

Abstract
The β-adrenoceptor blockers exhibit a well-characterized anti-apoptotic property in the heart and kidney while less is known about the effect of this class of drugs on neuronal apoptosis. We studied the effects of three β- adrenoceptor blockers propranolol (1-(isoproplyamino)-3-(naphthalene-1-yloxy)propan-2-ol), atenolol (2-[4-[2-hydroxy-3-(1-methylethylamino)propoxyl]phenyl]ehanamide), and ICI 118551 (1-[2,3-(dihydro-7-methyl-1H-iden-4-yl)oxy]-3-[(1-methylethyl)amino]-2-butanol), against staurosporine-induced apoptosis in SH-SY5Y human neuroblastoma cells. Staurosporine increased caspase 3-like activity, DNA fragmentation, PARP cleavage, and the number of TUNEL positive cells consistent with the induction of apoptosis. Propranolol and ICI 118551, but not atenolol, demonstrated a concentration-dependent inhibition of caspase 3-like activity. Propranolol and ICI 118551 directly inhibited the enzymatic activity of recombinant caspase 9 while atenolol did not; however, none of the β- adrenoceptor blockers that were examined directly blocked caspase 2 or 3 activity. In isolated mitochondria, propranolol and ICI 118551 inhibited staurosporine-induced cytochrome c release while atenolol did not. We conclude that propranolol and ICI 118551 protect SH-SY5Y cells against staurosporine-induced apoptosis through a dual action on the mitochondria and on caspase 9 in a cell type and an apoptotic paradigm where the conventional inhibitors of mitochondrial permeability transition such as cyclosporin A and bongkrekic acid demonstrate no protection.
Keywords: β-adrenoceptor blockers, apoptosis, staurosporine, SH-SY5Y, caspases
1. Introduction
Brain neurons undergo apoptotic cell death after acute injury and in chronic neurodegenerative disorders (Dickson, 2004; Dirnagl et al., 1999; Verma, 2000). Therefore, much effort has been put into the development of clinically acceptable therapeutics targeting the apoptotic cascade. Voltage gated calcium channel blockers, free radical scavengers, inhibitors of mitochondrial permeability pore, and caspase inhibitors are some examples of molecules explored as anti-apoptotic agents in the brain (reviewed in Merenda and Bullock, 2006). Although helpful in experimental investigations, none of the anti-apoptotic therapeutics has made much progress into the clinical arena. Furthermore, the conventional mitochondrial permeability transition inhibitor cyclosporin A demonstrates little anti-apoptotic activity in neurons (reviewed in Zoratti et al., 2005 but see Hansson et al., 2004) highlighting the importance of identifying a new class of anti-apoptotic drugs active in the brain.
β-adrenoceptor blockers are a clinically well-accepted and widely used class of drugs (Lopez-Sendon et al., 2004) with a well documented anti-apoptotic effect on the heart both in vivo and in vitro (Burniston et al., 2005; Communal et al., 1999; Zaugg et al., 2000). In contrast to the well-studied effects of β- adrenoceptor blockers on the heart and kidney, little is known about the action of these drugs on neurons. β-adrenoceptors are widely expressed in the brain and thought to mediate physiological responses to catecholamines. In the brain, β1-adrenoceptors are expressed mainly in neurons whereas β2-adrenoceptors are largely restricted to the glial cells (Nicholas et al., 1996). In the intact brain, both non-selective and β1- adrenoceptor selective blockers have been shown to decrease infarct volume and enhance functional recovery in a transient focal ischemia model (Goyagi et al., 2006; and references cited therein). However, the cellular mechanism responsible for such apparent neuro-protection by β-adrenoceptor blockers is not known. A recent study demonstrated that propranolol, a non-receptor subtype selective β-adrenoceptor antagonist, inhibits Bax- and Bcl-2 homology 3 (BH3) peptide-induced cytochrome c release from isolated adult rat brain mitochondria (Polster et al., 2003). If β- adrenoceptor blockers have a similar effect on the mitochondrial function in a cellular context, this could explain the observed neuroprotective effect in the intact brain.
In this study, we investigated the potential anti-apoptotic activity of β-adrenoceptor blockers in a model system using the human SH-SY5Y cells. These cells are derived from a human catecholaminergic neuroblastoma but have been proposed as a useful in vitro model of normal neurons and often used to study neuronal death (Abramova et al., 2002; McGinnis et al., 1999; Moriya et al., 2000; Tang et al., 2005; Tieu et al., 1999). In addition, caspase 8 is not expressed in this cell line (Hopkins-Donaldson et al., 2000), therefore the extrinsic apoptotic pathway contributes little to cell death (Lopez and Ferrer, 2000), enabling us to focus only on the effects of β-adrenoceptor blockers on the intrinsic apoptotic pathway. We specifically asked whether all β-adrenoceptor blockers exhibit an anti-apoptotic property and whether the drugs possess a direct caspase-inhibitor-like property in addition to the previously described mitochondrial stabilization effect.
We report that propranolol (1-(isoproplyamino)-3-(naphthalene-1-yloxy)propan-2-ol) as well as ICI 118551 (1-[2,3-(dihydro-7-methyl-1H-iden-4-yl)oxy]-3-[(1-methylethyl)amino]-2-butanol), an experimental β2- adrenoceptor selective antagonist, was protective against staurosporine-induced apoptosis while atenolol (2-[4-[2-hydroxy-3-(1-methylethylamino)propoxyl]phenyl]ehanamide), a relatively β1-adrenoceptor selective antagonist, was not. Propranolol and ICI 118551 demonstrated direct inhibition of caspase 9 activity but at higher concentrations than necessary for its anti-apoptotic effect on intact cells. Propranolol and ICI 118551 blocked opening of the cyclosporin-A-insensitive mitochondrial permeability transition pore (mPTP) and the release of cytochrome c while atenolol did not. The blockade of cytochrome c release and inhibition of caspase 9 may be two mechanisms by which some β-adrenoceptor antagonists protect neurons from apoptosis.
2. Materials & Methods
2.1. Cell culture
The SH-SY5Y human neuroblastoma cells were grown in RPMI supplemented with 10% fetal calf serum and antibiotics. Cells were passaged weekly at about 1:20 split after trypsin digestion to allow growth to near-confluence over 1 week. All experiments were done on cells between passages 10 – 20 after purchasing the original cells from ATCC (Manassas, VA, USA).
For induction of apoptosis, SH-SY5Y cells plated on a poly-d-lysine-coated 6-well cell culture plate were serum-starved overnight and replaced with a serum-free medium supplemented in the morning with 10 µM staurosporine. Preliminary studies demonstrated maximum caspase 3-like activity at 5–7 hours after initiation of staurosporine treatment with a decline in the detectable caspase 3-like activity at longer times (Supplemental data Fig. 1S). Therefore, all subsequent experiments were assayed after 6 hours of staurosporine treatment. The test β-adrenoceptor blockers were applied at the same time with the staurosporine-containing solution without pre-incubation. 2 µM cyclosporin A and 10 µM bongkrekic acid were applied 30 min before staurosporine or 5 µM p-trifluoromethoxy-carbonyl-cyanide-phenyl-hydrazone (FCCP) treatments. Cells treated with vehicle (dimethyl sulfoxide (DMSO) 0.1%) were prepared as control.
2.2. Caspase-like activity assay
Cells treated with staurosporine and incubated for variable times as dictated by the experiment were washed in phosphate buffered saline, lysed in ice-cold caspase lysis buffer (10 mM Tris-HCl, 10 mM NaH2PO4, 130 mM NaCl, 1% Triton X-100, 10 mM NaPPi, pH 7.5) for 15 min, and clarified by centrifugation. Protein concentration was standardized to 1 mg/ ml in caspase lysis buffer and 50 µg of protein was used for the assays. The caspase enzymatic reactions were carried out at 37°C in 20 mM PIPES, 100 mM KCl, 0.5 mM EDTA, 0.05% CHAPS, 5 mM dithio-threitol (pH 7.2) containing either 0.1 mM acyl-Asp-Glu-Val-Asp (Ac-DEVD) -7-amino-4-methylcoumarin (AMC) (caspase 3-like) or acyl-Leu-Glu-His-Asp (Ac-LEHD) -AMC (caspase 2- or 9-like) substrates. The rate of fluorescence increase was measured kinetically over 1 hour (ex: 360 nm, em: 460 nm) using a Synergy II microplate reader (Bio-Tek Instruments, Winooski, VT, USA) and converted to AMC released/ min/ µg protein using the calibration standard correlating fluorescence to AMC. Purified recombinant caspases, 2 units (caspase 2), 5 units (caspase 3) or 40 units (caspase 9) in ice-cold caspase lysis buffer described above, were used for the in vitro assays. Different units of caspases were used to compensate for the different efficacy of the reporter substrate. β–adrenoceptor blockers were added and the caspase enzymatic reactions carried out as described above with the exception that the read out was in fluorescent units/ min. Ac-LEHD-AMC was used to measure caspase 2 and 9, and Ac-DEVD-AMC for caspase 3 activity, respectively.
2.3. Apoptosis Assay
The TdT-mediated dUTP nick-end labeling (TUNEL) assay was carried out as per manufacturer’s instructions (Dead End Fluorometric TUNEL Assay, Promega, Madison, WI, USA). At least 6 random fields from 3 independent experiments (> 600 cells) were photographed and counted.
To detect DNA fragmentation, cells were grown in a 6 cm culture plate and treated with vehicle, staurosporine or 100 µM propranolol for 7 hours. Cells were collected by scraping and centrifuged, then the pellet was washed with phosphate buffered saline. The lysis buffer (1% Nonidet P40, 20 mM EDTA, 50 mM Tris-HCl, 1% sodium dodecyl sulfate (SDS), pH7.5) containing 5 µg/ µl RNAse A was added and incubated for 2 hours at 56 °C. After subsequent incubation with 2.5 µg/ µl Proteinase K for 2 hours at 37 °C, fragmented DNA was precipitated by adding ½ volume of 10 M ammonium acetate and 2.5 volume of absolute ethanol and then isolated by centrifugation at 10,000 × g for 10 min at 4 °C. The DNA pellet was dissolved in 20 µl of nuclease-free water. The DNA samples were subjected to electrophoresis on 2 % agarose gel, and visualized under ultraviolet light after staining with ethidium bromide.
Cells with membrane externalization of phosphatidylserine was detected via binding of annexin-V-fluorescein isothiocyanate (FITC) using the ApoAlert Annexin-V kit (Clontech, Montain View, CA, USA) and a Quanta SC flow cytometer (Beckman Coulter, Miami, FL, USA) as described previously (Lee et al., 2007). Non-viable (necrotic or late apoptotic) cells were detected with propidium iodide staining.
2.4. Western Blotting
Western blotting was carried out by standard methods. Samples were lysed in an ice-cold lysis buffer (1% Nonidet P40, 10 mM Tris pH 7.6, 50 mM NaCl, 30 mM NaPPi, 50 mM NaF) containing freshly added protease inhibitor (Complete, Roche Biochemicals, Indianapolis, IN, USA) for 30 min on ice and sonicated (50% power for 2 second × 10 pulses, Cole Palmer Model 130 Ultrasonic Processor). Lysed samples were centrifuged at 5000 × g for 15 min at 4 °C and the supernatant was combined with SDS sample buffer and incubated at 100 °C for 5 min before loading on a 10% SDS-polyacrylamide gel. Proteins were separated by electrophoresis, and transferred to a nitrocellulose membrane (BioRad, Hercules, CA, USA). The membrane was blocked in 3% milk-Tris buffered saline (50 mM Tris-HCl, pH8.0, 100 mM NaCl) supplemented with 0.1% Tween 20 (TBST), probed with the following primary antibodies (all 1:1000 – 1:5000 dilution): poly-ADP-ribose polymerase (PARP) (#556494, BD Pharmingen, San Diego, CA, USA), cytochrome c (S2050, Clontech, Mountain View, CA, USA), actin (Sigma-Aldrich, St. Louis, MO, USA), all in 1% milk-TBST, and reacted with the horseradish peroxidase-conjugated secondary antibody (1:2000) in 1% milk-TBST. After reaction with the Western Lightning chemiluminescence reagent (NEN Life Science Products, Boston, MA, USA), the images were captured on the EpiChemi Darkroom System (UVP Inc, Upland, CA, USA). Densitometric quantitation was done using the LabWorks 4.0 software (UVP Inc).
2.5. Isolation of mitochondrial fraction and determination of cytochrome c release
Mitochondrial pellets were prepared as described by Kim et al. (2007). Pellets were resuspended in a mannitol-sucrose buffer consisting of 225 mM mannitol, 25 mM sucrose, 10 mM HEPES, 1 mM EDTA, pH 7.4, protease inhibitors supplemented with 5 mM succinate, 3 mM ATP and 3 mM ADP. Staurosporine and β-adrenoceptor blockers were added and incubated at 30 °C. At the end of the stated incubation duration, the mitochondria were pelleted by centrifugation at 16,000 × g for 20 min, and the supernatant assayed for the presence of released cytochrome c by western blotting.
2.6. Detection of β- adrenoceptor in SH-SY5Y cells
For detection of β-adrenoceptor mRNA, 1 µg of total RNA isolated (RNAeasy, Invitrogen, Carlsbad, CA, USA) from SH-SY5Y cells was subjected to a single tube reverse transcription polymerase chain reaction (rt-PCR) (Superscript III, Invitrogen) using the primer pairs (β1: agcatcgagaccctgtgtgtcaat and agttgaagaagacgaagaggcggt (658 bp); β2: ggtcatcacagccattgccaagtt and aggtaaggcctgacacaatccaca (335 bp); β3: tctctgctggttgcccttctttct and tcattctgaacagaggccagaggt (621 bp)) specific for the human β- adrenoceptor subtypes. Negative controls were tubes subjected to the identical condition except for omitting the reverse transcription step. Positive controls were PCR reactions carried out with the same primer pairs using a human transcriptome cDNA library (#790000, Stratagene, La Jolla, CA, USA) as the template. Immunocytochemistry was performed with standard methods on 4% paraformaldehyde fixed cells using anti-β1 adrenoceptor (sc568, Santa Cruz Biotech, Santa Cruz, CA, USA) or anti-β2 adrenoceptor (sc569, Santa Cruz Biotech) polyclonal antibody both at 1:200 dilution in phosphate buffered saline with 0.2% Tween 20 and 2% normal goat serum. Images were captured on an epifluorescent microscope with a 100× oil immersion objective lens after reacting with an Alexa 594 -conjugated goat anti-rabbit secondary antibody.
2.7. Chemicals
ICI 118551 was purchased from Tocris Bioscience (Ellisville, MO, USA). Staurosporine, Ac-DEVD-AMC, Ac-LEHD-AMC, benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (zVAD-fmk), and human recombinant caspases were from Biomol International (Plymouth Meeting, PA, USA). All other chemicals were purchased from Sigma-Aldrich.
2.8. Data analysis
Data are presented as mean ± S.E.M. and the statistical significance between the control and a treatment group determined by the Mann-Whitney nonparametric test (SigmaStat v3.5, Systat Software Inc., Point Richmond, CA, USA) at the indicated P-level.
3. Results
Staurosporine induced apoptosis in SH-SY5Y neuroblastoma cells as assessed by caspase 3-like activity in a concentration and time-dependent manner, detection of PARP cleavage product by Western blot, and by externalization of phosphatidylserine as determined by flow cytometer (Supplemental Fig. 1–Supplemental Fig. 2). All subsequent experiments were performed at a fixed staurosporine concentration of 10 µM and assays performed at 6 – 7 hours at the peak of caspase 3-like activity. We examined the effect of propranolol, atenolol, and ICI 118551, on staurosporine-induced apoptosis. The anti-apoptotic property of propranolol at the 7 hour time point was confirmed by its ability to reduce the number of TUNEL positive cells (Fig. 1 and Table I). At the 24 hour time point, there was no statistically significant difference in the number of TUNEL positive cells between the staurosporine and the staurosporine + propranolol groups suggesting that propranolol did not inhibit but delayed the onset of apoptosis (Table I). The anti-apoptotic effect of propranolol was confirmed by the decrease in staurosporine-induced DNA fragmentation and PARP cleavage (Fig. 2A, B) while propranolol by itself had no effect on any of the indicators of apoptosis examined. Examination of the PARP cleavage over an extended time after staurosporine stimulation also suggested that propranolol delayed the onset of apoptosis (Fig. 2C), however, this interpretation must be viewed with caution since prolonged exposure of the cells to the serum starved experimental condition in itself induces apoptosis. Propranolol and ICI 118551 inhibited the staurosporine induction of caspase 3-like activity with an IC50 of 30 ± 2.8 µM and 2.5 ± 1.0 µM, respectively, while atenolol did not (Fig. 3).
Fig. 1. Propranolol, a clinically used β-adrenoceptor blocker, decreased the number of TUNEL positive cells.

A phase contrast (left), nuclear stain (middle), and TUNEL stain (right) images of the same field of view for cells subjected to the noted conditions. Staurosporine increased the number of TUNEL positive cells while propranolol decreased this effect.
Table I. Propranolol decreases the number of TUNEL positive cells induced by staurosporine.
The numbers are percentage (mean ± S.E.M.) of TUNEL positive cells at 7 or 24 hours after initiation of the staurosporine (10 µM) treatment with or without propranolol (100 µM). * P<0.05, ns=not significant.
| 7 hours | 24 hours | |||
|---|---|---|---|---|
| Control (DMSO) | 1.8±0.6 | 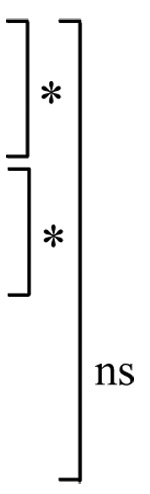 |
2.6±0.5 | 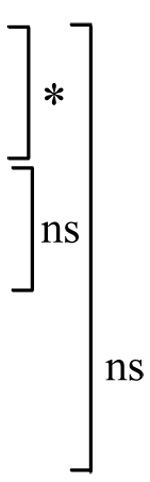 |
| Staurosporine | 12.1±1.1 | 21.9±3.8 | ||
| Staurosporine + Propranolol | 5.0±1.1 | 16.0±2.3 | ||
| Propranolol | 2.9±0.7 | 1.9±0.4 |
Fig. 2. Propranolol decreased staurosporine-induced DNA fragmentation and PARP cleavage.

A. A 2% agarose gel demonstrating DNA fragmentation and reversal of this effect by propranolol (lanes 1: DMSO, 2: staurosporine (10 µM), 3: staurosporine + propranolol (100 µM), and 4: propranolol alone (100 µM). B. A western blot of the cytosolic fraction probed with anti-PARP antibody demonstrating the cleavage of the PARP protein (85 kD). Anti-actin antibody probing of the same membrane confirmed equal loading of the lanes. The lanes correspond to the same conditions as above and assayed at 7 hours after initiation of staurosporine treatment. C. Same condition as above except assayed at later time points. Propranolol was unable to inhibit PARP cleavage at 24–48 hours indicating that the onset of staurosporine-induced apoptotic program was delayed but not inhibited.
Fig. 3. Not all of the β-adrenoceptor blockers inhibited staurosporine-induced increase in caspase3-like activity.

Inhibition concentration-response for the various β-adrenoceptor blockers with caspase-3-like activity reported as FU (fluorescence unit)/ min/ µg protein. Data fit with y=y0+A(IC50 n/(IC50 n+xn)) where IC50= half-inhibition concentration, n= Hill slope, gave IC50 of 30 ± 2.8 and 2.5 ± 1.0 µM, and n of 1.3 ± 0.4 and 1.4 ± 0.4, for propranolol and ICI 118551, respectively. Results from 3 experiments each with triplicate measurements.
Staurosporine, a protein kinase C inhibitor, induces apoptosis in a wide variety of cells. Its major site of action is the intrinsic pathway, although components of the extrinsic pathway also have been implicated (Stepczynska et al., 2001; Zhang et al., 2004). In SH-SY5Y cells devoid of caspase 8, staurosporine induces apoptosis mostly via activation of the upstream initiator caspases 2 and 9, with a consequent activation of the downstream executor caspase 3 (Lopez and Ferrer, 2000). The β-adrenoceptor blockers could inhibit staurosporine-induced apoptosis by interfering with caspase activation. In fact, the broad-spectrum caspase inhibitor zVAD-fmk inhibited staurosporine-induced caspase 3-like and caspase 9-like activity in a concentration-dependent manner much like the effect of propranolol as assessed by the substrates DEVD-AMC and LEHD-AMC (Fig. 4). However, it is not possible to convincingly examine the effect of propranolol on the individual caspases within a cellular context since no currently available caspase inhibitors (e.g. Ac-DEVD-aldehyde, Ac-LEHD-aldehyde, Ac-VDVAD-aldehyde) are strictly selective (Talanian et al., 1997). Therefore, we examined the effect of propranolol on the purified recombinant caspases activity in an in vitro experiment (Fig. 5). Caspase 2, 3, and 9 were chosen because these three caspases largely mediate staurosporine-induced apoptosis in SH-SY5Y cells (Lopez and Ferrer, 2000). Propranolol, atenolol, or ICI 118551 up to 100 µM did not have any significant direct effect on either caspase 2 or caspase 3 activities. In contrast, caspase 9 was directly inhibited by ICI 118551 and propranolol albeit at concentrations approximately one order of magnitude greater than the apoptosis-inhibitory activity of these β-adrenoceptor blockers observed within the in vivo cellular context. zVAD-fmk inhibited all in vitro caspase activities in a concentration-dependent manner as expected.
Fig. 4. Propranolol inhibited staurosporine-induced activation of caspase 3 and 9-like activities.

Propranolol (prop) (100 µM) inhibits staurosporine-activation of both caspase 3 and its upstream initiator caspase 9-like activities much like the broad-spectrum caspase inhibitor zVAD-fmk (10, 1, 0.1 µM). Results from two independent experiments each with triplicate measurements.
Fig. 5. Propranolol and ICI 118551 inhibited staurosporine activation of caspase 9 in vitro.

The direct effect of β- adrenoceptor blockers on purified recombinant caspases 9, 2, and 3 activities were examined in vitro. ICI 118551 (10 – 100 µM) and propranolol (100 – 1000 µM) directly inhibited caspase 9 activity. All caspases were inhibited by zVAD-fmk (0.1 – 10 µM). Representative of 3 independent experiments each with triplicate measurements. The ordinate is presented as fluorescence unit (FU) / min since the amount of recombinant caspase used for the assay was defined by the activity unit rather than µg protein (see Methods).
Polster et al. (2003) demonstrated that propranolol inhibited BH3 peptide and Bax -induced cytochrome c release from isolated brain mitochondria. In SH-SY5Y cells, staurosporine similarly induced cytochrome c release from isolated mitochondria in a time dependent manner with a reciprocal decrease in the mitochondrial cytochrome c consistent with an earlier report (Kim et al., 2007), and propranolol significantly reduced this release (Fig. 6A, B). ICI 118551 also reduced the staurosporine-induced cytochrome c release from the mitochondria but to a lesser degree than propranolol, suggesting that the two β-adrenoceptor blockers may utilize different mechanisms to manifest the anti-apoptotic effect. FCCP, a mitochondrial depolarizer and a pro-apoptotic agent (Gautier et al., 2000), induced caspase 3-like activation in SH-SY5Y cells. Staurosporine-induced cytochrome c release has been reported to depend on opening of the mPTP in some preparations but not in others (Scarlett et al., 2000; Wigdal et al., 2002). We examined whether the staurosporine-induced caspase 3 activation in SH-SY5Y was inhibited by the classical mPTP blockers cyclosporin A and bongkrekic acid. Staurosporine-induced caspase 3 activation in SH-SY5Y cells was not blocked by cyclosporin A or bongkrekic acid (Fig. 6C, left) but rather paradoxically increased. The caspase-3 activation by FCCP was not blocked by cyclosporin A or bongkrekic acid either (Fig. 6C, right). However, propranolol inhibited the caspase 3 activity induced by both staurosporine and FCCP indicating that this β- adrenoceptor blocker demonstrated anti-apoptotic activity in a cell type where the conventional mPTP blockers were unable to provide protection.
Fig. 6. Cytochrome c release from mitochondria induced by staurosporine was blocked by propranolol.

A. Time-dependent increase in cytosolic- and a reciprocal decrease in the mitochondrial- cytochrome c induced by 10 µM staurosporine (A1) and the effect of β-adrenoceptor blockers on this release at 60 min (A2). The concentration of the drugs were staurosporine (STS) 10 µM, propranolol (prop) 100 µM, ICI 118551 (ICI) 10 µM, and atenolol (aten) 100 µM. The cytosolic and mitochondrial cytochrome c appeared differently on the Western blot due to the different extraction buffers used for the two cellular fractions (see Methods). B. A bar plot of densitometric quantification of immunoblots of cytosolic cytochrome c normalized to the STS only condition from four separate experiments. The relative intensities (in arbitrary units) of propranolol-treated (0.62 ± 0.02) and ICI 118551–treated (0.88 ± 0.03) samples were significantly reduced from staurosporine alone while atenolol had no effect (0.96 ± 0.02). C. Propranolol inhibited both STS- and FCCP-induced caspase 3-like activation that was not blocked by 2 µM cyclosporin A (CysA) or 10 µM bongkrekic acid (BA). The relative caspase 3-like activity was normalized to the STS-only (left) or p-trifluoromethoxy-carbonyl-cyanide-phenyl-hydrazone (FCCP)-only conditions (right). Statistical significance at * P<0.05 and *** P<0.001. n.s. = not significant. Number of independent measurements in parentheses.
Lastly, we sought to demonstrate the presence of β-adrenoceptor in SH-SY5Y cells. Rt-PCR demonstrated the presence of both β1- and β2- adrenoceptor mRNA (Fig. 7A) and immunocytochemistry confirmed the presence of immunoreactivity to both anti-β1- adrenoceptor and anti-β2- adrenoceptor antibodies consistent with both β- adrenoceptor proteins being expressed in these cells (Fig. 7B). However, the immunoreactivity was mostly peri-nuclear to diffusely cytocolic and not strictly plasma membrane limited. Direct β- adrenoceptor stimulation with norepinephrine, epinephrine, or isoproterenol did not increase the number of TUNEL positive cells or caspase 3-like activation, indicating that these receptors do not serve an acute pro-apoptotic role in the SH-SY5Y cells (Fig. 8). Thus the observed anti-apoptotic effect of the β- adrenoceptor antagonists was most likely not mediated by inhibition of an occult stimulation of these receptors.
Fig. 7. β-adrenoceptors exist in SH-SY5Y cells.

A. rt-PCR confirmation of the presence of β1- and β2- adrenoceptor mRNA. rt-PCR was performed on total RNA isolated from the cells demonstrating products of the expected size. The negative rt control (-RT) where the reverse transcription step was omitted resulted in no product. The same primer pairs using a cDNA brain library control template served as a positive control for the PCR reaction. B. Immunocytochemistry with β1- (left) or β2- (middle) adrenoceptor selective antibodies demonstrated immunoreactivity consistent with the expression of the receptor protein. Fluorescent antibody signal (upper panels) and nuclear staining (lower panels) views of the same field are shown. Omission of the primary antibody (right) gave no signal.
Fig. 8. Direct catecholamine stimulation does not increase the number of TUNEL positive cells or caspase 3-like activity in SH-SY5Y cells.

A. A histogram summary of TUNEL positive cells (mean ± S.E.M. from triplicate counts). TUNEL labeling was performed in cells with no treatment (None) or 8 hours after treatment with 100 µM norepinephrine (NE), epinephrine (Epi), isoproterenol (Iso), or 10 µM staurosporine (STS). B. Similar experiments but assayed for caspase 3-like activity. Results from N=3 independent experiments. * P < 0.01 and no statistically significant difference between the other 4 groups.
4. Discussion
In this study, we demonstrated that the β-adrenoceptor blockers propranolol and ICI 118551 protect SH-SY5Y cells against staurosporine-induced apoptosis. Both β-adrenoceptor blockers directly inhibited recombinant caspase 9 activity in vitro; however, they did so at concentrations greater than necessary for the manifestation of their cytoprotective effect in intact cells. Therefore, it is unlikely that the direct inhibition of caspase 9 activity alone plays a significant role in the drugs’ anti-apoptotic effect. Although β-adrenoceptors are present in these cells, the anti-apoptotic effect of β- adrenoceptor blockers is unlikely to be mediated by the actual blockade of the receptors because direct stimulation of the β-adrenoceptors by catecholamines does not induce apoptosis. Furthermore, concentrations of β-adrenoceptor blockers necessary for the anti-apoptotic (1 – 100 µM) effect were orders of magnitude greater than the binding affinity of these ligands for the β- adrenoceptors (1 – 100 nM) (Hoffman et al., 2004).
Polster et al. (2003) suggested that amphiphilic molecules such as propranolol and dibucaine inhibit cytochrome c release by acting on events downstream of Bax incorporation into the outer mitochondrial membrane, perhaps by blocking Bax-induced changes in the lipid structure (Basanez et al., 2002). In their model of isolated brain mitochondria, the BH3 peptide and Bax-induced release of cytochrome c was not due to the opening of conventional mPTP since cyclosporin A did not block this release. Our experiments on intact SH-SY5Y cells demonstrated a similar non-cyclosporin A and non-bongkrekic acid-blockable activation of the apoptotic cascade by staurosporine or FCCP which was blocked by two of the three β- adrenoceptor antagonists tested. Cyclosporin A and bongkrekic acid are well known inhibitors of apoptosis in many cell types (reviewed in Bouchier-Hayes et al., 2005; Zoratti et al., 2005). Cyclosporin A is thought to acts through inhibition of association between the matrix cyclophilin D and the inner membrane adenine nucleotide translocator while bongkrekic acid acts by binding to the adenine nucleotide translocator fixing its conformational state to the closed state. Both adenine nucleotide translocator and cyclophilin D are thought to be components of the putative mPTP. However, the ability of these classical mPTP inhibitors to prevent opening of the mPTP and the subsequent induction of apoptosis in SH-SY5Y cells appears to depend on the pro-apoptotic stimulus employed (Abramova et al., 2002; McGinnis et al., 1999; Moriya et al., 2000) and we confirmed the earlier report by McGinnis et al. (1999) that staurosporine-induced apoptosis in SH-SY5Y cells was not blocked by the classical mPTP inhibitors. It is likely that the β- adrenoceptor blockers are inhibiting the initiation of cyclosporin A and bongkrekic acid insensitive mitochondria-mediated apoptotic cascade in the intact SH-SY5Y cells similar to that described in the isolated brain mitochondria (Polster et al., 2003). The site of action of the β- adrenoceptor blockers remains undefined at this time but most likely not mediated by the β- adrenoceptors. The opening of the mPTP is a complex process with many different inducers, modulators and inhibitors, dependent on the cellular model (reviewed by Zoratti et al., 2005). Therefore our finding may not be generalizable, however, β-adrenoceptor blockers may prove to be better anti-apoptotic drugs than the conventional mPTP inhibitors when targeting neurons.
The role of reactive oxygen species in the opening of mPTP is well-known (He and Lemasters, 2002; Kowaltowski et al., 1996), and some β-adrenoceptor blockers have been reported to exhibit anti-oxidant properties (Jaboureck-Bouttier et al., 1999; Nishina et al., 2001). Carvedilol, an α- and β- adrenoceptor antagonist, demonstrated neuroprotection in an in vitro model of free radical-mediated neuronal injury (Yue et al., 1994), in vivo global ischemic model (Lysco et al., 1992), and in vivo focal cerebral ischemia model (Savitz et al., 2000) through its anti-oxidant effect. However, the IC50 required to inhibit lipid peroxidation in the swine brain homogenate for propranolol and atenolol were >3000 and 1000 µM, respectively (Yue et al., 1994), thus much higher than the concentrations used in our experiment. Therefore it is likely that the anti-apoptotic effect of propranolol was not due to its antioxidant property.
Although we focused on the β- adrenoceptor blockers’ effect on caspases and mitochondrial integrity, staurosporine induces cell death in many ways. Decrease in the expression of the anti-apoptotic Bcl 2 and increase in the expression of the pro-apoptotic p53 (Tieu et al., 1999), Bax translocation to the mitochondria secondary to intracellular alkalinization (Tafani et al., 2002), and increase in the phosphorylation of glycogen synthase kinase-3β (Bhat et al., 2000) are some of the other reported staurosporine-induced cell death mechanisms. Further studies will be required to rule out the possibility that the β- adrenoceptor blockers’ anti-apoptotic effect may be mediated by effects on any of the above mechanisms.
As a class of drugs, β- adrenoceptor blockers share common molecular characteristics but also individually possess unique structural signatures. Regardless of the precise mechanism responsible for the anti-apoptotic action, further structure-activity analysis common to propranolol and ICI 118551 exhibiting an anti-apoptotic property, but not shared with atenolol devoid of the protective effect, may lead to identification of critical molecular signatures essential to the development of a more potent clinically acceptable novel anti-apoptotic small molecule derivative of the β- adrenoceptor blockers.
Supplementary Material
A. Cells were treated with different concentrations of staurosporine and caspase 3-like activity assayed at 5 hours after administration of the drug (EC50 = 1.02 µM, n=1.51) or at different time points after 10 µM staurosporine (B). Mean ± S.E.M. results from triplicate experiments. FU=fluorescence unit.
A. Cells were treated with 10 µM staurosporine for the indicated times and analyzed by flow cytometer for early apoptosis (annexin V-positive but propidium iodide (PI)-negative) and late apoptosis (annexin V-positive and PI-positive). B. A western blot of cell lysates obtained from cells treated with DMSO or staurosporine (10 µM) for the indicated times. The 85kD band represents the cleaved PARP. Equal loading of the lanes was confirmed by immunoblotting with the anti-actin antibody. Representative of three independent experiments.
Acknowledgments
This work was partly supported by NIH RO1 GM071485 (JY) and funds from the Department of Anesthesiology at Columbia University. We thank Judy Choi and Peter Shaughnessy for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abramova NA, Cassarino DS, Khan SM, Painter TW, Bennett JP., Jr Inhibition by R(+) or S(−) pramipexole of caspase activation and cell death induced by methylpyridinium ion or beta amyloid peptide in SH-SY5Y neuroblastoma. J. Neurosci. Res. 2002;67:494–500. doi: 10.1002/jnr.10127. [DOI] [PubMed] [Google Scholar]
- Basanez G, Sharpe JC, Galanis J, Brandt TB, Hardwick JM, Zimmerberg J. Bax-type apoptotic proteins porate pure lipid bilayers through a mechanism sensitive to intrinsic monolayer curvature. J. Biol. Chem. 2002;277:49360–49365. doi: 10.1074/jbc.M206069200. [DOI] [PubMed] [Google Scholar]
- Bhat RV, Shanley J, Correll MP, Fieles WE, Keith RA, Scott CW, Lee CM. Regulation and localization of tyrosine 216 phosphorylation of glycogen synthase kinase-3β in cellular and animal models of neuronal degeneration. Proc. Natl. Acad. Sci. 2000;97:11074–11079. doi: 10.1073/pnas.190297597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchier-Hayes L, Lartigue L, Newmeyer DD. Mitochondria: pharmacological manipulation of cell death. J Clin. Invest. 2005;115:2640–2647. doi: 10.1172/JCI26274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burniston JG, Tan LB, Goldspink DF. β2-adrenergic receptor stimulation in vivo induces apoptosis in the rat heart and soleus muscle. J. Appl. Physiol. 2005;98:1379–1386. doi: 10.1152/japplphysiol.00642.2004. [DOI] [PubMed] [Google Scholar]
- Communal C, Singh K, Sawyer DB, Colucci WS. Opposing effects of β1- and β2-adrenergic receptors on cardiac myocyte apoptosis. Circulation. 1999;100:2210–2212. doi: 10.1161/01.cir.100.22.2210. [DOI] [PubMed] [Google Scholar]
- Dickson DW. Apoptotic mechanisms in Alzheimer neurofibrillary degeneration: cause or effect? J. Clin. Invest. 2004;114:23–27. doi: 10.1172/JCI22317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Gautier I, Geeraert V, Coppey J, Coppey-Moisan M, Durieux C. A moderate but not total decrease of mitochondrial membrane potential triggers apoptosis in neuron-like cells. Neuroreport. 2000;11:2953–2956. doi: 10.1097/00001756-200009110-00024. [DOI] [PubMed] [Google Scholar]
- Goyagi T, Kimura T, Nishikawa T, Tobe Y, Masaki Y. β-adrenoreceptor antagonists attenuate brain injury after transient focal ischemia in rats. Anesth. Analg. 2006;103:658–663. doi: 10.1213/01.ane.0000228859.95126.69. [DOI] [PubMed] [Google Scholar]
- Hansson MJ, Mansson R, Mattiasson G, Ohlsson J, Karlsson J, Keep MF, Elmer E. Brain-derived respiring mitochondria exhibit homogeneous, complete and cyclosporine-sensitive permeability transition. J. Neurochem. 2004;89:715–729. doi: 10.1111/j.1471-4159.2004.02400.x. [DOI] [PubMed] [Google Scholar]
- He L, Lemasters JJ. Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS Letters. 2002;512:1–7. doi: 10.1016/s0014-5793(01)03314-2. [DOI] [PubMed] [Google Scholar]
- Hoffmann C, Leitz MR, Oberdorf-Maass S, Lohse MJ, Klotz KN. Comparative pharmacology of human β-adrenergic receptor subtypes- characterization of stably transfected receptors in CHO cells. Naunyn-Schm. Arch. Pharmacol. 2004;369:151–159. doi: 10.1007/s00210-003-0860-y. [DOI] [PubMed] [Google Scholar]
- Hopkins-Donaldson S, Bodmer JL, Bourloud KB, Brognara CB, Tschopp J, Gross N. Loss of Caspase-8 expression in highly malignant human neuroblastoma cells correlates with resistance to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Cancer Res. 2000;60:4315–4319. [PubMed] [Google Scholar]
- Jaboureck-Bouttier R, Gressier B, Dine T, Brunet C, Luyckx M, Harfaut P, Ballester L, Cazin M, Cazin JC. Effects of two antihypertensive agents, labetalol and metprolol, on the production of reactive oxygen species by normal polymorphonuclear leukocytes in vitro. Hyperten. Pregnancy. 1999;18:239–247. doi: 10.3109/10641959909016197. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Yune TY, Han CT, Kim YC, Oh YJ, Markelonis GJ, Oh TH. Mitochondrial isocitrate dehydrogenase protects human neuroblastoma SH-SY5Y cells against oxidative stress. J. Neurosci. Res. 2007;85:139–152. doi: 10.1002/jnr.21106. [DOI] [PubMed] [Google Scholar]
- Kowaltowski AJ, Castilho RF, Vercesi AE. Opening of the mitochondrial permeability transition pore by uncoupling or inorganic phosphate in the presence of Ca2+ is dependent on mitochondrial-generated reactive oxygen species. FEBS Letters. 1996;378:150–152. doi: 10.1016/0014-5793(95)01449-7. [DOI] [PubMed] [Google Scholar]
- Lee HT, Kim M, Kim J, Kim N, Emala CW. TGF-beta1 release by volatile anesthetics mediates protection against renal proximal tubule cell necrosis. Amer. J. Nephrol. 2007;27:416–424. doi: 10.1159/000105124. [DOI] [PubMed] [Google Scholar]
- Lopez E, Ferrer I. Staurosporine- and H-7-induced cell death in SH-SY5Y neuroblastoma cells is associated with caspase-2 and caspase-3 activation, but not with activation of the FAS/FAS-L-caspase-8 signaling pathway. Mol. Brain Res. 2000;85:61–67. doi: 10.1016/s0169-328x(00)00235-7. [DOI] [PubMed] [Google Scholar]
- Lopez-Sendon J, Swedberg K, McMurray J, Tamargo J, Maggioni AP, Dargie H, Tendera M, Waagstein F, Kjekshus J, Lechat P, Torp-Pedersen C. Expert consensus document on β-adrenergic receptor blockers. Eur. Heart J. 2004;25:1341–1362. doi: 10.1016/j.ehj.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Lysko PG, Lysko KA, Yue TL, Webb CL, Gu JL, Feuerstein G. Neuroprotective effects of carvedilol, a new antihypertensive agent, in cultured rat cerebellar neurons and in gerbil global brain ischemia. Stroke. 1992;23:1630–1636. doi: 10.1161/01.str.23.11.1630. [DOI] [PubMed] [Google Scholar]
- McGinnis KM, Gnegy ME, Wang KKW. Endogenous Bax translocation in SH-SY5Y human neuroblastoma cells and cerebellar granule neurons undergoing apoptosis. J. Neurochem. 1999;72:1899–1906. doi: 10.1046/j.1471-4159.1999.0721899.x. [DOI] [PubMed] [Google Scholar]
- Merenda A, Bullock R. Clinical treatments for mitochondrial dysfunctions after brain injury. Curr. Opin. Crit. Care. 2006;12:90–96. doi: 10.1097/01.ccx.0000216573.26686.27. [DOI] [PubMed] [Google Scholar]
- Moriya R, Uehara T, Nomura Y. Mechanism of nitric oxide-induced apoptosis in human neuroblastoma SH-SY5Y cells. FEBS Letters. 2000;484:253–260. doi: 10.1016/s0014-5793(00)02167-0. [DOI] [PubMed] [Google Scholar]
- Nicholas AP, Hokfelt T, Pieribone VA. The distribution and significance of CNS adrenoceptors examined with in situ hybridization. Trends Pharmacol. Sci. 1996;17:245–255. doi: 10.1016/0165-6147(96)10022-5. [DOI] [PubMed] [Google Scholar]
- Nishina K, Akamatsu H, Mikawa K, Shiga M, Obara H, Niwa Y. A comparison of atenolol, labetalol, esmolol, and landiolol for altering human neutrophil functions. Anesth. Analg. 2001;93:641–644. doi: 10.1097/00000539-200109000-00021. [DOI] [PubMed] [Google Scholar]
- Polster BM, Basanez G, Young M, Suzuki M, Fiskum G. Inhibition of Bax-induced cytochrome c release from neural cell and brain mitochondria by dibucaine and propranolol. J. Neurosci. 2003;23:2735–2743. doi: 10.1523/JNEUROSCI.23-07-02735.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitz SI, Erhardt JA, Anthony JV, Gupta G, Li X, Barone FC, Rosenbaum DM. The novel β-blocker, carvedilol, provides neuroprotection in transient focal stroke. J. Cereb. Blood Flow Metab. 2000;20:1197–1204. doi: 10.1097/00004647-200008000-00005. [DOI] [PubMed] [Google Scholar]
- Scarlett JL, Sheard PW, Hughes G, Ledgerwood EC, Ku HH, Murphy MP. Changes in mitochondrial membrane potential during staurosporine-induced apoptosis in Jurkat cells. FEBS Letters. 2000;475:267–272. doi: 10.1016/s0014-5793(00)01681-1. [DOI] [PubMed] [Google Scholar]
- Stepczynska A, Lauber K, Engels IH, Janssen O, Kabelitz D, Wesselborg S, Schulze-Osthoff K. Staurosporine and conventional anticancer drugs induce overlapping, yet distinct pathways of apoptosis and caspase activation. Oncogene. 2001;20:1193–1202. doi: 10.1038/sj.onc.1204221. [DOI] [PubMed] [Google Scholar]
- Tafani M, Cohn JA, Karpinich NO, Rothman RJ, Russo MA, Farber JL. Regulation of intracellular pH mediates Bax activation in Hela cells treated with staurosporine or tumor necrosis factor-α. J. Biol. Chem. 2002;277:49569–49576. doi: 10.1074/jbc.M208915200. [DOI] [PubMed] [Google Scholar]
- Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, Ghayur T, Brady KD, Wong WW. Substrate specificities of caspase family proteases. J. Biol. Chem. 1997;272:9677–9682. doi: 10.1074/jbc.272.15.9677. [DOI] [PubMed] [Google Scholar]
- Tang LL, Wang R, Tang XC. Huperzine A protects SHSY5Y neuroblastoma cells against oxidative stress damage via nerve growth factor production. Eur. J. Pharmacol. 2005;519:9–15. doi: 10.1016/j.ejphar.2005.06.026. [DOI] [PubMed] [Google Scholar]
- Tieu K, Zuo DM, Yu PH. Differential effects of staurosporine and retinoic acid on the vulnerability of the SH-SY5Y neuroblastoma cells: Involvement of Bcl-2 and p53 proteins. J. Neurosci. Res. 1999;58:426–435. [PubMed] [Google Scholar]
- Verma A. Opportunities for neuroprotection in traumatic brain injury. J. Head Trauma Rehab. 2000;15:1149–1161. doi: 10.1097/00001199-200010000-00008. [DOI] [PubMed] [Google Scholar]
- Wigdal SS, Kirkland RA, Franklin JL, Haak-Frendscho M. Cytochrome c release precedes mitochondrial membrane potential loss in cerebellar granule neuron apoptosis: lack of mitochondrial swelling. J. Neurochem. 2002;82:1029–1038. doi: 10.1046/j.1471-4159.2002.01049.x. [DOI] [PubMed] [Google Scholar]
- Yue TL, Lysko PG, Barone FC, Gu JL, Ruffolo RR, Jr, Feuerstein GZ. Carvedilol, a new antihypertensive drug with unique antioxidant activity: potential role in cerebroprotection. Ann. NY Acad. Sci. 1994;738:230–242. doi: 10.1111/j.1749-6632.1994.tb21808.x. [DOI] [PubMed] [Google Scholar]
- Zaugg M, Xu W, Lucchinetti E, Shafiq SA, Jamali NZ, Siddiqui MAQ. β-adrenergic receptor subtypes differentially affect apoptosis in adult rat ventricular myocytes. Circulation. 2000;102:344–350. doi: 10.1161/01.cir.102.3.344. [DOI] [PubMed] [Google Scholar]
- Zhang XD, Gillespie SK, Hersey P. Staurosporine induces apoptosis of melanoma by both caspase-dependent and –independent apoptotic pathways. Mol. Cancer Ther. 2004;3:187–197. [PubMed] [Google Scholar]
- Zoratti M, Szabo I, De Marchi U. Mitochondrial permeability transitions: how many doors to the house? Biochim. Biophys. Acta. 2005;1706:40–52. doi: 10.1016/j.bbabio.2004.10.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. Cells were treated with different concentrations of staurosporine and caspase 3-like activity assayed at 5 hours after administration of the drug (EC50 = 1.02 µM, n=1.51) or at different time points after 10 µM staurosporine (B). Mean ± S.E.M. results from triplicate experiments. FU=fluorescence unit.
A. Cells were treated with 10 µM staurosporine for the indicated times and analyzed by flow cytometer for early apoptosis (annexin V-positive but propidium iodide (PI)-negative) and late apoptosis (annexin V-positive and PI-positive). B. A western blot of cell lysates obtained from cells treated with DMSO or staurosporine (10 µM) for the indicated times. The 85kD band represents the cleaved PARP. Equal loading of the lanes was confirmed by immunoblotting with the anti-actin antibody. Representative of three independent experiments.