA in THF at reflux with 2-aminobenzoate.22 However, this protocol employs a fairly unusual aryne precursor, and it also suffers from the moisture-sensitive reagents and conditions. Methyl 2-aminobenzoate (8a) was first prepared and allowed to react with aryne precursor 2a, and a 50% yield of the acridone product 9a was obtained (Table 4, entry 1). Methyl 2-(N-methylamino)benzoate (8b) was then allowed to react with aryne precursor 2a. After 1 day of reaction, a 72% yield of acridone 9b was isolated by flash chromatography (entry 2). However, the reaction employing methyl 2-(N-phenylamino)benzoate (8c) was very sluggish; after 2 days of reaction, only a 7% yield of the desired product 9c was observed by GC-MS analysis. The low yield is presumably due to the steric hindrance introduced by the presence of the phenyl substituent (entry 3). Interestingly, the reaction of methyl 2-(N,N-dimethylamino)benzoate (8d) affords a 65% yield of acridone product 9b, which indicates that even tertiary amines can be successfully employed in this transformation (entry 4). Apparently the anticipated ammonium-containing product undergoes demethylation under the reaction conditions. Several halogen-substituted benzoates (8e-8g) have also been prepared from the corresponding acids and employed in this process (entries 5-7). Yields of 48-71% of the corresponding acridone products (9d-9f) have been obtained. We have not employed protecting groups on nitrogen since that could well lead to C-N insertion products.14,15
Table 4.
Synthesis of Acridones.a
| entry | benzoate | aryne | product(s) | % yield | ||||
|---|---|---|---|---|---|---|---|---|
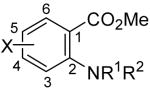
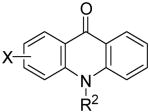
|
|
|||||||
| R1 | R2 | X | ||||||
| 1 | H | H | H | 8a | 2a | 9a | 50 | |
| 2 | H | Me | H | 8b | 2a | 9b | 72 | |
| 3 | H | Ph | H | 8c | 2a | 9c | 7b | |
| 4 | Me | Me | H | 8d | 2a | 9b | 65 | |
| 5 | H | Me | 4-F | 8e | 2a | 9d | 48 | |
| 6 | H | Me | 5-F | 8f | 2a | 9e | 71 | |
| 7 | H | Me | 5-Br | 8g | 2a | 9f | 61 | |
| 8 | H | Me | H | 8b | 2b |
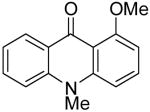
|
9g | tracec |
| 9 | H | Me | H | 8b | 2d |
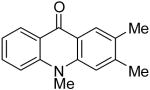
|
9h | 27c |
| 10 | H | Me | H | 8b | 2e |
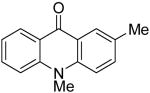
|
9i | 51c (1:1) |
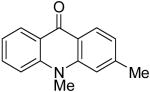
|
9j | |||||||
| 11 | H | Me | H | 8b | 2f |
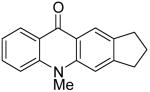
|
9k | 35c |
All reactions were conducted on a 0.25 mmol scale in the presence of 4 equiv of CsF and 1.1 equiv of the silylaryl triflate in 10 mL of THF at 65 °C.
The yield was determined by GC-MS analysis.
The reaction was conducted in 10 mL of THF at 90 °C (sealed tube). The ratio in parentheses has been determined by 1H NMR spectroscopy.
At this point, we examined the effect of the aryne structure on the yield of acridone. When silylaryl triflate 2b was employed with benzoate 8b, the reaction was very sluggish and only a trace amount of the desired acridone product 9g was observed by GC-MS analysis (entry 8). Aryne precursor 2d afforded only a 27% yield of the product 9h and this reaction had to be run at 90 °C (entry 9). The reaction of aryne precursor 2e with benzoate 8a afforded a 51% yield of two isomeric acridones, 9i and 9j, in a 1:1 ratio (entry 10). A 35% yield of acridone product 9k was obtained when aryne precursor 2f was employed (entry 11). These last two reactions also had to be run at 90 °C.
A plausible mechanism for these aminobenzoate reactions is proposed in Scheme 4. The benzoate bearing an amino group presumably first undergoes nucleophilic attack on the aryne generated in situ from the silylaryl triflate. When R is a proton, the actual nucleophile involved could be either the neutral amine or the anionic intermediate D generated by hydrogen abstraction from the amine by CsF. However, when the tertiary amine 8d is employed, although no proton is available for abstraction, this reaction still works well, suggesting that the neutral amine itself is nucleophilic enough for this transformation. Therefore, the reaction mechanism which proceeds via intermediates F and G seems more likely, although we cannot rule out possible anionic nucleophilic attack on the aryne, which proceeds via intermediates D and E. Subsequent intramolecular cyclization should afford the final acridone products.
Conclusions
A general one-pot synthesis of biologically-interesting xanthones, thioxanthones and acridones has been developed. This chemistry presumably proceeds by a tandem intermolecular nucleophilic coupling of the substituted benzoates and arynes and subsequent intramolecular electrophilic cyclization. The mild reaction conditions and generally high reaction efficiency provide advantages over previously reported multi-step procedures. In generally, this strategy tolerates both electron-donating and electron-withdrawing functionalities on the benzoate ring, but substituents on the aryne ring appear to lower the yields of the desired products.
Experimental Section
Representative procedure for the coupling-cyclization of arynes and salicylates
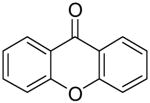
CsF (1.0 mmol), the salicylate (0.25 mmol), and the silylaryl triflate (0.28 mmol) in 5 mL of anhydrous THF were stirred at 65 or 90 °C for 24 h. The reaction mixture was allowed to cool to room temperature, diluted with diethyl ether (25 mL) and washed with brine (25 mL). The aqueous layer was re-extracted with diethyl ether (2 × 25 mL). The organic layers were combined, dried (MgSO4), filtered, and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel. 9H-Xanthen-9-one (4a). 1H NMR (CDCl3) δ 7.37 (t, J = 6.0 Hz, 2H), 7.48 (d, J = 6.4 Hz, 2H), 7.70-7.74 (m, 2H), 8.33 (dd, J = 6.0, 1.2 Hz, 2H); 13C NMR (CDCl3) 118.2, 122.1, 124.1, 126.9, 135.0, 156.4, 177.4; IR (CDCl3) 2914, 2874, 1654, 1456 cm-1; HRMS m/z 196.0527 (calcd for C13H8O2, 196.0524).
Representative procedure for the coupling-cyclization of arynes and thiosalicylates
CsF (1.0 mmol), the thiosalicylate (0.25 mmol) and the silylaryl triflate (0.28 mmol) were added to 10 mL of anhydrous THF, and the reaction vial was flushed with Ar. The whole reaction solution was then stirred at 65 or 90 °C for 24 h and worked up as described previously. 9H-Thioxanthen-9-one (7a). 1H NMR (CDCl3) δ 7.48 (td, J = 6.7, 1.5 Hz, 2H), 7.56-7.65 (m, 4H), 8.62 (dd, J = 7.4, 0.8 Hz, 2H); 13C NMR (CDCl3) 126.2, 126.5, 129.4, 130.1, 132.5, 137.5, 180.2; IR (CDCl3) 2971, 2919, 1684, 1459 cm-1; HRMS m/z 212.0299 (calcd for C13H8OS, 212.0296).
Representative procedure for the coupling-cyclization of arynes and 2-aminobenzoates
CsF (1.0 mmol), the 2-aminobenzoate (0.25 mmol), and the silylaryl triflate (0.28 mmol) in 10 mL of anhydrous THF were stirred at 65 or 90 °C for 24 h. The reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate (25 mL) and washed with brine (25 mL). The aqueous layer was re-extracted with ethyl acetate (2 × 25 mL). The organic layers were combined, dried (MgSO4), filtered, and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel. 10-Methyl-10H-acridin-9-one (9b). 1H NMR (CDCl3) δ 3.88 (s, 3H), 7.28 (t, J = 7.2 Hz, 2H), 7.50 (d, J = 8.7 Hz, 2H), 7.69-7.73 (m, 2H), 8.55 (dd, J = 8.0, 1.2 Hz, 2H); 13C NMR (CDCl3) 33.9, 115.0, 121.5, 122.7, 128.0, 134.0, 142.8, 178.3; IR (CDCl3) 2917, 2850, 1637 cm-1; HRMS m/z 209.0843 (calcd for C14H11NO, 209.0841).
Supplementary Material
Preparation and characterization of the xanthones, thioxanthones and acridones. This material is available free of charge via the internet at http://pubs.acs.org.
Scheme 4.

Plausible Mechanism for the Acridone Synthesis.
Acknowledgments
We gratefully acknowledge the financial support of this work by the National Institutes of Health Kansas University Chemical Methodologies and Library Development Center of Excellence (P50 GM 069663).
References
- 1.Cardona ML, Fernandez MI, Pedro JR, Serrano A. Phytochemistry. 1990;29:3003. [Google Scholar]
- 2.For a recent review on naturally-occurring xanthones, see: Peres V, Nagem TJ, Faustino de Oliveira F. Phytochemistry. 2000;55:683. doi: 10.1016/s0031-9422(00)00303-4.Schwaebe MK, Moran TJ, Whitten JP. Tetrahedron Lett. 2005;46:827.Kenji M, Yukihiro A, Hong Y, Kenji O, Tetsuro I, Toshiyuki T, Emi K, Munekazu I, Yoshinori N.Bioorg Med Chem 200412579915498656Pedro M, Cerqueira F, Sousa ME, Nascimento MSJ, Pinto M. Bioorg Med Chem. 2002;10:3725. doi: 10.1016/s0968-0896(02)00379-6.
- 3.(a) Mahabusarakam W, Nuangnaowarat W, Taylor WC. Phytochemistry. 2006;67:470. doi: 10.1016/j.phytochem.2005.10.008. [DOI] [PubMed] [Google Scholar]; (b) Gnerre C, Thull U, Gailland P, Carrupt PA, Testa B, Fernandes E, Silva F, Pinto M, Pinto M, Wolfender JL, Hostettmann K, Cruciani G. Helv Chim Acta. 2001;84:552. [Google Scholar]
- 4.Poondru S, Zhou S, Rake J, Shackleton G, Corbett TH, Parchment R, Jasti BR. J Chrom B. 2001;759:175. doi: 10.1016/s0378-4347(01)00217-1. and references therein. [DOI] [PubMed] [Google Scholar]
- 5.(a) Grover PK, Shah GD, Shah RC. J Chem Soc. 1955:3982. [Google Scholar]; (b) Quillinan AJ, Scheinmann F. J Chem Soc, Perkin Trans. 1973:1329. [Google Scholar]; (c) Jackson WT, Robert JB, Froelich LL, Gapinski DM, Mallett BE, Sawyer JS. J Med Chem. 1993;36:1726. doi: 10.1021/jm00064a006. [DOI] [PubMed] [Google Scholar]; (d) Familoni OB, Ionica I, Bower JF, Snieckus V. Synlett. 1997:1081. [Google Scholar]; (e) Hassal CH, Lewis JR. J Chem Soc. 1961;2:2312. [Google Scholar]
- 6.Tabarrini O, Manfroni G, Fravolini A, Cecchetti V, Sabatini S, De Clercq E, Rozenski J, Canard B, Dutartre H, Paeshuyse J, Neyts J. J Med Chem. 2006;49:2621. doi: 10.1021/jm051250z. and references therein.Taraporewala IB, Cessac JW, Chanh TC, Delgado AV, Schinazi RF. J Med Chem. 1992;35:2744. doi: 10.1021/jm00093a005.MacNeil SL, Wilson BJ, Snieckus V. Org Lett. 2006;8:1133. doi: 10.1021/ol053162e.Harrison RJ, Reszka AP, Haider SM, Romagnoli B, Morrell J, Read MA, Gowan SM, Incles CM, Kellandc LR, Neidle S. Bioorg Med Chem Lett. 2004;14:5845. doi: 10.1016/j.bmcl.2004.09.037.
- 7.(a) Nishio R, Wessely S, Sugiura M, Kobayashi S. J Comb Chem. 2006;8:459. doi: 10.1021/cc060011+. [DOI] [PubMed] [Google Scholar]; (b) Goodell JR, Madhok AA, Hiasa H, Ferguson DM. Bioorg Med Chem. 2006;14:5467. doi: 10.1016/j.bmc.2006.04.044. [DOI] [PubMed] [Google Scholar]
- 8.For a recent review, see: Sander W. Acc Chem Res. 1999;32:669.Wittig G. Naturwissenschaften. 1942:696.
- 9.Roberts JD, Semenow DA, Simmons HE, Jr, Carlsmith LA. J Am Chem Soc. 1956;78:601. [Google Scholar]
- 10.(a) Huisgen R, Sauer J. Angew Chem. 1960;72:91. [Google Scholar]; (b) Biehl ER, Nieh E, Hsu KC. J Org Chem. 1969;34:3595. [Google Scholar]; (c) Bunnett JF, Kim JK. J Am Chem Soc. 1973;95:2254. [Google Scholar]
- 11.(a) Wittig G, Pohmer L. Chem Ber. 1956;89:1334. [Google Scholar]; (b) Wittig G. Org Synth. 1963;IV:964. [Google Scholar]
- 12.Sapountzis I, Lin W, Fischer M, Knochel P. Angew Chem, Int Ed. 2004;43:4364. doi: 10.1002/anie.200460417. and references therein. [DOI] [PubMed] [Google Scholar]
- 13.Himeshima Y, Sonoda T, Kobayashi H. Chem Lett. 1983:1211. [Google Scholar]
- 14.Yoshida H, Shirakawa E, Honda Y, Hiyama T. Angew Chem, Int Ed. 2002;41:3247. doi: 10.1002/1521-3773(20020902)41:17<3247::AID-ANIE3247>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 15.Liu Z, Larock RC. J Am Chem Soc. 2006;128:13112. doi: 10.1021/ja054079p. [DOI] [PubMed] [Google Scholar]
- 16.Tambar UK, Stoltz BM. J Am Chem Soc. 2005;127:5340. doi: 10.1021/ja050859m. [DOI] [PubMed] [Google Scholar]
- 17.Zhao J, Larock RC. Org Lett. 2005;7:4273. doi: 10.1021/ol0517731. [DOI] [PubMed] [Google Scholar]
- 18.Liu Z, Larock RC. Org Lett. 2004;6:99. doi: 10.1021/ol0361406. [DOI] [PubMed] [Google Scholar]
- 19.Alonso F, Beletskaya IP, Yus M. Tetrahedron. 2005;61:11771.Nicolaou KC, Bulger PG, Sarlah D. Angew Chem, Int Ed. 2005;44:4442. doi: 10.1002/anie.200500368.Negishi E. Handbook of Organopalladium Chemistry for Organic Synthesis. 2002;1:1223. and references therein.Tsuji J. Palladium Reagents and Catalysts. John Wiley & Sons; England: 2004. p. 135.
- 20.Tofi M, Georgiou T, Montagnon T, Georgios V. Org Lett. 2005;7:3347. doi: 10.1021/ol051205l. [DOI] [PubMed] [Google Scholar]
- 21.Shah STA, Khan KM, Fecker M, Voelter W. Tetrahedron Lett. 2003;44:6789. and references therein. [Google Scholar]
- 22.Yoon K, Ha SM, Kim K. J Org Chem. 2005;70:5741. doi: 10.1021/jo050420c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Preparation and characterization of the xanthones, thioxanthones and acridones. This material is available free of charge via the internet at http://pubs.acs.org.