

Abstract
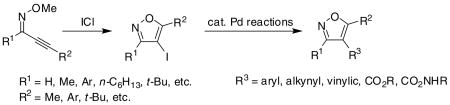
A large number of functionally-substituted 2-alkyn-1-one O-methyl oximes have been cyclized under mild reaction conditions in the presence of ICl to give the corresponding 4-iodoisoxazoles in moderate to excellent yields. The resulting 4-iodoisoxazoles undergo various palladium-catalyzed reactions to yield 3,4,5-trisubstituted isoxazoles, including valdecoxib.
Introduction
The isoxazole skeleton has been the focus of many biological studies in recent years.1 Our group and others have reported that the electrophilic cyclization of functionally-substituted acetylenes is a powerful synthetic tool for constructing a diverse assortment of ring systems, including benzo[b]thiophenes,2 isoquinolines and naphthyridines,3 isocoumarins and α-pyrones,4 benzofurans,5 furans,6 indoles,7 furopyridines,8 cyclic carbonates,9 2,3-dihydropyrroles and pyrroles,10 pyrilium salts and isochromenes,11 bicyclic β-lactams,12 2H-benzopyrans,13 naphthalenes and 2-naphthols,14 chromones,15 isoindolin-1-ones16 and benzo[b]selenophenes.17
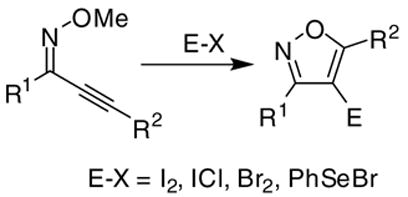
We recently reported the synthesis of numerous highly substituted isoxazoles through the cyclization of various 2-alkyn-1-one O-methyl oximes (eq 1)18 by a variety of electrophiles. The yields of the desired Z-O-methyl oximes from the ynones are generally good, and these compounds are easily isolated by column chromatography on silica gel. We now report the full details of our work on the ICl-induced cyclization of 2-alkyn-1-one O-methyl oximes, which provides a very efficient synthesis of 4-iodoisoxazoles. Numerous examples are reported. The resulting 4-iodoisoxazoles are readily elaborated by conventional palladium-catalyzed processes to afford highly substituted isoxazoles, including valdecoxib.
(1).
Results and Discussion
The requisite ynones can be easily prepared (Scheme 1) by the palladium/copper-catalyzed Sonogashira coupling of an acid chloride with a terminal acetylene19 or by Pd-catalyzed carbonylative coupling of terminal acetylenes with aryl iodides.20 The ynones can also be prepared by allowing a lithium acetylide to react with an aldehyde, followed by oxidation of the resulting secondary alcohol.21 In addition, ynones can be conveniently prepared by the treatment of silyl acetylenes with an acid chloride in the presence of aluminum chloride.22
Scheme 1.

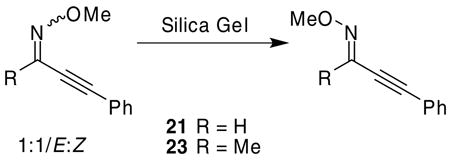
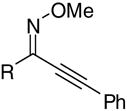
The O-methyl oximes required in our isoxazole synthesis are readily prepared by stirring the ynone in the presence of methoxylamine hydrochloride, pyridine, and Na2SO4 or MgSO4 at room temperature using methanol as the solvent.23 Heating is sometimes required, especially when R1 is an aromatic ring bearing electron-donating groups. When R1 is a bulky group relative to the alkyne moiety, the desired Z isomer is the predominant product. However, if R1 is significantly less bulky, a mixture of isomers often results and the desired isomer must be separated by column chromatography. Upon preparation of the 2-alkyn-1-one O-methyl oximes 21 and 23, a 1:1 mixture of E:Z O-methyl oximes, observable by 1H NMR spectroscopy, was obtained. Our attempts at separating these isomers on a silica gel column led to the unwanted E isomer exclusively (eq 2). The 1:1 mixture of E:Z O-methyl oximes 21 and 23 isomerized exclusively to their E isomers. This isomerization only occurred when R was either a proton or a methyl group, but was not observed in any other cases. Nevertheless, in the cases when R is equal to a proton (21) or methyl (23), the desired Z-O-methyl oxime could be isolated by utilizing a basic alumina column, although the yields were only modest. It was discovered that only the Z isomer cyclized when subjected to our usual ICl cyclization conditions. Our attempts at simultaneous isomerization and cyclization of the E isomer were unsuccessful. Consequently, it is essential that the Z-O-methyl oxime isomer be employed in the cyclization process.
(2).
With the desired Z-O-methyl oximes in hand, we studied the scope of the cyclization methodology (Table 1). In our earlier studies,18 we discovered that using ICl as the source of electrophilic iodine provided the best results for the formation of isoxazoles. I2 may also be used to induce the cyclization of compound 1. However, the reactions are slower and higher molar equivalents of I2 are required to achieve yields comparable to those of ICl (Table 1, compare entries 1 and 2).
Table 1.
The Synthesis of 4-Iodoisoxazoles by Electrophilic Cyclizationa
| entry | O-methyl oxime | electrophile | time (h) | product | yield (%)b |
|---|---|---|---|---|---|
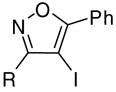
|
|
||||
| 1 | R = Ph (1) | 3.0 I2c | 1.0 | (2) | 77 |
| 2 | 1.2 ICl | 0.5 | 86 | ||
| 3 | R = Me (3) | 1.2 ICl | 0.75 | (4) | 99 |
| 4 | R = n-Bu (5) | 1.2 ICl | 0.75 | (6) | 91 |
| 5 | R = t-Bu (7) | 1.2 ICl | 0.75 | (8) | 90 |
| 6 |
|
1.2 ICl | 0.5 | (10) | 87 |
| 7 |
|
1.2 ICl | 0.5 | (12) | 89 |
| 8 |
 (13) (13) |
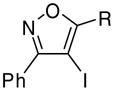
1.2 ICl | 0.5 | (14) | 82 |
|
|
||||
| 9 |
|
1.2 ICl | 1.0 | (16) | 94 |
| 10 |
|
1.2 ICl | 1.0 | (18) | 93 |
| 11 |
|
2.2 ICl | 6.0 | (20) | 98 |
| 12 | R = H (21) | 1.2 ICl | 0.5 | (22) | 83 |
| 13 | R = Me (23) | 1.2 ICl | 0.75 | (24) | 79 |
| 14 | R = n-C6H13 (25) | 1.2 ICl | 0.75 | (26) | 94 |
| 15 | R = t-Bu (27) | 1.2 ICl | 0.75 | (28) | 100 |
| 16 |
 (29) (29) |
1.2 ICl | 0.5 | (30) | 93 |
| 17 |
 (31) (31) |
1.2 ICl | 1.0 | (32) | 89 |
| 18 |
 (33) (33) |
1.2 ICl | 1.0 | (34) | 82 |
| 19 |
 (35) (35) |
1.2 ICl | 0.5 | (36) | 86 |
| 20 |
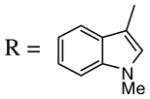 (37) (37) |
1.2 ICl | 0.5 | (38) | 55 |
| 21 |
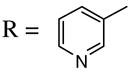 (39) (39) |
3.0 ICl | 3.0 | (40) | 68 |
| 22 |
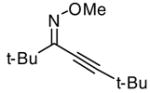
(41) |
1.2 ICl | 2.0 |
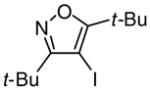 (42) (42) |
87 |
| 23 |
 (43) (43) |
1.2 ICl | 1.0 |
 (44) (44) |
96 |
All reactions were carried out in CH2Cl2 (10 mL/mmol) at room temperature using 0.25 mmol of starting material unless otherwise specified.
Isolated yields after column chromatography.
The reaction was carried out in CH3CN.
When the terminus of the alkynyl moiety was substituted with aliphatic substituents ranging from compact to bulky, the yields were excellent (entries 3–5). Furthermore, the introduction of substituents on the aryl group, has little effect on the yield of the reaction. The electron-withdrawing group CO2Et (9) and the electron-donating group OMe (11), both in the para position of the phenyl ring, did not significantly change the reaction yields compared to the parent system (entries 6 and 7). Furthermore, substituting the alkynyl unit with a thiophene heterocycle (13) lowered the yield only slightly (entry 8).
The effect of changing the substituents in the 1 position of the 2-alkyn-1-one O-methyl oxime was also studied. The para electron-withdrawing groups in compounds 15 and 17 provided the corresponding isoxazoles 16 and 18 in excellent yields (entries 9 and 10). The electron-donating group NMe2 in the para position of compound 19 required a dramatic increase in reaction time and 2.2 mole equivalents of ICl had to be used to achieve an excellent yield of 20. 1.2 Mole equivalents of ICl provided only a trace of 20 and the reaction suffered from low conversion. No further iodinated products were observed in the reaction.
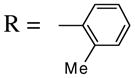
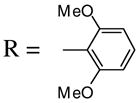
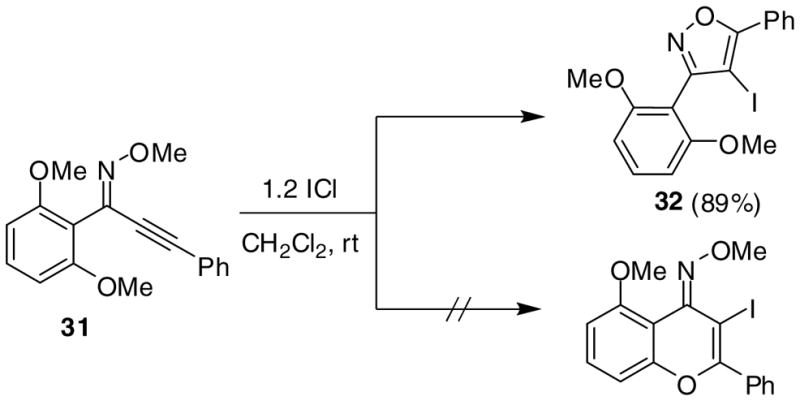
Other substituents in the 1 position of the 1-alkynone were generally quite successful. For example, compounds 21 and 23, as the pure Z-isomers, afforded the expected product with only a slight decrease in yields (entries 12 and 13) using our standard procedure, while compound 25 provided an excellent yield of 26 (entry 14). Introducing a bulky t-Bu group (27) into the alkynone provided a quantitative yield of the desired isoxazole 28 (entry 15). Compounds 29 and 31 introduce steric bulk at the ortho positions of the parent phenyl rings. This did not hinder the reaction and high yields of the isoxazole products 30 and 32 were obtained using short reaction times (entries 16 and 17). Thus, steric effects appear to be minimal in this process. It is noteworthy that compound 31 cyclized exclusively to the desired 5-endo dig isoxazole product 32 and the possible 6-exo dig chromone oxime product was not observed, even though we have successfully cyclized closely related ketones to the corresponding chromones15 (Scheme 2).
Scheme 2.
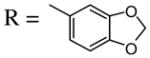
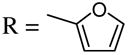
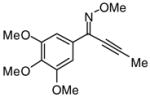
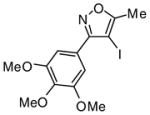
Introduction of the oxygen-containing heterocycles 33 and 35 into the alkynone also provided isoxazoles in good yields (entries 18 and 19). The nitrogen-containing heterocycles 37 and 39 also afforded the desired isoxazoles, although the yields were only modest (entries 20 and 21). The cyclization of compound 39 required the use of 3 equivalents of ICl, as well as an increased reaction time. Compound 41 was synthesized to study the effect of having two sterically bulky t-Bu substituents present in the same substrate. The reaction required a slight increase in time. Nevertheless, isoxazole 42 was obtained in a very good yield (entry 22). The highly substituted aryl group of O-methyl oxime 43 also provided an excellent yield of isoxazole 44 (entry 23). Scaled up reactions, including multi -gram preparations of compounds 2, 4, and 44, did not affect the yield or outcome of the methodology. This is important to note in cases where one wishes to produce a large quantity of product for use in total synthesis or library generation.
To demonstrate the value of the 4-iodoisoxazole products generated in this methodology, a number of reactions were carried out utilizing the iodine handle. We reasoned that we could approach the highly potent COX-2 inhibitor 4-(5-methyl-3-phenyl-4-isoxazolyl) benzenesulfonamide (valdecoxib) (46)24,1g by a Suzuki-Miyaura cross-coupling of isoxazole 4 with the appropriate boronic acid ester.25 Using our isoxazole methodology to prepare 4 and a Suzuki-Miyaura coupling with the commercially available benzenesulfonamide-4-boronic acid pinacol ester, we were able to develop a very efficient route to valdecoxib (Scheme 3). Starting with ynone 45, O-methyl oxime 3 was obtained in a 92% yield. Compound 3 was subjected to our ICl cyclization conditions to afford isoxazole 4 in nearly a quantitative yield. After several protocols were screened, Suzuki cross-coupling was best accomplished using 5 mol % PdCl2 catalyst, 1.4 equiv of KHCO3 in 4:1 DMF:H2O at 85 °C for 3 h to provide valdecoxib (46) in an 81% isolated yield, which constitutes a 74% overall yield from ynone 45.
Scheme 3.

This synthetic route to valdecoxib provides a high overall yield and utilizes very mild reaction conditions compared to those previously reported. Earlier reports on the synthesis of valdecoxib required the use of strong bases, such as n-BuLi,1g,24,26 or the preparation of boronic acids in situ using air and water sensitive triisopropyl borate (i-PrO)3B.26 These previous processes also suffer from poor overall yields. In addition to the aforementioned advantages of this synthesis, one may construct many analogues of valdecoxib by choosing from the wide array of commercially available starting materialsthat are appropriate for this methodology.
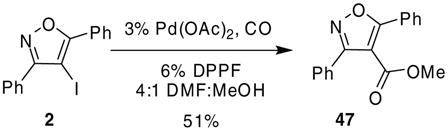
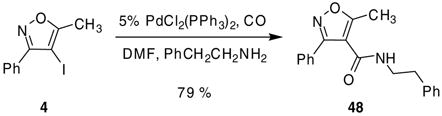
Other palladium-catalyzed methodology has also proven useful for further elaboration of these iodoisoxazoles. For example, we were able to convert 4-iodo-3,5-diphenylisoxazole (2) to the corresponding methyl ester by allowing 2 to react in the presence of catalytic amounts of Pd(OAc)2 plus a ferrocene ligand, carbon monoxide and methanol to afford methyl ester 47 in a 51% yield (eq 3).27 Reduction of compound 2 to 3,5-diphenylisoxazole was an observed minor side product. In addition, we were able to convert 4-iodo-5-methyl-3-phenylisoxazole (4) to the corresponding phenethylamide by a similar approach using a modified literature procedure.28 By allowing compound 4 to react in the presence of catalytic amounts of PdCl2(PPh3)2, carbon monoxide and 2-phenethyl amine, we were able to obtain 48 cleanly in good yield (eq 4).
(3).
(4).
Additionally, we were able to affect Heck and Sonogashira cross-couplings on 4-iodo-5-methyl-3-phenylisoxazole (4) (Scheme 4). By allowing compound 4 to react under standard Sonogashira29 conditions in the presence of 1.2 equiv of phenyl acetylene, alkyne 49 was obtained in a good yield. Also, allowing compound 4 to react under Heck30 reaction conditions in the presence of N-acryloylmorpholine, provided the desired α,β-unsaturated amide 50 in excellent yield.
Scheme 4.

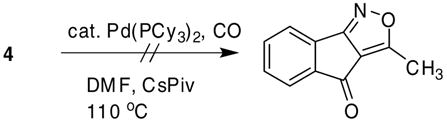
Unfortunately, the palladium-catalyzed carbonylative cyclization31 of isoxazole 4 failed and reduction of the starting material was the only observed product (eq 5).
(5).
Conclusions
3,4,5-Trisubstituted isoxazoles have been generated in high yields under mild reaction conditions by the electrophilic cyclization of Z-O-methyl oximes of 2-alkyn-1-ones. Our methodology tolerates a wide variety of functional groups, including heterocycles and sterically cumbersome substrates. This process can be scaled up to provide multi-gram quantities of the desired product without sacrificing the yield or outcome of the methodology. One can construct libraries of highly substituted isoxazoles by invoking the appropriate starting materials in an orderly fashion. Furthermore, the iodine handle of the products provides an opportunity for further functionalization, as demonstrated by the ester and amide products 47 and 48, the Sonogashira product 49 and the Heck product 50.
Experimental Section
General procedure for the preparation of alkynones from acyl chlorides
To a 25 mL flask were added CuI (0.05 mmol), PdCl2(PPh3)2 (0.01 mmol) and triethylamine (5 mL). The flask was flushed with argon and the terminal acetylene (2.5 mmol) was added to the stirred suspension, followed by immediate dropwise addition of the acyl chloride (3.25 mmol, 1.3 equiv). When the acyl chloride was a solid, it was added as a solution in THF. The resulting mixture was allowed to stir at room temperature overnight, water (5 mL) was added, and the aqueous layer was extracted with ethyl acetate. The organic layers were combined, dried and concentrated under reduced pressure. The residue was then purified by column chromatography on silica gel to afford the desired alkynone.
1,3-Diphenylprop-2-yn-1-one
Purification by flash chromatography (hexanes/EtOAc) afforded 433 mg (84%) of the product as a yellow liquid with spectral properties identical to those previously reported.19
General procedure for preparation of the O-methyl oximes
The alkynone (3.5 mmol), methoxylamine hydrochloride (7.0 mmol, 579 mg), Na2SO4 (7.0 mmol, 994 mg) and pyridine (1 mL) in methanol (10 mL) were stirred at room temperature. The reaction was monitored by TLC until the reaction was complete. The mixture was diluted with water (25 mL) and extracted with EtOAc (3 × 5 mL). The organic layer was washed with brine, dried and evaporated. The residue was then purified by column chromatography on silica gel to afford the desired O-methyl oxime.
(Z)-1,3-Diphenylprop-2-yn-1-one O-methyl oxime (1)
Purification by flash chromatography (40:1 hexanes/EtOAc) afforded 617 mg (75%) of the product as an off white solid: mp 40–42 °C; 1H NMR (CDCl3) δ 4.14 (s, 3H), 7.37–7.40 (m, 6H), 7.60–7.63 (m, 2H), 7.90–7.93 (m, 2H); 13C NMR (CDCl3) δ 63.4, 79.7, 101.4, 122.0, 126.8, 128.6, 128.7, 129.8, 129.9, 132.4, 133.8, 140.2; IR (neat, cm−1) 3052, 2935, 1598; HRMS Calcd for C16H13NO: 235.0997. Found: 235.1000.
General procedure for iodocyclization using Icl
To a stirred solution of the appropriate O-methyl oxime (0.25 mmol) in CH2Cl2 (2.5 mL) was added ICl (1M in CH2Cl2, 1.2 equiv) dropwise and the solution was allowed to stir at room temperature. The reaction was monitored by TLC to establish completion. The excess ICl was removed by washing with a satd aq soln of Na2S2O3. The aqueous solution was then extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were dried over anhydrous MgSO4 and concentrated under a vacuum to yield the crude product, which was purified by flash chromatography on silica gel using hexanes/EtOAc as the eluent.
Procedure for iodocyclization using I2
To a stirred solution of the O-methyl oxime (0.25 mmol) in CH3CN (2.5 mL) was added I2 (0.75 mmol, 191 mg) and the solution was allowed to stir at room temperature for 1 h. The excess I2 was removed by washing with a satd aq soln of Na2S2O3. The aqueous solution was extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were dried over anhydrous MgSO4 and concentrated under reduced pressure to yield the crude product. Purification by flash chromatography afforded the product.
3,5-Diphenyl-4-iodoisoxazole (2)
The product was obtained as a colorless solid: mp 176–178 °C (lit.32 mp 176.5–177.5 °C). The spectral properties were identical to those previously reported. 32
Supplementary Material
General experimental procedures and spectral data for all previously unreported starting materials and products. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank the National Institute of General Medical Sciences (R01 GM070620 and R01 GM079593) and the National Institutes of Health Kansas University Chemical Methodologies and Library Development Center of Excellence (P50 GM069663) for support of this research; Johnson Matthey, Inc. and Kawaken Fine Chemicals Co., Ltd. for donations of palladium catalysts ; and Frontier Scientific and Synthonix for donations of the phenylboronic acid.
References
- 1.For a brief review, see: Carlsen L, Döpp D, Döpp H, Duus F, Hartmann H, Lang-Fugmann S, Schulze B, Smalley RK, Wakefield BJ. In: Houben-Weyl, Methods in Organic Chemistry. Schaumann E, editor. E8a. Georg Thieme Verlag; Stuttgart, Germany: 1992. pp. 45–204.Rowley M, Broughton HB, Collins I, Baker R, Emms F, Marwood R, Patel S, Ragan CI. J Med Chem. 1996;39:1943. doi: 10.1021/jm960072u.Frolund B, Jorgensen AT, Tagmose L, Stensbol TB, Vestergaard HT, Engblom C, Kristiansen U, Sanchez C, Krogsgaard-Larsen P, Liljefors T.J Med Chem 200245245412036354Daidone G, Raffa D, Maggio B, Plescia F, Cutuli VMC, Mangano NG, Caruso A. Arch Pharm Pharm Med Chem. 1999;332:50. doi: 10.1002/(sici)1521-4184(19993)332:2<50::aid-ardp50>3.0.co;2-s.Tomita K, Takahi Y, Ishizuka R, Kamamura S, Nakagawa M, Ando M, Nakanishi T, Nakamura T, Udaira H. Ann Sankyo Res Lab. 1973;1:25.Chem Abstr. 1974;80:120808.Talley JJ. Prog Med Chem. 1999;13:201. doi: 10.1016/s0079-6468(08)70048-1.Talley JJ, Brown DL, Carter JS, Graneto MJ, Koboldt CM, Masferrer JL, Perkins WE, Rogers RS, Shaffer AF, Zhang YY, Zweifel BS, Seibert K. J Med Chem. 2000;43:775. doi: 10.1021/jm990577v.Giovannoni MP, Vergelli C, Ghelardini C, Galeotti N, Bartolini A, Kal Piaz V. J Med Chem. 2003;46:1055. doi: 10.1021/jm021057u.Li WT, Hwang DR, Chen CP, Shen CW, Huang CL, Chen TW, Lin CH, Chang YL, Chang YY, Lo YK, Tseng HY, Lin CC, Song JS, Chen HC, Chen SJ, Wu SH, Chen CT. J Med Chem. 2003;46:1706. doi: 10.1021/jm020471r.
- 2.(a) Larock RC, Yue D. Tetrahedron Lett. 2001;42:6011. [Google Scholar]; (b) Yue D, Larock RC. J Org Chem. 2002;67:1905. doi: 10.1021/jo011016q. [DOI] [PubMed] [Google Scholar]; (c) Flynn BL, Verdier-Pinard P, Hamel E. Org Lett. 2001;3:651. doi: 10.1021/ol0067179. [DOI] [PubMed] [Google Scholar]
- 3.(a) Huang Q, Hunter JA, Larock RC. Org Lett. 2001;3:2973. doi: 10.1021/ol010136h. [DOI] [PubMed] [Google Scholar]; (b) Huang Q, Hunter JA, Larock RC. J Org Chem. 2002;67:3437. doi: 10.1021/jo020020e. [DOI] [PubMed] [Google Scholar]
- 4.(a) Yao T, Larock RC. Tetrahedron Lett. 2002;43:7401. [Google Scholar]; (b) Yao T, Larock RC. J Org Chem. 2003;68:5936. doi: 10.1021/jo034308v. [DOI] [PubMed] [Google Scholar]; (c) Oliver MA, Gandour RD. J Org Chem. 1984;49:558. [Google Scholar]; (d) Biagetti M, Bellina F, Carpita A, Stabile P, Rossi R. Tetrahedron. 2002;58:5023. [Google Scholar]; (e) Rossi R, Carpita A, Bellina F, Stabile P, Mannina L. Tetrahedron. 2003;59:2067. [Google Scholar]
- 5.(a) Arcadi A, Cacchi S, Fabrizi G, Marinelli F, Moro L. Synlett. 1999:1432. [Google Scholar]; (b) Yue D, Yao T, Larock RC. J Org Chem. 2005;70:10292. doi: 10.1021/jo051299c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Bew SP, Knight DW. J Chem Soc, Chem Commun. 1996:1007. [Google Scholar]; (b) Djuardi E, McNelis E. Tetrahedron Lett. 1999;40:7193. [Google Scholar]; (c) Sniady A, Wheeler KA, Dembinski R. Org Lett. 2005;7:1769. doi: 10.1021/ol050372i. [DOI] [PubMed] [Google Scholar]; (d) Yao T, Zhang X, Larock RC. J Am Chem Soc. 2004;126:11164. doi: 10.1021/ja0466964. [DOI] [PubMed] [Google Scholar]; (e) Yao T, Zhang X, Larock RC. J Org Chem. 2005;70:7679. doi: 10.1021/jo0510585. [DOI] [PubMed] [Google Scholar]
- 7.(a) Barluenga J, Trincado M, Rublio E, Gonzalez JM. Angew Chem, Int Ed. 2003;42:2406. doi: 10.1002/anie.200351303. [DOI] [PubMed] [Google Scholar]; (b) Muhammad A, Knight DW. Tetrahedron Lett. 2004;45:539. [Google Scholar]; (c) Yue D, Larock RC. Org Lett. 2004;6:1037. doi: 10.1021/ol0498996. [DOI] [PubMed] [Google Scholar]; (d) Yue D, Yao T, Larock RC. J Org Chem. 2006;71:62. doi: 10.1021/jo051549p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arcadi A, Cacchi S, Di Giuseppe S, Fabrizi G, Marinelli F. Org Lett. 2002;4:2409. doi: 10.1021/ol0261581. [DOI] [PubMed] [Google Scholar]
- 9.Marshall JA, Yanik MM. J Org Chem. 1999;64:3798. [Google Scholar]
- 10.Knight DW, Redfern AL, Gilmore J. J Chem Soc, Chem Commun. 1998;2207 [Google Scholar]
- 11.(a) Barluenga J, Vazque-Villa H, Ballesteros A, Gonzalez JM. J Am Chem Soc. 2003;125:9028. doi: 10.1021/ja0355372. [DOI] [PubMed] [Google Scholar]; (b) Yue D, Della Cà N, Larock RC. Org Lett. 2004;6:1581. doi: 10.1021/ol049690s. [DOI] [PubMed] [Google Scholar]; (c) Yue D, Della Cà N, Larock RC. J Org Chem. 2006;71:3381. doi: 10.1021/jo0524573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ren XF, Konaklieva MI, Shi H, Dickey S, Lim DV, Gonzalez J, Turos E. J Org Chem. 1998;63:8898. [Google Scholar]
- 13.Worlikar SA, Kesharwani T, Yao T, Larock RC. J Org Chem. 2007;72:1347. doi: 10.1021/jo062234s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang X, Sarkar S, Larock RC. J Org Chem. 2006;71:236. doi: 10.1021/jo051948k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou C, Dubrovsky AV, Larock RC. J Org Chem. 2006;71:1626. doi: 10.1021/jo0523722. [DOI] [PubMed] [Google Scholar]
- 16.Yao T, Larock RC. J Org Chem. 2005;70:1432. doi: 10.1021/jo048007c. [DOI] [PubMed] [Google Scholar]
- 17.Kesharwani T, Worlikar SA, Larock RC. J Org Chem. 2006;71:2307. doi: 10.1021/jo0524268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waldo JP, Larock RC. Org Lett. 2005;23:5203. doi: 10.1021/ol052027z. [DOI] [PubMed] [Google Scholar]
- 19.Tohda Y, Sonogashira K, Hagihara N. Synthesis. 1977:777. [Google Scholar]
- 20.(a) Kobayashi T, Tanaka M. J Chem Soc, Chem Commun. 1981:333. [Google Scholar]; (b) Mohamed Ahmed MS, Mori A. Org Lett. 2003;5:3057. doi: 10.1021/ol035007a. [DOI] [PubMed] [Google Scholar]
- 21.Lin CF, Lu W-D, Wang IW, Wu MJ. Synlett. 2003:2057. [Google Scholar]
- 22.Birkofer L, Ritter A, Uhlenbrauck H. Chem Ber. 1963;96:3280. [Google Scholar]
- 23.Beak P, Basha A, Kokko B, Loo D. J Am Chem Soc. 1986;108:6016. doi: 10.1021/ja00279a058. [DOI] [PubMed] [Google Scholar]
- 24.(a) Talley JJ. G. D. Searle & Company; U.S. Patent 5,859,257. 1999; Chem Abstr. 1999;130:110269. [Google Scholar]; (b) Talley JJ. Prog Med Chem. 1999;13:201. doi: 10.1016/s0079-6468(08)70048-1. [DOI] [PubMed] [Google Scholar]
- 25.(a) Miyaura N, Suzuki A. Chem Rev. 1995;95:2457. [Google Scholar]; (b) Suzuki A. J Organomet Chem. 1999;576:147. and references therein. [Google Scholar]
- 26.Kumar JS, Ho MM, Leung JM, Toyokuni T. Adv Synth Catal. 2002;344:1146. [Google Scholar]
- 27.Friesen RW, Ducharme Y, Ball RG, Blouin M, Boulet L, Cote B, Frenette R, Girard M, Guay D, Huang Z, Jones TR, Laliberte F, Lynch JJ, Mancini J, Martins E, Masson P, Muise E, Pon DJ, Siegl PKS, Styhler A, Tsou NN, Turner MJ, Young RN, Girard Y. J Med Chem. 2003;46:2413. doi: 10.1021/jm0204542. [DOI] [PubMed] [Google Scholar]
- 28.Schoenberg A, Heck RF. J Org Chem. 1974;39:3327. [Google Scholar]
- 29.Sonogashira K. Chapter 5. In: Diederich F, Stang PJ, editors. Metal-Catalyzed Cross-Coupling Reactions. Wiley-VCH; Weinheim, Germany: 1998. pp. 203–229. [Google Scholar]; (b) Sonogashira K, Tohda Y, Hagihara N. Tetrahedron Lett. 1975:4467. [Google Scholar]
- 30.(a) Alonso F, Beletskaya IP, Yus M. Tetrahedron. 2005;61:11771. [Google Scholar]; (b) Nicolaou KC, Bulger PG, Sarlah D. Angew Chem, Int Ed. 2005;44:4442. doi: 10.1002/anie.200500368. [DOI] [PubMed] [Google Scholar]; (c) Negishi E, de Meijere A, editors. Handbook of Organopalladium Chemistry for Organic Synthesis. Vol. 1. Wiley-Interscience; New York: 2002. p. 1223. and references therein. [Google Scholar]; (d) Tsuji J. Palladium Reagents and Catalysts. John Wiley & Sons; Chichester, England: 2004. p. 135. [Google Scholar]
- 31.(a) Campo MA, Larock RC. Org Lett. 2000;2:3675. doi: 10.1021/ol006585j. [DOI] [PubMed] [Google Scholar]; (b) Campo MA, Larock RC. J Org Chem. 2002;67:5616. doi: 10.1021/jo020140m. [DOI] [PubMed] [Google Scholar]
- 32.Day RA, Blake JA, Stephens CE. Synthesis. 2003:1586. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
General experimental procedures and spectral data for all previously unreported starting materials and products. This material is available free of charge via the Internet at http://pubs.acs.org.