

1. Isolation and Structure Determination
In three papers that appeared in the early 1990’s, Blackman and coworkers described the isolation of a family of eleven structurally related tricyclic alkaloids produced by the marine ascidian (sea squirt) Clavelina cylindrica, collected off the east coast of Tasmania.1 The most abundant of these alkaloids are cylindricines A (1) and B (7), whose structures were established by spectral analysis, along with X-ray crystallography of the corresponding picrates (Figure 1). The former compound has a pyrrolo[2,1-j]quinoline tricyclic framework, whereas the latter possesses a C-ring-expanded pyrido[2,1-j]quinoline system. Interestingly, after standing for six days in solution, these compounds produce the same 3:2 equilibrium mixture of 1 and 7. Since this equilibration occurs only with the free bases of the alkaloids (the corresponding picrates are stable), it was presumed that the interconversion probably occurs via the aziridinium intermediate 6.1a
FIGURE 1.
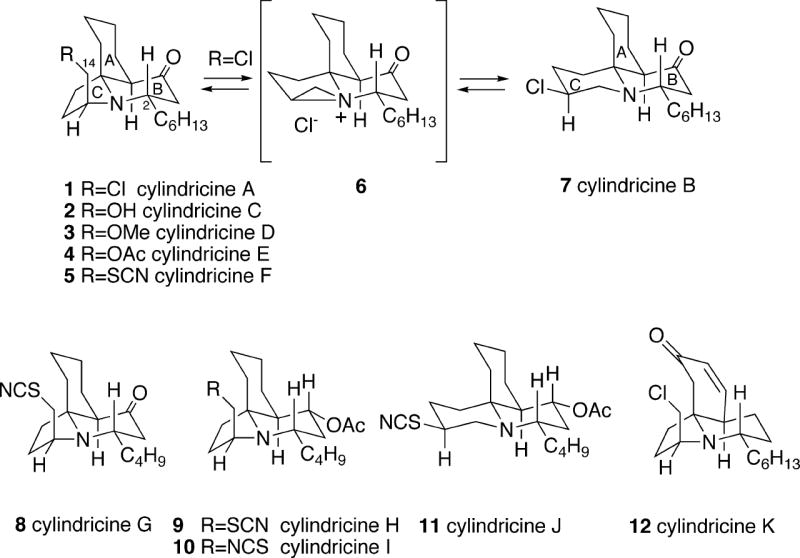
Subsequent investigations by the Blackman group led to the isolation of some additional minor compounds possessing the cylindricine A pyrroloquinoline framework and stereochemistry, but differing only in the functionality at C(14). The structures of these metabolites were secured mainly by NMR analysis, as well as by a few simple chemical interconversions. Examples of these alkaloids include cylindricines C (2), D (3), E (4) and F (5). In addition, a few cylindricine A-type alkaloids were found possessing a butyl chain at C(2) rather than the hexyl group (e.g. cylindricines G (8), H (9) and I (10)). Similarly, a second compound exists in the cylindricine B pyridoquinoline series, but has a C(2) butyl appendage (i.e. cylindricine J (11)). Cylindricine K (12) is related to cylindricine A, and is the only alkaloid isolated to date with functionality in the A-ring. All of the cylindricine alkaloids possess a cis-fused 1-azadecalin A/B-ring system and prefer to exist in the conformations shown in Figure 1, as evidenced by X-ray crystal structure and NMR data, as well as molecular mechanics calculations.1c
The absolute configurations of these alkaloids were not established during the structural studies. However, recent enantioselective syntheses of lepadiformine have proven its absolute configuration to be antipodal to those drawn in 1-11 (see sections 5.2.4 and 5.2.5), and this alkaloid is at present the only member of the family for which absolute stereochemistry is definitively known. One might reasonably assume that all the other alkaloids of this family have the same absolute stereochemistry as lepadiformine, but this supposition has not yet been proven. Based upon this assumption and the synthetic work outlined below, structures 1-11 arbitrarily used by Blackman in fact would have the unnatural configuration and correspond to the (-)-cylindricines. The (+)-alkaloids in the natural series would be enantiomeric to the above structures. Unfortunately, optical rotations of the natural cylindricine alkaloids were not taken during the structure work and thus direct comparison with enantiomerically pure synthetic compounds is not possible. Until reisolation of these metabolites, the question of their absolute configurations will remain unanswered. For convenience and consistency throughout this review, the enantiomeric series shown by Blackman in the original papers is used for discussions of the racemic syntheses, and the actual antipode which was prepared is shown in the enantioselective syntheses.
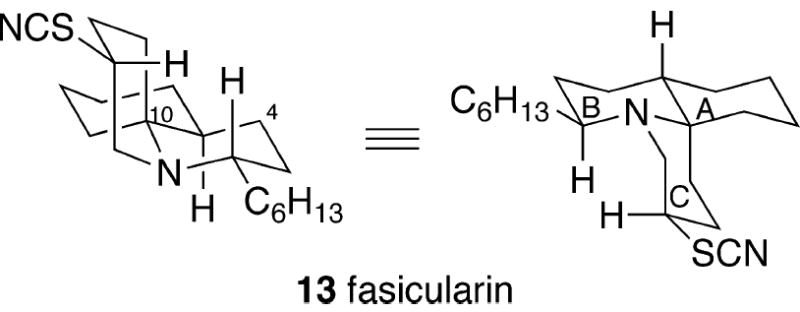
In 1997, the tricyclic alkaloid fasicularin (13) was isolated by a SmithKline Beecham group from the ascidian Neptheis fasicularis collected in Pohnpei, Micronesia (Figure 2).2 NMR and NOE experiments led to the assignment of the structure, relative stereochemistry and conformation of fasicularin as depicted in 13. This alkaloid is closely related to the cylindricine B series of pyridoquinolines, but is epimeric at the C(10) quaternary center, thereby rendering a trans-1-azadecalin A/B ring fusion. Moreover, this compound lacks the C(4) oxygenation found in all but one of the cylindricine alkaloids. As is the case in the cylindricines, the optical rotation of natural fasicularin was not measured, and thus it has not been possible to compare enantiomerically pure synthetic material with the natural alkaloid to establish its absolute configuration.
FIGURE 2.
In 1994, Biard and coworkers described the isolation of a new marine alkaloid, lepadiformine,3 obtained by HCl extraction of the methylene chloride-soluble portion of the marine tunicate Clavelina lepadiformis (Muller) collected in the Mediterranean off the coast of Tunisia,3a and later isolated from Clavelina moluccensis (Sluiter) obtained near Djibouti.3b Based on proton and carbon NMR analysis, it was suggested that lepadiformine has tricyclic structure 14 which contains an unprecedented zwitterionic vicinal amino alcohol moiety (Figure 3). It should be noted that the A/B-ring system in putative structure 14 is a cis-1-azadecalin, thus putting it in the cylindricine class. In addition, the NMR NOE experiments on lepadiformine were interpreted to suggest that this alkaloid had the conformation shown in 14a. However, based upon synthetic work outlined below, it was found that structure 14 is incorrect, nor does this molecule exist as a zwitterion.4 Moreover, it was also determined by synthesis that lepadiformine is not a C(2) or C(13) epimer of structure 14.5 The constitution and relative configuration of lepadiformine was finally established as shown in 15a by total synthesis (vide infra).6,7 Lepadiformine, therefore, is in the fasicularin trans-1-azadecalin series, but has a pyrroloquinoline rather than a pyridoquinoline ring system. Moreover, it was found by X-ray crystallography of synthetic material that the alkaloid exists with the B-ring in a boat conformation, thereby avoiding having the hexyl chain in an axial position. As noted above, two subsequent enantioselective total syntheses of lepadiformine determined that the molecule has the absolute configuration shown in 15a.
FIGURE 3.
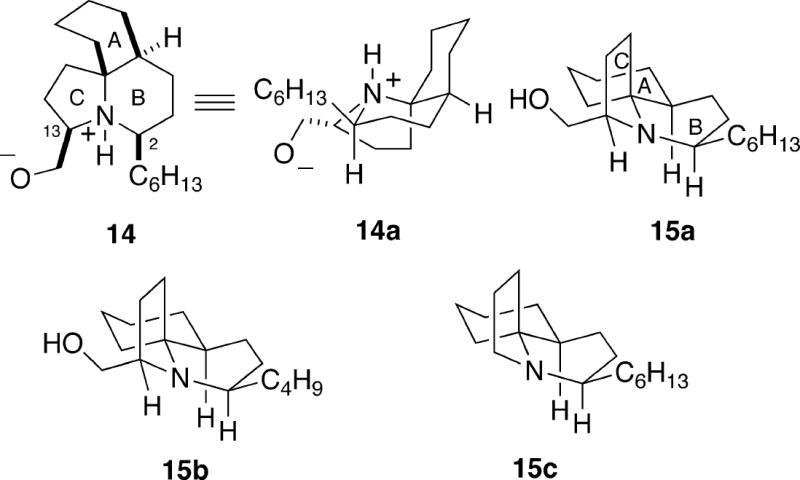
Very recently, two additional alkaloids closely related to lepadiformine were isolated from C. moluccensis.3c These componds include lepadiformine B (15b) which bears a butyl group rather than hexyl at C(2), and lepadiformine C (15c), which has lost the hydroxymethyl group at C(13). With these new compounds being isolated, it was proposed that the name of lepadiformine (15a) be changed to lepadiformine A. However, since all of the synthetic work described here was done before this latest paper appeared, the name lepadiformine is used throughout to refer to 15a.
2. Biological Activity
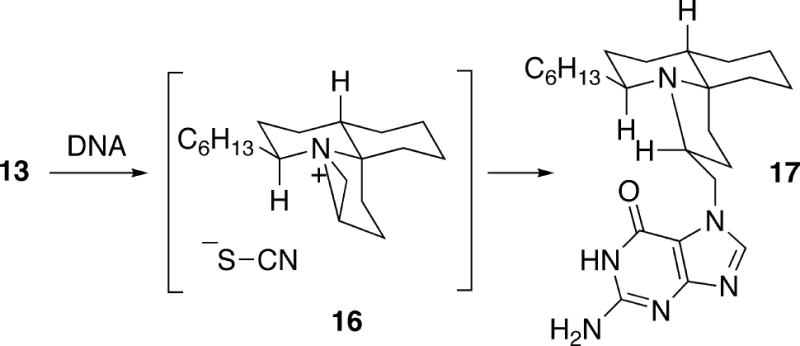
No significant biological activity has been reported for any of the cylindricines other than toxicity in a brine shrimp assay. However, fasicularin (13) was found to have biological activity against a DNA repair-deficient strain of yeast, as well as cytotoxicity against Vero cells.2 Recently, Gates et al. investigated the ability of fasicularin to damage DNA by acting as an alkylating agent.8 In this experiment, mixed-sequence duplex DNA was treated with racemic synthetic fasicularin, and after thermal treatment the guanine alkylation product 17 could be detected (Scheme 1). The assumption was that fasicularin is first converted to the aziridinium ion 16, analogous to that proposed for interconversion of cylindricines A and B (Cf. 6, Figure 1; see also section 4.2). Subsequent attack on intermediate 16 by N(7) of the guanine base led to the observed alkylation product 17. Although the regiochemistry of the opening of aziridinium ion 16 was not actually proven, it was assumed that nucleophilic attack occured at the primary carbon as shown in the scheme to give 17 based on results in the cylindricine A and B series.1b It was also found in control experiments that the thiocyanate anion formed in the process does not cause DNA strand cleavage.
Scheme 1.
Lepadiformine (15a) has been reported to have moderate in vitro cytotoxicity against nasopharynx carcinoma (KB) and non-small-cell lung carcinoma (NSCLC-N6).3a In addition, the alkaloid has been investigated in vivo for its cardiovascular effects. The alkaloid caused a variety of effects when tested in rats, including induction of bradycardia, prolongation of ECG parameters, and led to a transient fall of blood pressure.3b It was also suggested that the alkaloid may have antiarrythmic properties. Recently, it was reported that alkaloids 15a-c block cardiac muscle Kir channel.3c
3. Synthetic Approaches to the Cylindricines
3.1 Snider Synthesis of (±)-Cylindricines A, D and E
The first total synthesis of a cylindricine alkaloid was reported by Snider and Liu in 1997.9 In this approach, key steps included a double Michael reaction of ammonia with a dienone to form the fused A/B-ring system, and a copper catalyzed N-chloroamine/olefin radical cyclization to construct the C-ring. The synthesis commenced with ketone 18, which underwent addition of 3-butenylmagnesium bromide followed by acid hydrolysis to produce enal 19 (Scheme 2). Addition of 1-octynyllithium to aldehyde 19, reduction of the resulting propargylic alcohol and allylic alcohol oxidation led to the requisite dienone substrate 20. It was found that heating this dienone with ammonium hydroxide in methanol under various pH conditions led to only three of the four possible diastereomeric Michael addition products. At high pH, 56% of the desired cylindricine stereoisomer 22 was formed along with 19% of 21 and 6% of 23. However, at lower pH’s, controlled by addition of ammonium chloride to the reaction mixture, the amount of the undesired trans-fused 1-azadecalin system 21 increased at the expense of the requisite cylindricine isomer 22. Based upon some equilibration studies along with molecular mechanics calculations, it was postulated that the ratio of the three products 21-23 is determined by the second kinetically controlled intramolecular Michael step. However, no speculation about the actual details of this process was offered.
Scheme 2.
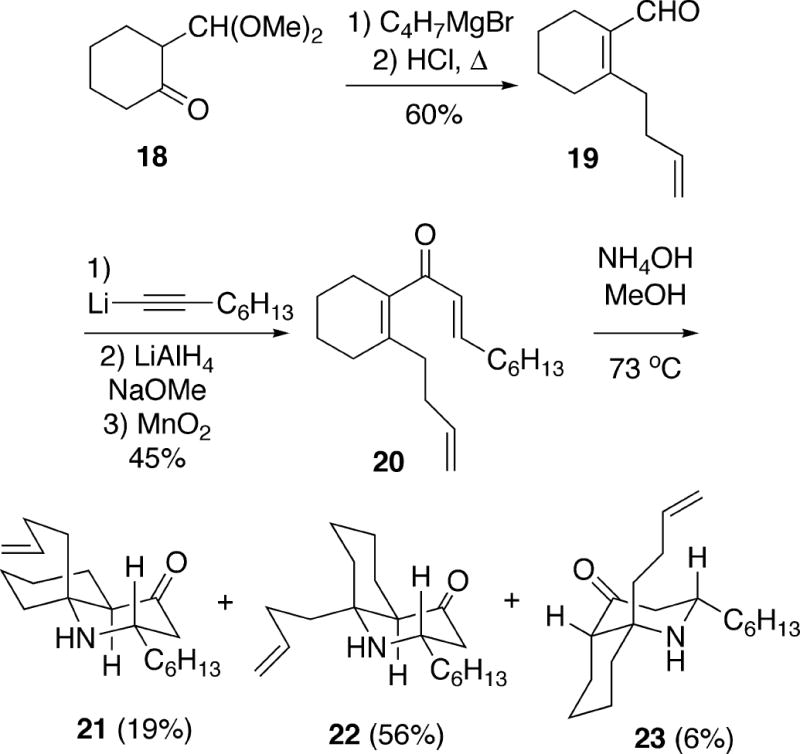
To complete the synthesis, bicyclic amine 22 was first treated with NCS, which provided the N-chloroamine 24 in high yield (Scheme 3). Exposure of this compound to the Stella conditions for generating aminyl radicals10 induced a 5-exo cyclization which was non-stereoselective, affording a 1:1 mixture of racemic cylindricine A (1) and the undesired C(12) epimer 25. It was possible, however, to recycle unwanted chloride 25 back to amino olefin 22 by a zinc/HCl reduction. Using procedures previously developed by Blackman during structural studies,1b it was also possible to convert synthetic (±)-cylindricine A (1) to (±)-cylindricine D (3) by treatment with sodium methoxide in methanol, and to (±)-cylindricine E (4) with sodium acetate in methanol.
Scheme 3.
3.2 Heathcock Synthesis of (±)-Cylindricines A and B
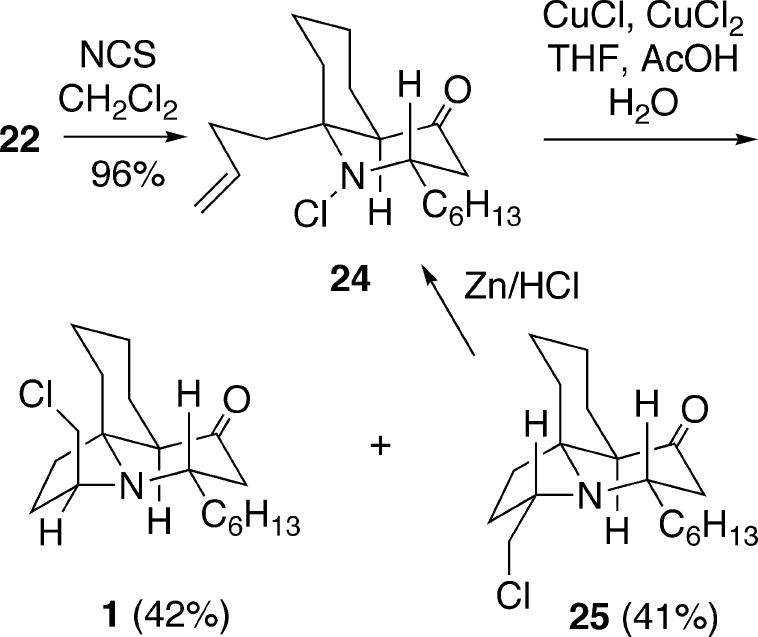
The Liu/Heathcock approach to the cylindricines is very closely related to the strategy devised by Snider.11 In fact, their first generation approach was identical to that shown above in Scheme 2, in that it involved the double Michael addition of ammonia to dienone 20 to form a mixture of 1-azadecalins 21-23 in the same proportions as observed by Snider and Liu. Since the stereoselectivity in this process is low, a more efficient second generation strategy was therefore developed. Enol triflate 26 was first coupled with the second order cuprate derived from 3-butenyllithium to generate ester 27, which was then converted into β-ketophosphonate 28 (Scheme 4). This intermediate was next deprotonated with NaH and condensed with formaldehyde to form dienone 29. Heating this compound with ammonia/ammonium hydroxide in ethanol resulted in a double Michael reaction to afford the desired 1-azadecalin 30 in high yield as a 1:1 mixture of cis and trans isomers. This mixture was N-acylated to generate the Teoc-protected system 31, which was first converted to the corresponding TIPS enol ether and then oxidized with ceric ammonium nitrate to the vinylogous amide 32.
Scheme 4.
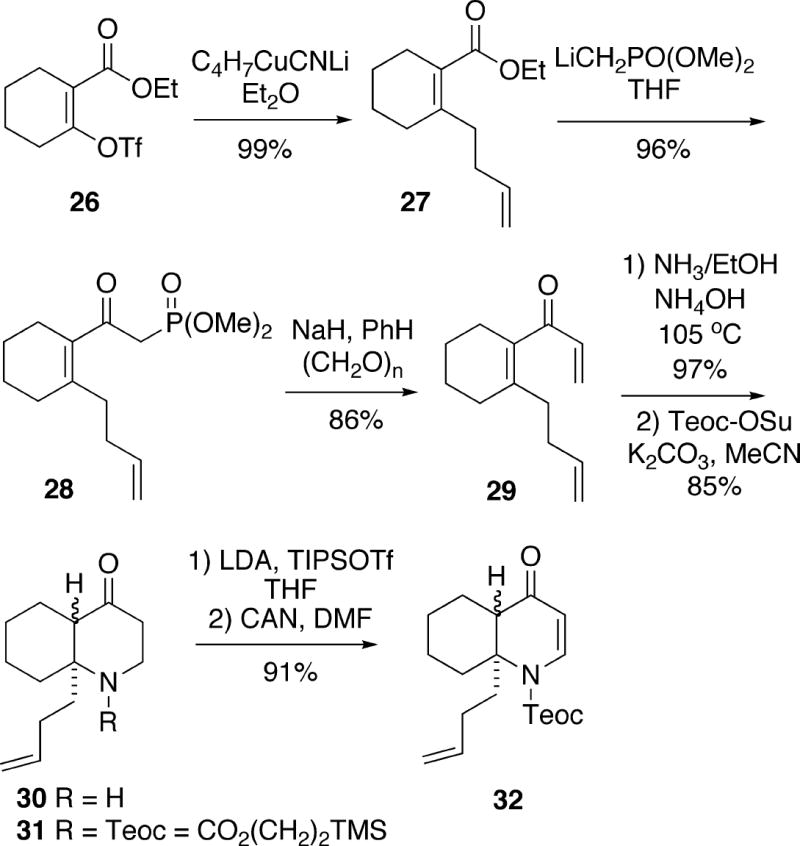
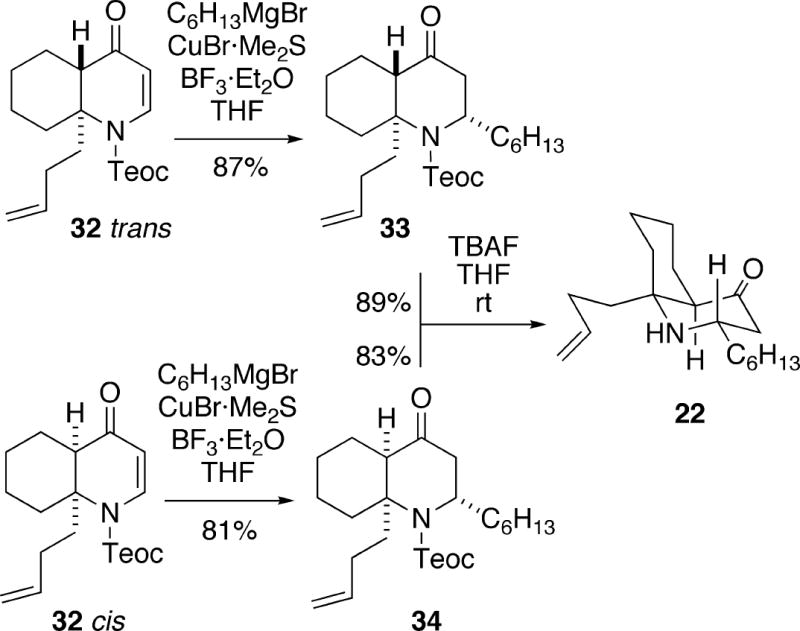
At this point, the cis and trans isomers could be separated by HPLC, and each was individually alkylated using the methodology of Comins.12 Thus, conjugate addition of the cuprate derived from hexylmagnesium bromide to trans-1-azadecalin 32 was highly stereoselective and afforded adduct 33 via axial attack of the organometallic reagent (Scheme 5). Similarly, the addition to cis isomer 32 gave adduct 34, once again via axial attack. Removal of the Teoc group from both trans-fused isomer 33 and cis-fused isomer 34 with tetrabutylammonium fluoride led to the more stable cis-1-azadecalin 22. Conversion of amino olefin 22 to a mixture of (±)-cylindricine A (1) and its C(12) epimer 25 was effected as previously done by Snider (Cf. Scheme 3). Synthetic cylindricine A, when dissolved in C6D6, underwent equilibration to a mixture of cylindricines A and B.1
Scheme 5.
3.3 Molander Synthesis of (-)-Cylindricine C
In 1999, Molander and Ronn described an enantioselective total synthesis of (-)-cylindricine C (2) which featured an intramolecular double Michael addition of an amine to a dienone.13 Starting with the known14 (S)-1,2,4-butanetriol-derived tosylate 35 as the source of absolute chirality, the lithium anion of the N,N-dimethylhydrazone derivative of cyclohexanone could be alkylated to form ketone 36 (Scheme 6). This ketone was then transformed as shown in three steps to dienone 37. Hydrolysis of the ketal moiety in 37 proved difficult, but it was found that a palladium-mediated cleavage led to the desired diol, which was processed in three steps to afford azide 38.
Scheme 6.
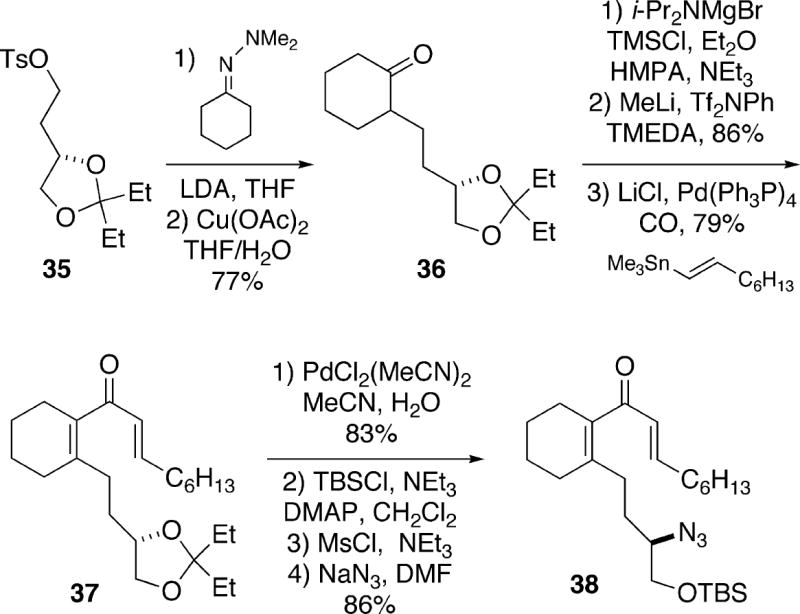
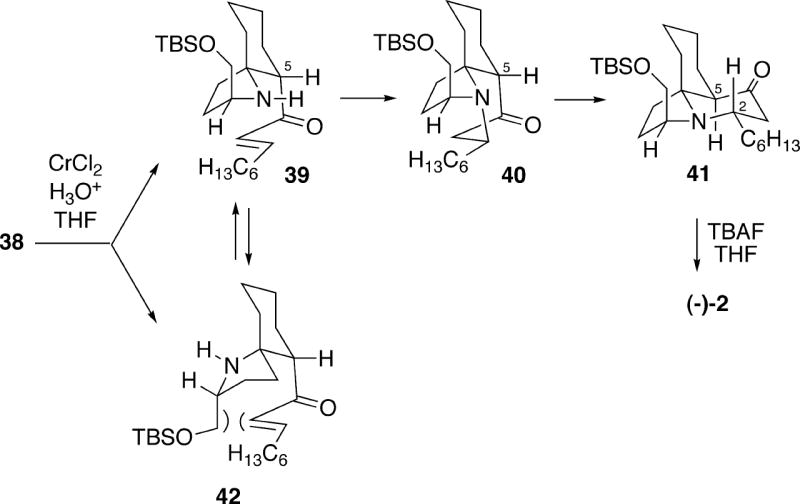
Treatment of azide 38 with chromium(II) chloride under acidic conditions, followed by desilylation of the crude product with tetrabutylammonium fluoride, led to (-)-cylindricine C (2) in 37-55% yields. The course of this complex cyclization sequence can best be rationalized as shown in Scheme 7. Initial Michael reaction of the amine derived from reduction of azide 38 can lead to diastereomeric spirocycles 39 and 42. For stereoelectronic reasons, conjugate addition of the amino group in enone 39 occurs to form tricycle 40 in which the B-ring is a boat, but which has the requisite configuration at C(2) for the cylindricines. Such a ring system is actually relatively stable, as is known to be the case in lepadiformine 15 (vide supra). Subsequent epimerization of ketone 40 at C(5) then produces the thermodynamically more stable cylindricine C system 41 (for a further discussion of the relative stabilities of these ring systems see section 3.5). It is highly unlikely that enone 39 epimerizes at C(5) prior to cyclization since Weinreb and coworkers have shown that this isomeric compound in fact undergoes conjugate addition to produce the C(2) epimer of tricycle 41 (see Scheme 30 below). In the case of diastereomeric spirocycle 42, the second Michael addition may be precluded for steric reasons due to an unfavorable interaction between the enone and the siloxymethyl group. This intermediate could in principle be reversibly converted to spirocycle 39, but under these particular reaction conditions may simply decompose, explaining the moderate yields of tricyclic product 2.
Scheme 7.
Scheme 30.
3.4 Trost Synthesis of (+)-Cylindricines C, D and E
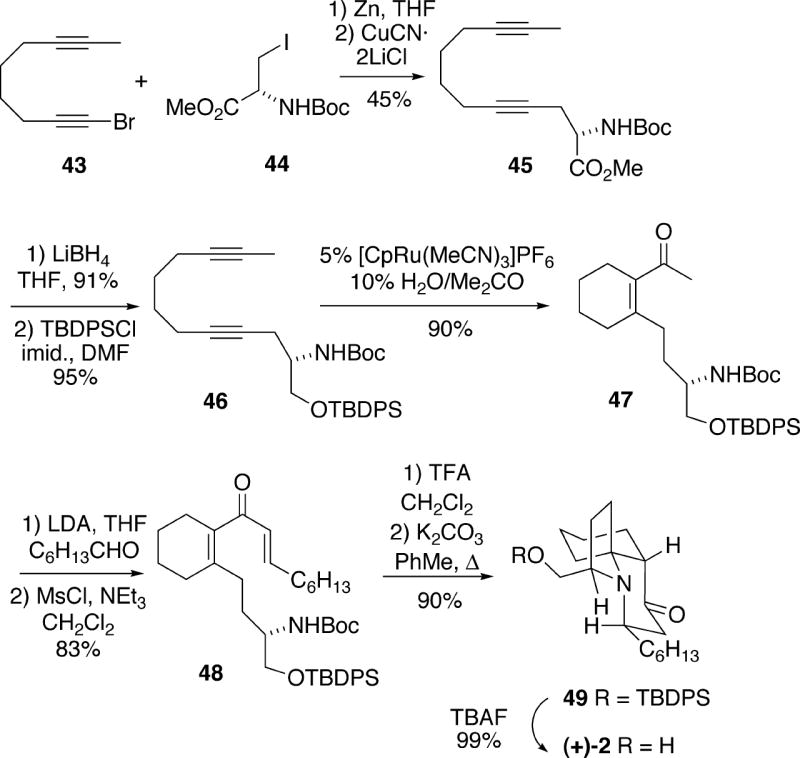
The strategy in the cylindricine synthesis reported by Trost and Rudd involved an intramolecular double Michael cyclization of an amino dienone which was virtually identical to that of Molander, but in the antipodal series (Cf. Scheme 7).15 The Trost approach made use of an efficient ruthenium-catalyzed diyne cyclization to generate the dienone substrate.
Using methodology of Knochel,16 readily prepared bromo diyne 43 was first coupled with enantiomerically pure iodide 44 derived from (S)-serine to produce 45 (Scheme 8). The ester functionality in 45 was then reduced with lithium borohydride, and the resulting alcohol was protected as the TBDPS ether 46. Using methodology previously developed in the Trost lab,17 this diyne was found to undergo chemoselective ruthenium-catalyzed hydrative cyclization to afford the desired enone 47 in high yield. This product could next be condensed with heptanal in an aldol reaction and dehydration to yield the requisite dienone 48. Removal of the Boc protecting group of dienone 48 with TFA, followed by heating the resulting amine salt with potassium carbonate in toluene resulted in a 90% yield of tricyclic product 49. Subsequent cleavage of the silyl protecting group of this material afforded (+)-cylindricine C (2). Of interest is the fact that this cyclization resulted in a significantly higher yield of the desired tricycle 49 than was the case in the Molander synthesis. One possibility is that the reaction conditions used in the cyclization step in the Trost work allowed reversible equilibration of the undesired spirocycle 42 with 39 (Cf. Scheme 7). Using straightforward chemistry, (+)-cylindricine C could be converted to cylindricines D (3) and E (4).
Scheme 8.
3.5 Kibayashi Syntheses of (+)-Cylindricine C
The Kibayashi group has reported two different enantioselective total syntheses of (+)-cylindricine C. The first synthesis, which was published in 2004, began with lactam 50, derived from (S)-pyroglutamic acid, which underwent Grignard addition to produce ketone 51 (Scheme 9).18 Removal of the Boc protecting group of 51 with TFA caused cyclization of the resulting ketoamine to the imine, and also resulted in loss of the PMB group, affording the corresponding alcohol. Reprotection of the alcohol functionality as the TBDPS ether then produced ketimine 52. Based upon the results of some preliminary model studies, it was found that this imine undergoes a highly stereoselective addition of allylmagnesium bromide in the presence of boron trifluoride etherate to afford olefin 53. This product was processed via the simple four step sequence shown in the scheme to yield aldehyde tosylate 54.
Scheme 9.
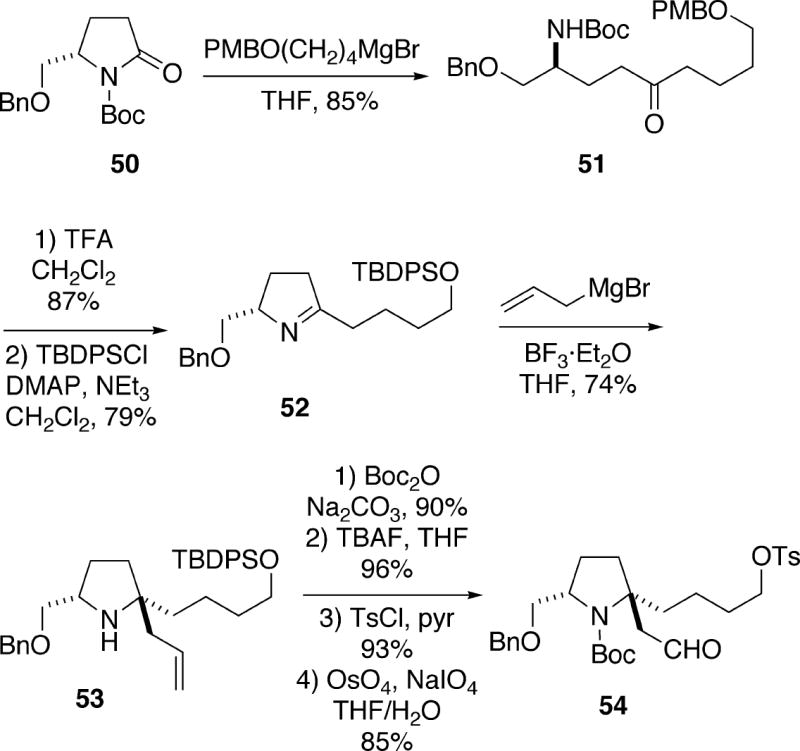
A number of attempts were made to cyclize the enolate derived from aldehyde 54, as well as the corresponding iodide, to produce spirocycle 56. However, substrate 54 is prone to undergo retro-Michael reaction under strongly basic conditions. Alternatively, it was discovered that treatment of aldehyde 54 with pyrrolidine leads to enamine 55 which successfully cyclized (Scheme 10). Hydrolysis of the resulting enamine cyclization product with aqueous acetic acid provided the desired spirocyclic aldehyde 56 in moderate yield as a single stereoisomer. To complete the synthesis, aldehyde 56 was first converted to ynone 57 in two steps, and this compound was then hydrogenated with Lindlar catalyst to produce the Z-enone. Removal of the Boc protecting group with TFA, followed by neutralization of the amine salt with aqueous sodium carbonate afforded cylindricine tricycle 59. This cyclization may in fact involve initial isomerization of the (Z)- to the (E)-enone, as well as formation of the trans- 1-azadecalin 58. Epimerization of 58 at C(5) provided the observed cis-1-azadecalin product 59, which upon reductive debenzylation yielded (+)-cylindricine C (2).
Scheme 10.
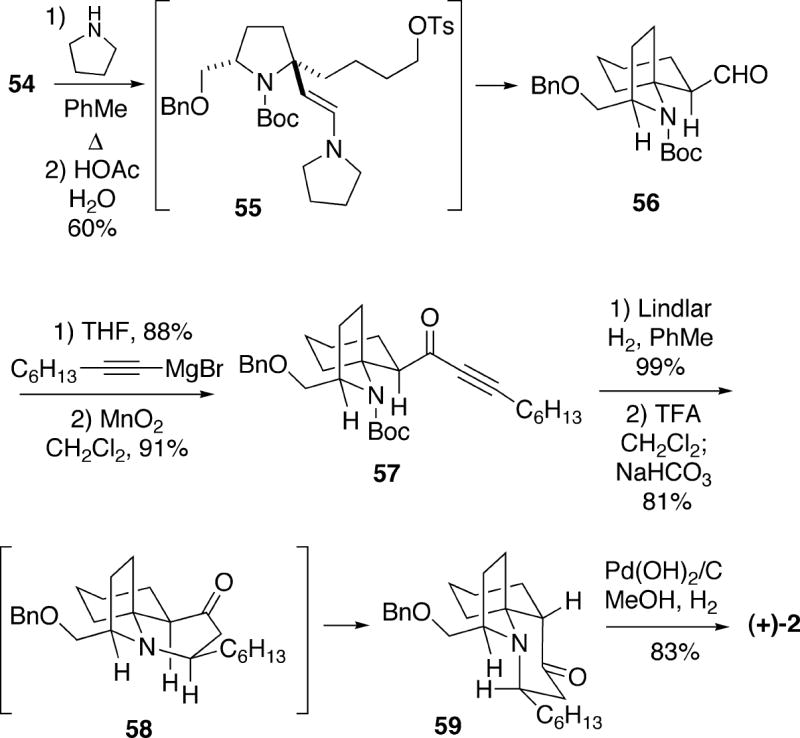
The second Kibayashi synthesis of (+)-cylindricine C involved the convergent preparation of a key advanced intermediate 63 which was also used for both (-)-fasicularin and (-)-lepadiformine (see sections 4.2 and 5..3.5 ).19 This approach also started with enantiomerically pure (S)-lactam 50, which was coupled with Grignard reagent 60 to form ketone 61 (Scheme 11). Exposure of 61 to formic acid presumably first generated N-acyliminium ion 62, which cyclized stereoselectively via a chair-like transition state to afford spirocycle 63 in excellent yield. Compound 63 was approximately a 1.6:1 mixture of stereoisomers favoring the α-formate. The mixture of esters 63 was hydrolyzed to a mixture of allylic alcohols, which was oxidized to enone 64. It was found that this ketone could be reduced stereoselectively with (S)-BINALH in high yield with a 97% diastereomeric excess in favor of the β-allylic alcohol 65.
Scheme 11.
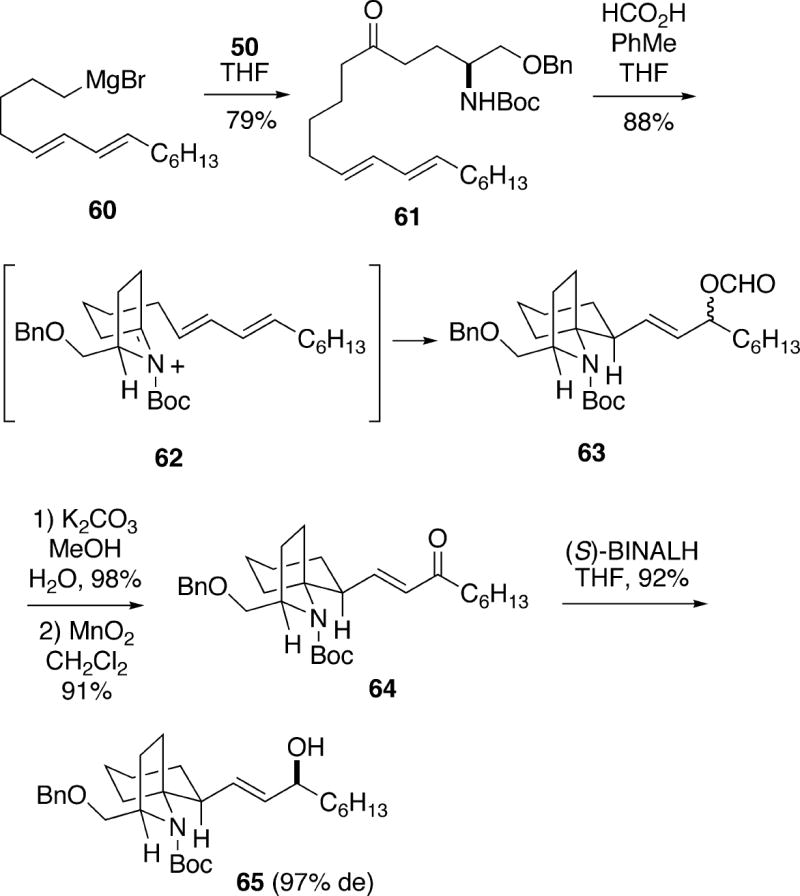
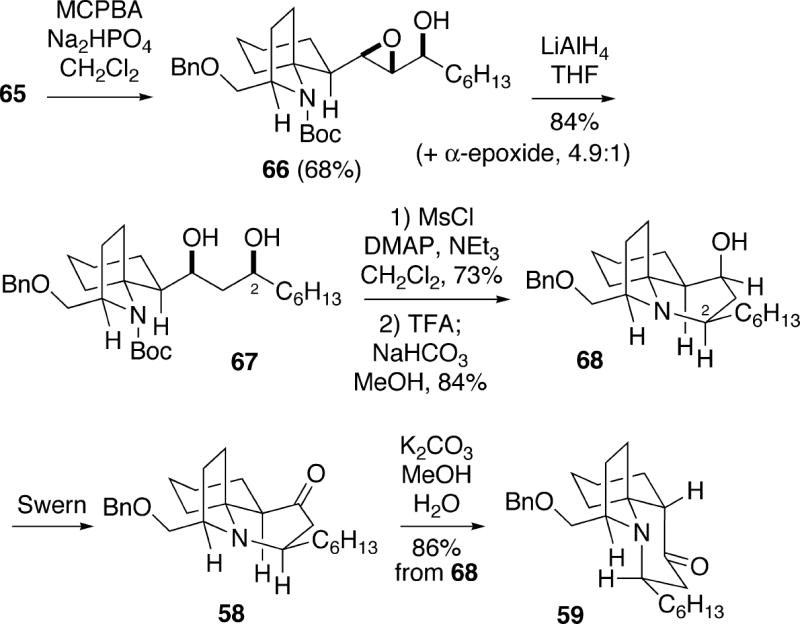
Hydroxyl-directed epoxidation of allylic alcohol 65 with m-chloroperbenzoic acid was moderately stereoselective, producing a 4.9:1 mixture of the β-epoxide 66 along with the α-isomer (Scheme 12). The desired major β-epoxide 66, which could be isolated in 68% yield, was reduced to 1,3-diol 67 with lithium aluminum hydride. It was possible to selectively form the mesylate of the less hindered C(2) alcohol group of 67, and following Boc removal, cyclization occurred with inversion to give tricyclic alcohol 68. Swern oxidation of this alcohol subsequently led to ketone 58. Molecular mechanics calculation on 58 indicate that the B-ring boat system in the trans-1-azadecalin is the lowest energy conformation. Moreover, it was calculated that the cis-1-azadecalin 59 is more stable than 58 by 5.5 kcal/mol. In fact, when ketone 58 was exposed to aqueous potassium carbonate, complete epimerization to 59 occurred. Cleavage of the benzyl ether protecting group of 59 as shown in Scheme 10 afforded (+)-cylindricine C (2).
Scheme 12.
3.6 Hsung Synthesis of (+)-Cylindricine C
In 2004, Hsung and coworkers reported an approach to (+)-cylindricine C involving a pivotal N- acyliminium ion/diene cyclization which is closely related to that of Kibayashi outlined in Scheme 11.20 In the successful route, iodide 69 was metallated and added to enantiomerically pure (S)-lactam 70 to form ketone 71 in moderate yield (Scheme 13). Formic acid induced cyclization of this compound proceeded as in the Kibayashi case (Cf. 61 to 63)19 to produce a mixture of spirocyclic formates which was hydrolyzed to a stereoisomeric mixture (ratio unspecified) of allylic alochols 72. This alcohol could then be oxidized to enone 73. All attempts, however, to epimerize this enone at C(5) with base to produce the cylindricine stereochemistry were unsuccessful. It was suggested that the difficulty here is the inability to properly align the C(5) hydrogen with the enone due to severe A1,3-strain in the conformations required for deprotonation.
Scheme 13.
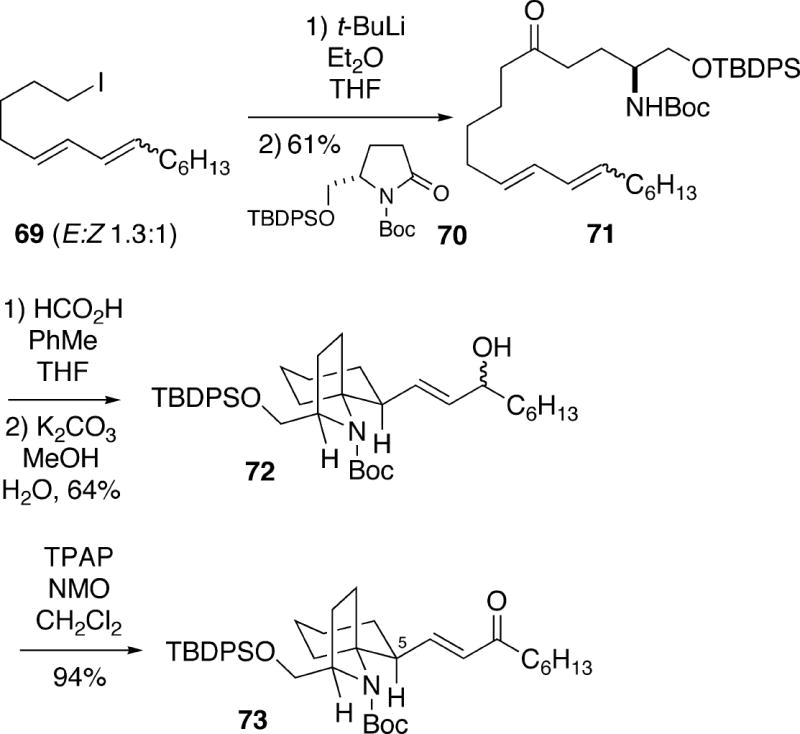
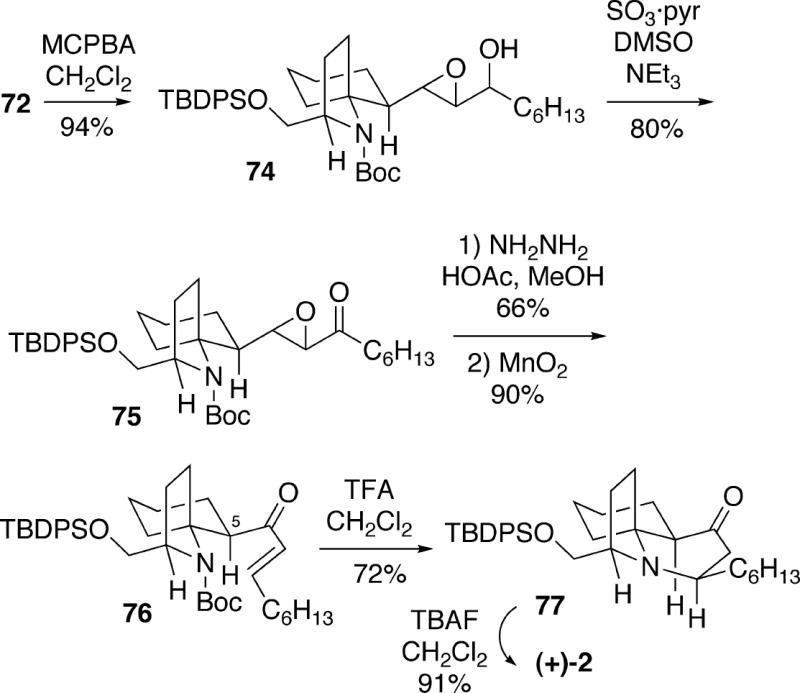
Alternatively, the mixture of alcohols 72 was epoxidized to produce 74, which was then oxidized to epoxyketone 75 (Scheme 14). This intermediate was subjected to a Wharton rearrangement21 to yield the transposed allylic alcohol, followed by oxidation to enone 76. Once again, some unsuccessful attempts were made to epimerize this enone at C(5) with various bases. However, removal of the Boc protecting group of 76 resulted in Michael cyclization of the amino enone as observed in other previously described syntheses (vide supra) to afford trans-1-azadecalin 77. It was found that upon removal of the silyl group of 77 with TBAF, C(5) epimerization also occurs leading to (+)-cylindricine C (2).
Scheme 14.
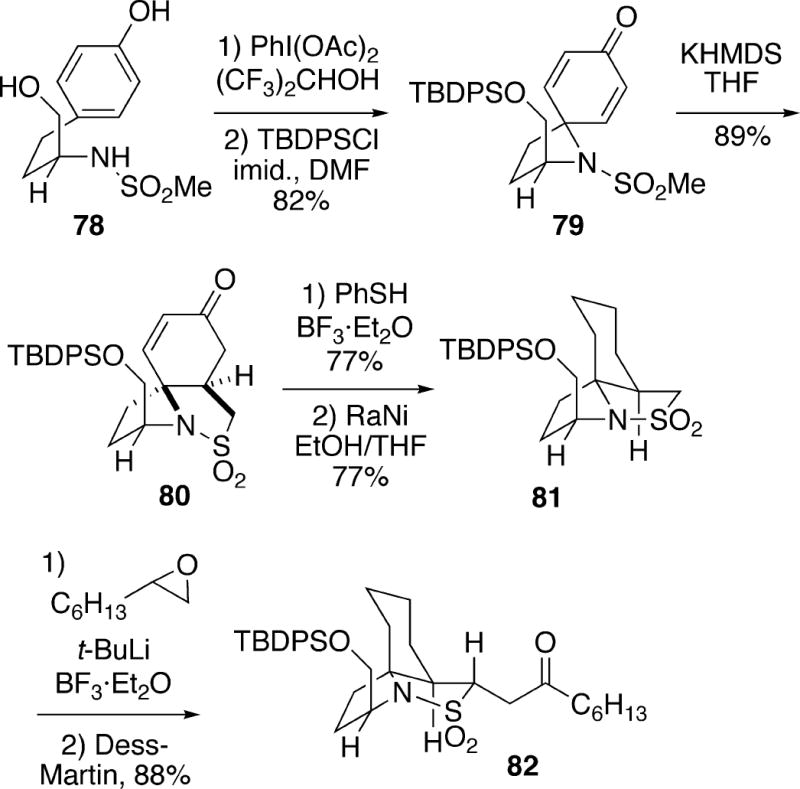
3.7 Ciufolini Synthesis of (-)-Cylindricine C and (-)-2-Epicylindricine C
The Ciufolini group has recently reported a unique synthetic approach to (-)-cylindricine C and its unnatural C(2)-epimer.22 The route began with (R)-homotyrosine derivative 78, which was oxidized with iodosobenzene diacetate, followed by alcohol protection, to afford the dienone 79 (Scheme 15). Treatment of 79 with KHMDS resulted in conjugate addition of the derived sulfonamide anion to produce adduct 80 with 7:1 diastereoselectivity. The major isomer 80 was then reduced in two steps to afford 81. The sulfonamide moiety in 81 was deprotonated with t-butyllithium, and the resulting anion was alkylated with 1-octene oxide leading to an alcohol which was subsequently oxidized to ketone 82.
Scheme 15.
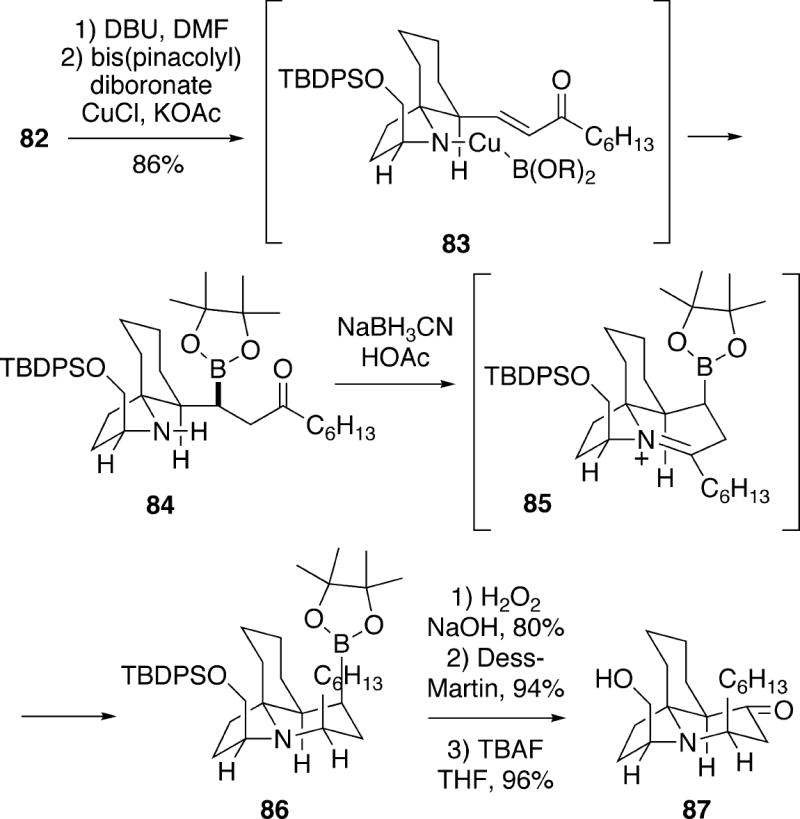
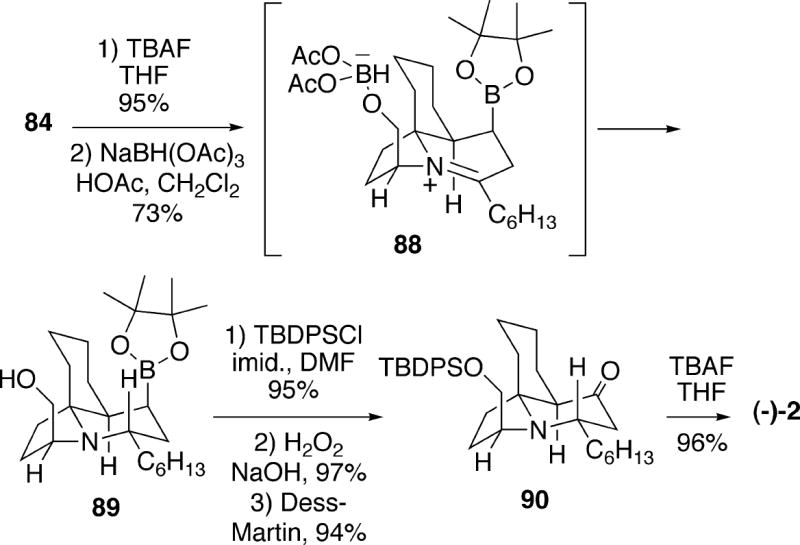
Continuing the synthesis, ketone 82 was exposed to DBU which effected elimination to an isolable α,β-unsaturated ketone, and subjection of this compound to the Miyaura borylation conditions23 afforded boronic ester 84 as a single stereoisomer (Scheme 16). The high stereoselectivity and the rapid rate observed for this reaction suggests that boron delivery to the Re-face of the enone occurs internally via a complex like 83. Reductive amination of aminoketone 84 could be effected with sodium cyanoborohydride, and this step occurred via reduction of iminium compound 85 from the least hindered face to afford tricycle 86, which is epimeric to cylindricine C at C(2). Finally, oxidative conversion of the boronate to the alcohol, followed by oxidation to the corresponding ketone and silyl group removal provided (-)-2-epicylindricine C (87), which had previously been prepared in racemic form by the Weinreb group (see section 5.1.1).4
Scheme 16.
Using advanced intermediate 84, it was also possible to synthesize (-)-cylindricine C as shown in Scheme 17. In this sequence, the silyl ether protecting group of 84 was first removed to provide the corresponding primary alcohol. Using the Evans protocol,24 this compound underwent hydroxyl-directed reductive amination (Cf. 85) to afford tricycle 89 having the desired cylindricine C configuration at C(2). This intermediate was processed as was done in the epimeric series to afford ketone 90, which upon desilylation yielded (-)-cylindricine C (2).
Scheme 17.
3.8 Ishibashi Synthesis of the Cylindricine Ring Skeleton
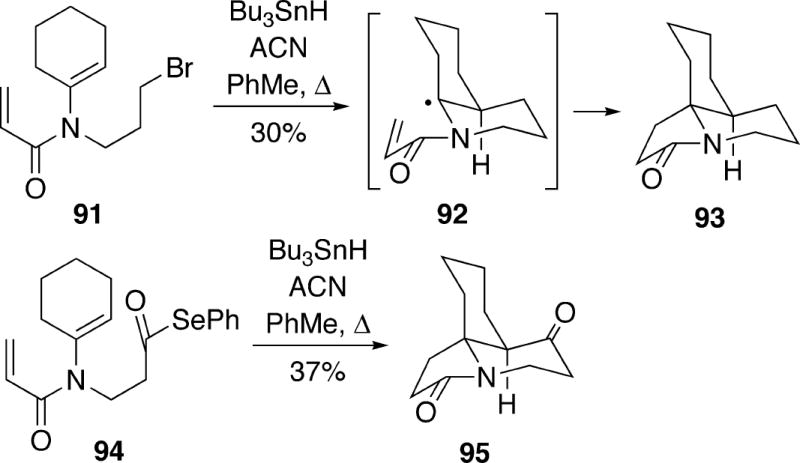
Ishibashi and coworkers have used a tandem radical cascade protocol to access the tricyclic system of the cylindricines, although none of the natural products were synthesized.25 Thus, treatment of bromide 91 with tributyltin hydride in the presence of azobis(cyclohexanecarbonitrile) (ACN) afforded tricycle 93 in modest yield as one stereoisomer (Scheme 18). This cyclization presumably occurs via radical intermediate 92, which undergoes a subsequent stereoselective 5-endo-trig cyclization via the conformation shown to provide the product which contains a cis-1-azadecalin framework. Similarly, an acyl radical could be employed in this tandem process. For example, cyclization of selenoester 94 was also stereoselective and afforded tricycle 95 in moderate yield.
Scheme 18.
3.9 Hunter Approach to the Cylindricine/Lepadiformine Ring System
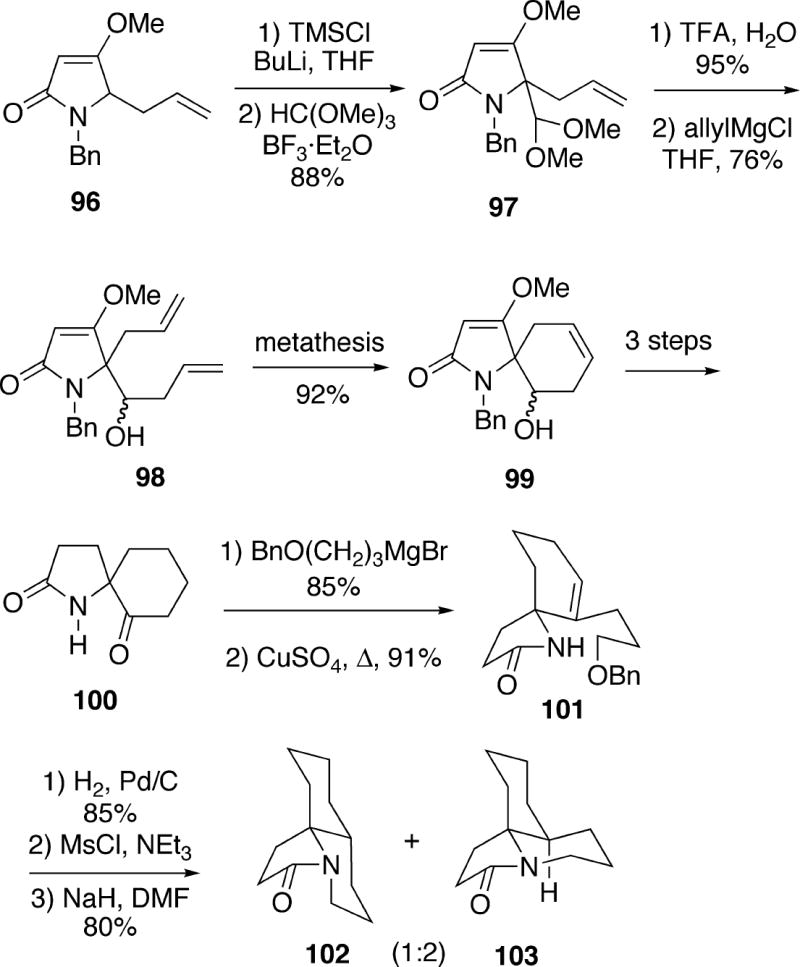
Hunter and Richards have reported an approach to the skeleton of the cylindricines and lepadiformine which proved to be lengthy and non-stereoselective.26 Lactam 96 was first converted to acetal 97, and following hydrolysis and addition of allylmagnesium chloride, diene alcohol 98 was produced (Scheme 19). Using the first generation Grubbs catalyst, diene 98 underwent ring closing metathesis in high yield to produce cyclohexene 99. This intermediate was then converted in three simple steps to spirocyclic ketone 100. Addition of 3-benzyloxypropylmagnesium bromide to this ketone, followed by dehydration yielded olefin 101. Catalytic hydrogenation of the double bond of 101 was non-stereoselective, and was also accompanied by debenzylation. The resulting mixture of alcohols was next converted to the mesylate and cyclized, resulting in a 2:1 mixture of trans- and cis-1-azadecalins 102 and 103.
Scheme 19.
4. Synthesis of Fasicularin
4.1 Kibayashi Synthesis of (±)-Fasicularin
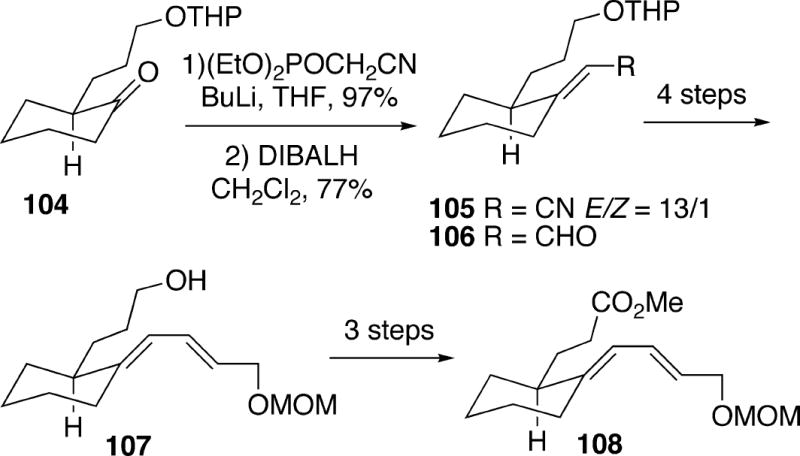
The Kibayashi group utilized an intramolecular acylnitroso Diels-Alder reaction as a key step in the first total synthesis of racemic fasicularin.6b The substrate needed for this cycloaddition was prepared in several steps starting from ketone 104 (Scheme 20). This compound was first converted to a 13:1 mixture of E:Z unsaturated nitriles 105, and the major geometric isomer was reduced to aldehyde 106. It was then possible to homologate 106 in four straightforward operations into diene alcohol 107. Three additional steps then served to deliver diene ester 108.
Scheme 20.
To set up the system for the hetero Diels-Alder step, ester 108 was first transformed into hydroxamic acid 109 (Scheme 21). This compound could be oxidized with tetrabutylammonium periodate in aqueous DMSO to produce a good yield of a 4.8:1 mixture of isomeric Diels-Alder adducts A/B-trans 111 and A/B-cis 114. The cycloadditon proved to be somewhat solvent dependent, and other combinations of aqueous solvents provided slightly poorer ratios of 111:114. In chloroform as the reaction solvent, the selectivity was significantly lower. The major product 111 of this reaction presumably arises via the transition state conformation shown in 110 where the acylnitroso group is endo to the diene but is facially anti. The endo-syn-facial conformers 112 and 113 would lead to the minor Diels-Alder product 114, but it was proposed that these conformations are destabilzed relative to 110 due to non-bonded interactions between the cyclohexane ring and the acylnitroso tether.
Scheme 21.
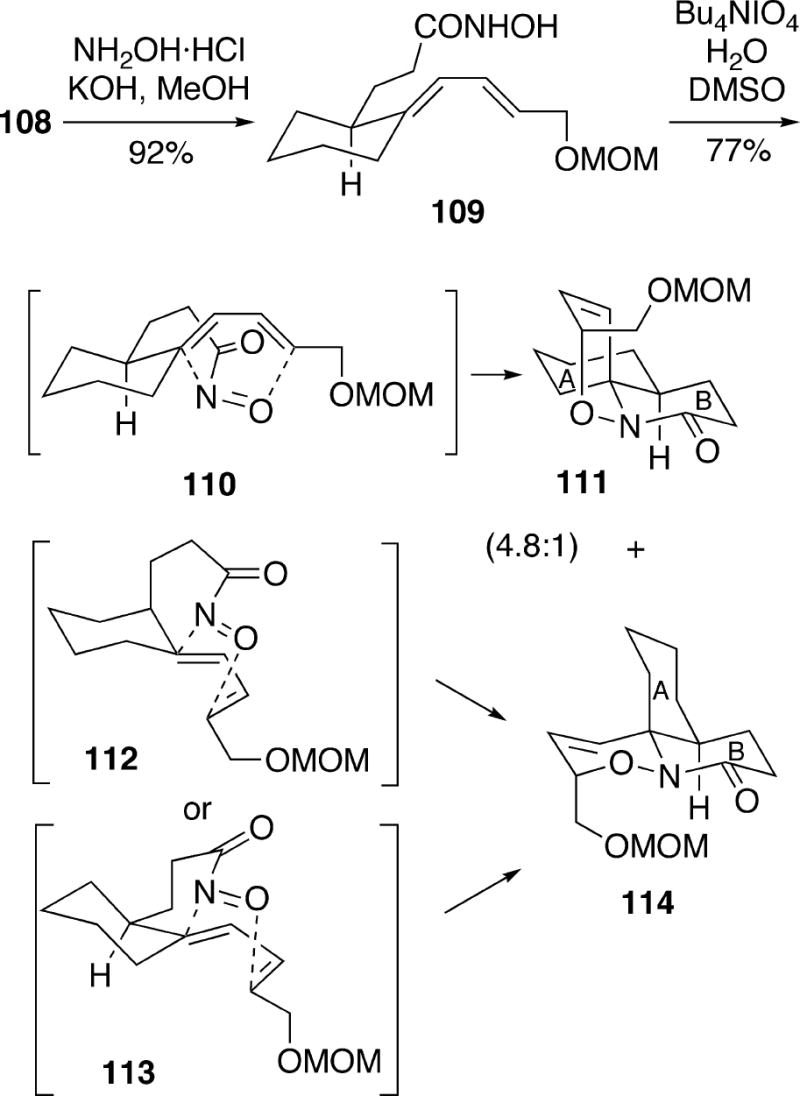
The synthesis of fasicularin continued from the major Diels-Alder adduct 111, which was hydrogenated to remove the olefinic double bond, leading to tricycle 115 (Scheme 22). At this point it was necessary to replace the MOM protecting group and therefore benzyl ether 116 was prepared. Cleavage of the N-O bond in tricycle 116 was performed with sodium amalgam, and the resulting alcohol was protected as the TBDPS ether 117. Removal of the benzyl ether and tosylation of the primary alcohol produced 118, which upon treatment with sodium hydride cyclized to afford 119.
Scheme 22.
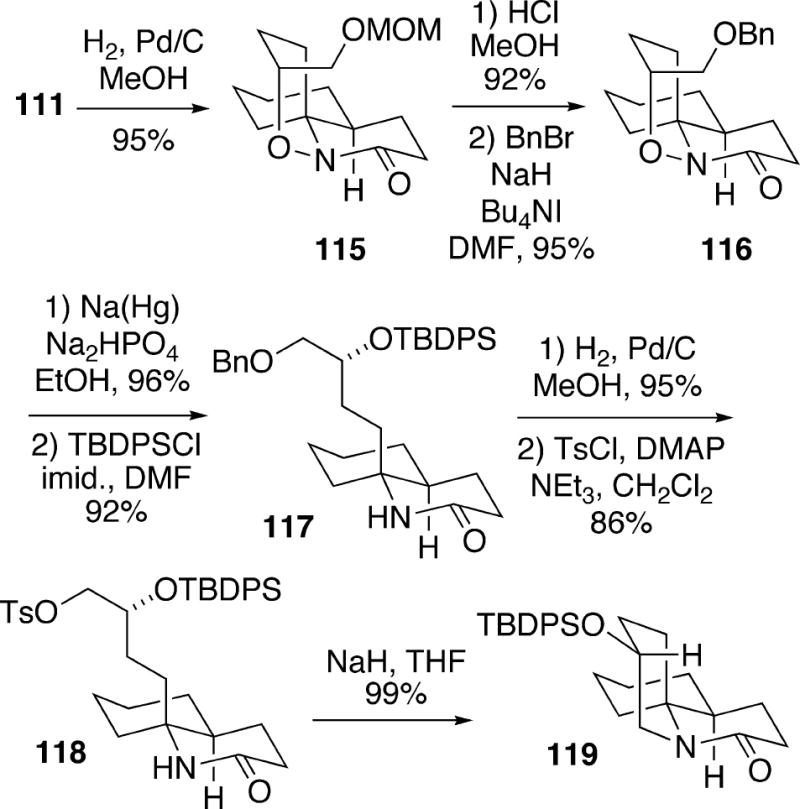
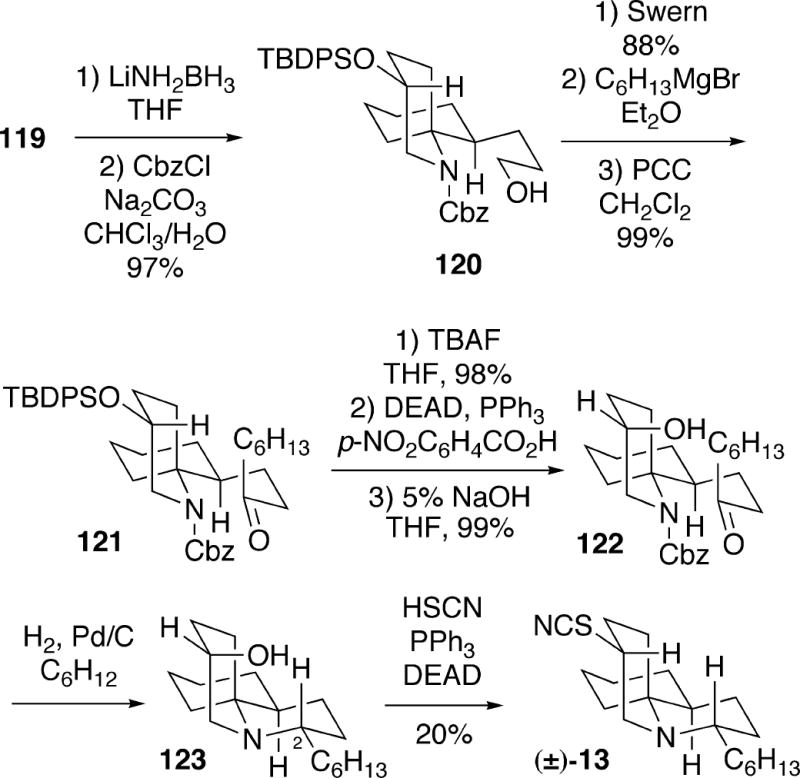
Tricyclic lactam 119 was reductively ring opened, and the amino group was protected to form alcohol Cbz-derivative 120 (Scheme 23). Oxidation of the primary alcohol to the corresponding aldehyde, addition of hexyl Grignard reagent and oxidation led to ketone 121. Removal of the Cbz group of 121 and reductive amination of the resulting aminoketone proved to be non-stereoselective. Therefore as an altermative, the silyl group of 121 was removed and the alcohol was inverted by a Mitsunobu procedure yielding alcohol 122. It was found that the stereochemical course of the catalytic hydogenation of aminoketone 122 was highly solvent dependent. Using cyclohexane as solvent, a 5.2:1 mixture favoring the desired tricycle 123 along with its C(2) epimer was produced. Presumably the hydroxyl group of 122 is involved in delivery of hydrogen to the syn-face of the intermediate iminium ion. With other solvents such as ethyl acetate, ethanol and benzene, the stereoselectivities were lower in the reduction step. To complete the synthesis, it was necessary to introduce a thiocyanate group, and this transformation proved to be surprisingly difficult to effect. For example, attempted displacement of the mesylate derived from alcohol 123 with KSCN gave only elimination and decomposition products. It was finally found that a Mitsunobu reaction of alcohol 123 with thiocyano acid produced racemic fasicularin (13) but in only 20% yield. Although successful, this synthesis of fasicularin was rather long, requiring approximately 29 steps.
Scheme 23.
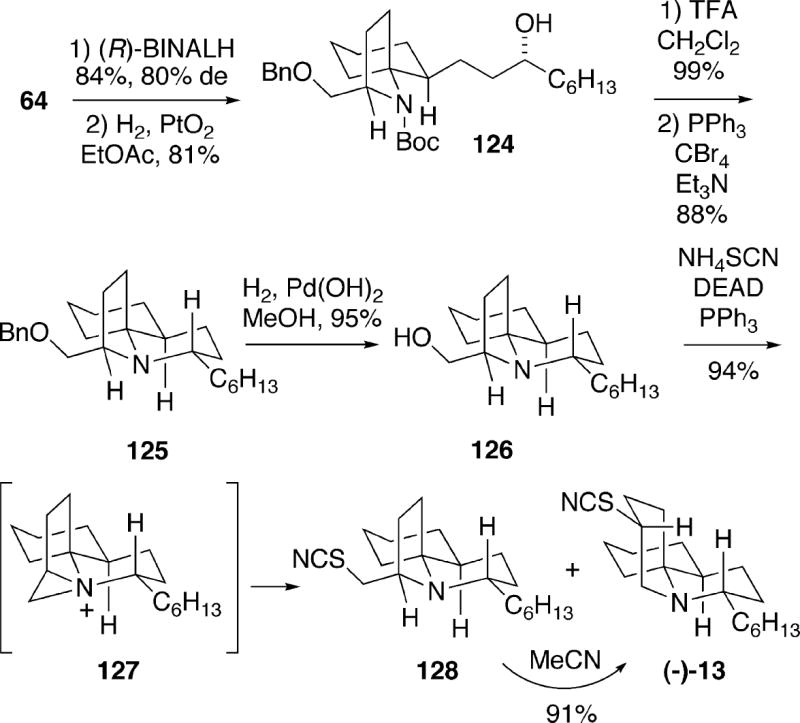
4.2 Kibayashi Synthesis of (-)-Fasicularin
Spirocyclic enone intermediate 64, used in the enantioselective synthesis of (+)-cylindricine C described in Schemes 11 and 12, was utilized in a short, efficient total synthesis of (-)-fasicularin.19 1,2-Reduction of enone 64 with (R)-BINALH proceeded with 9:1 stereoselectivity, and following double bond hydrogenation alcohol 124 was formed (Scheme 24). Removal of the Boc group of 124, followed by cyclocondensation of the resulting amino alcohol, provided tricycle 125. Hydrogenolysis of the benzyl ether protecting group of 125 then led to alcohol 126. To complete the synthesis, alcohol 126 was exposed to Mitsunobu conditions using ammonium thiocyanate to afford a 94% yield of a 1:1 mixture of 128 and (-)-fasicularin (13). This transformation presumably involves the aziridinium ion 127 as an intermediate (Cf. Scheme 1). If the undesired isomer 128 is allowed to stand in acetonitrile at room temperature for three days, it is converted into (-)-fasicularin in high yield, once again probably involving intermediate 127. This total synthesis of (-)-fasicularin (13) requires only about 11 steps from enantiomerically pure pyrrolidinone 50 (Scheme 11), and proceeds in 28% overall yield.
Scheme 24.
4.3 Funk Synthesis of (±)-Fasicularin
Funk and Maeng reported a total synthesis of racemic fasicularin which utilizes a Diels-Alder cycloaddition of a 2-amidoacrolein derivative as a key component of the synthetic strategy.27 Readily prepared triflamido-1,3-dioxin 129 underwent a retro-Diels-Alder cycloaddition upon heating in toluene to afford 2-triflamidoacrolein 130 along with acetone (Scheme 25). Using unsaturated aldehyde 130, a regio-and stereoselective [4+2]-cycloaddition with diene 131 could be effected at 12 kbar to afford a single Diels-Alder adduct 132. Lithium aluminum hydride reduction of 132 served to both reduce the aldehyde and remove the triflamide protecting group, providing amino alcohol 133. The olefinic double bond and N-benzyl group in 133 were then concomitantly removed by catalytic hydrogenation, and the resulting amino alcohol was cyclized with acid to oxazolidine 134.
Scheme 25.
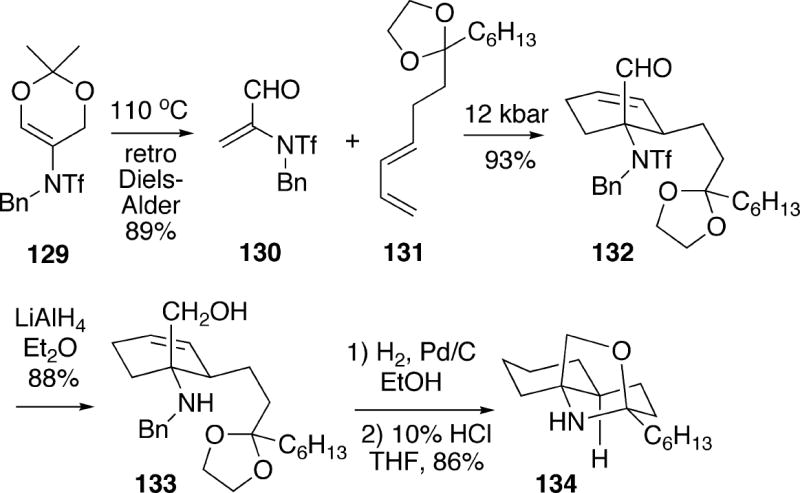
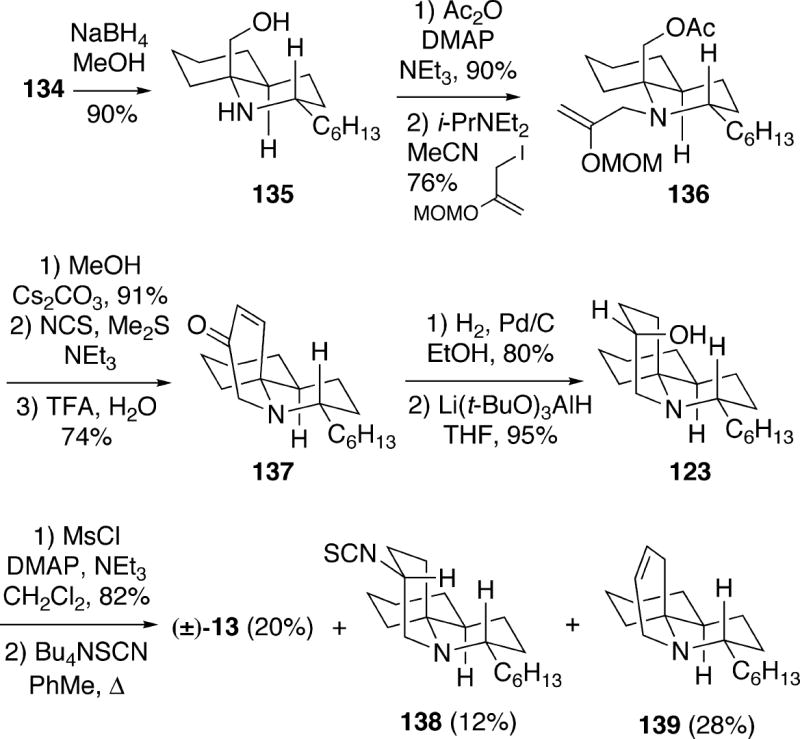
Reduction of oxazolidine 134 with sodium borohydride was found to be stereoselective, and proceeded via axial reduction of an intermediate iminium ion, affording amino alcohol 135 having the requisite C(2) stereochemistry for fasicularin (Scheme 26). In order to construct the C-ring of the natural product, intermediate 135 was first acetylated, and the amine was alkylated with 3-iodo-2-(methoxymethoxy)propene to afford 136. The acetate group of 136 was removed, and the primary alcohol was next oxidized to the corresponding aldehyde. This compound underwent hydrolysis/aldol condensation with aqueous TFA to provide tricyclic enone 137. Catalytic hydrogenation of the enone double bond of 137, followed by reduction of the ketone functionality with lithium tri-tert-butoxyaluminohydride afforded a 5.3:1 mixture of alcohols with the desired axial isomer 123 being the major product. As was the case in the Kibayashi synthesis (Scheme 23), conversion of 123 to fasicularin was problematic. In this case, the Mitsunobu procedure used by Kibayashi failed to provide the natural product. However, it was possible to displace the mesylate derived from alcohol 123 with tetrabutylammonium thiocyanate to afford racemic fasicularin (13), but only in low yield, along with by-products 138 and 139 which were observed also by Kibayashi.
Scheme 26.
4.4 Dake Enantioselective Formal Synthesis of Fasicularin
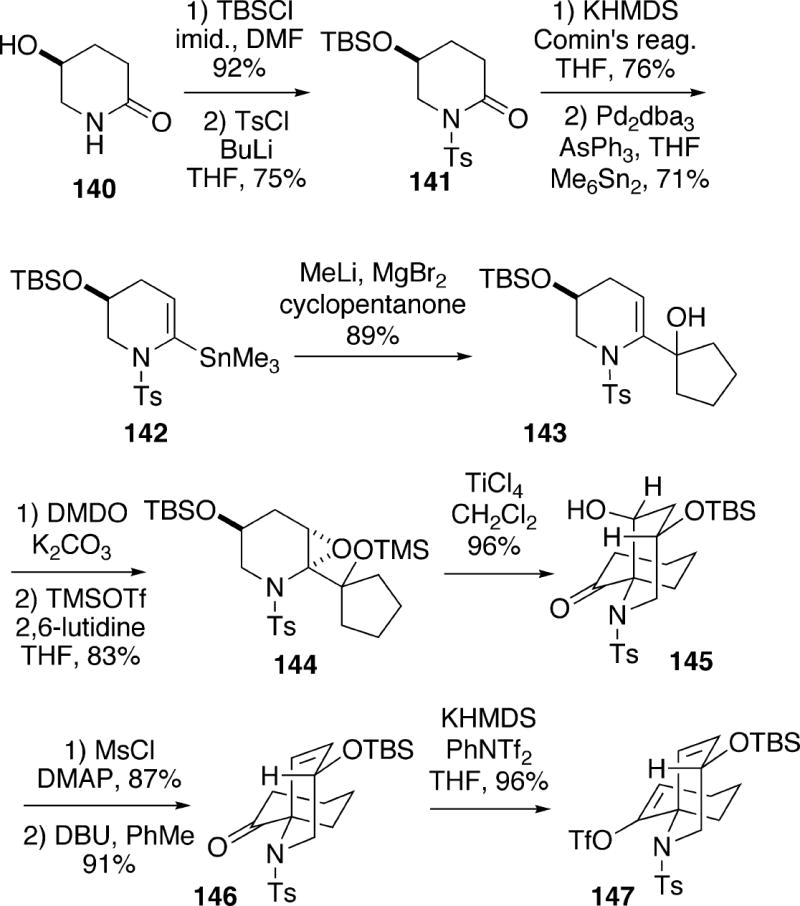
Dake and coworkers devised a novel approach to fasicularin which employs siloxyepoxide semipinacol rearrangement as a pivotal transformation.28,29 The synthesis began with hydroxylactam 140, which can be obtained in enantiomerically pure form from L-glutamic acid (Scheme 27).30 Thus, this synthesis is in the antipodal series relative to the Kibayashi work (Cf. Scheme 24). Compound 140 was protected as TBS ether N-tosyllactam 141, and after conversion of the lactam to the enol triflate, palladium(0)-catalyzed coupling was effected with hexamethyldistannane to produce vinyl stannane 142. Transmetallation of this stannane with methyllithium and magnesium bromide, followed by addition to cyclopentanone led to alcohol 143. To prepare for the key rearrangement step, enesulfonamide 143 was first stereoselectively epoxidized with dimethyldioxirane. This oxidation probably occurs via axial attack on a half chair conformation of enesulfonamide 143 by DMDO.29 The alcohol functionality was then silylated to generate 144. Exposure of compound 144 to titanium tetrachloride promoted a semipinacol rearrangement to afford the desired spirocyclic ketone 145 in high yield. It might be added that the alcohol corresponding to silyl ether 144 does not rearrange cleanly. The hydroxyl functionality of 145 was converted to the mesylate, and eliminated with DBU to alkene 146. The ketone moiety of 146 was next transformed to the enol triflate 147.
Scheme 27.
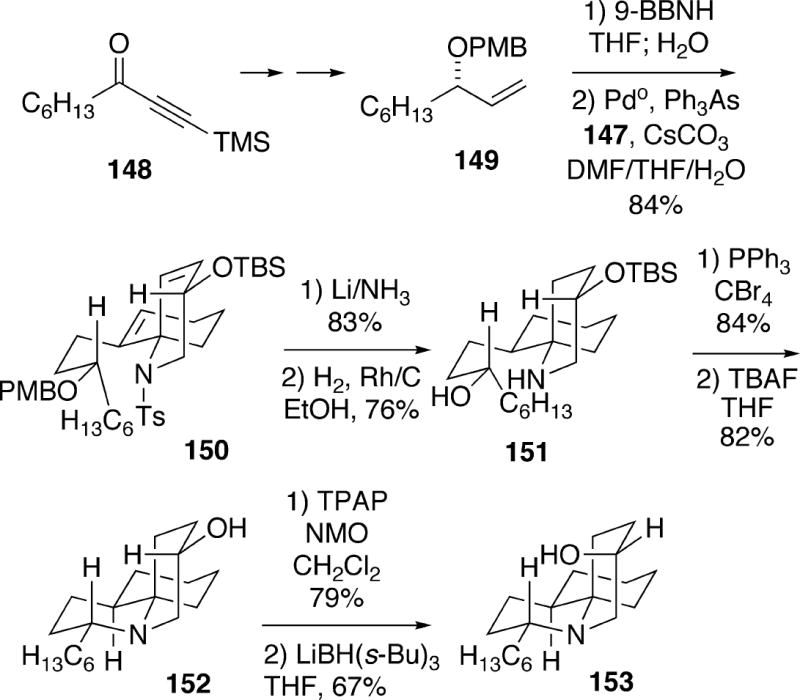
A number of metal-mediated coupling experiments were conducted with enol triflate 147 in order to incorporate a fragment for construction of the remaining B-ring of the alkaloid. The route which was ultimately successful involved utilization of (S)-allylic ether 149, which could be prepared in high ee via the Noyori transfer hydrogenation procedure31 from ynone 148, followed by some simple transformations (Scheme 28). The alkene 149 was initially hydroborated, and a subsequent Suzuki-Miyaura coupling could then be effected with triflate 147 to produce the requisite alkylated product 150 in good yield. The tosyl and PMB protecting groups of 150 were removed by a dissolving metal reduction to generate the corresponding amino alcohol. A number of hydrogenation conditions were then examined to reduce the double bonds, and it was found that rhodium on carbon was the best catalyst, leading to a 10.5:1 mixture of stereoisomers favoring the desired reduction product 151. This compound then cyclized with inversion to afford a tricycle, which upon removal of the silyl protecting group provided alcohol 152. The stereochemistry of this alcohol could be inverted by a two step process involving initial TPAP oxidation to the ketone followed by stereoselective reduction with L-Selectride to 153, which is enantiomeric to compound 123 synthesized by Kibayashi. Unfortunately, the Dake group was unable to reproduce either the Kibayashi (Scheme 23) or Funk (Scheme 26) procedures for conversion of alcohol 153 to the thiocyanate, which would have led to (+)-fasicularin (13).
Scheme 28.
5. Synthesis and Structure Determination of Lepadiformine
5.1 Approaches to the Putative Lepadiformine Structure 14 and Related Compounds
5.1.1 Weinreb Synthesis of Racemic Structure 14
In 1999, the Weinreb group reported the first synthesis of the structure 14 for lepadiformine which was suggested by Biard.4 The approach featured an intramolecular nitrone/diene 1,3-dipolar cycloaddition. The synthesis commenced with acetone oxime, which was deprotonated and alkylated with epoxide 154 to afford oxime alcohol 155 (Scheme 29). Oxime alcohol 155 was converted to the corresponding trianion, and alkylated with iodo diene 156 to yield 157. Cleavage of the oxime to the corresponding ketone with TiCl3 and silylation of the alcohol afforded 158. The carbonyl group in 158 was then ketalized, and the silyl group was cleaved to afford alcohol 159. Swern oxidation of 159 to the ketone, followed by oxime formation provided 160, and reduction of the oxime functionality with sodium cyanoborohydride afforded hydroxylamine 161.
Scheme 29.
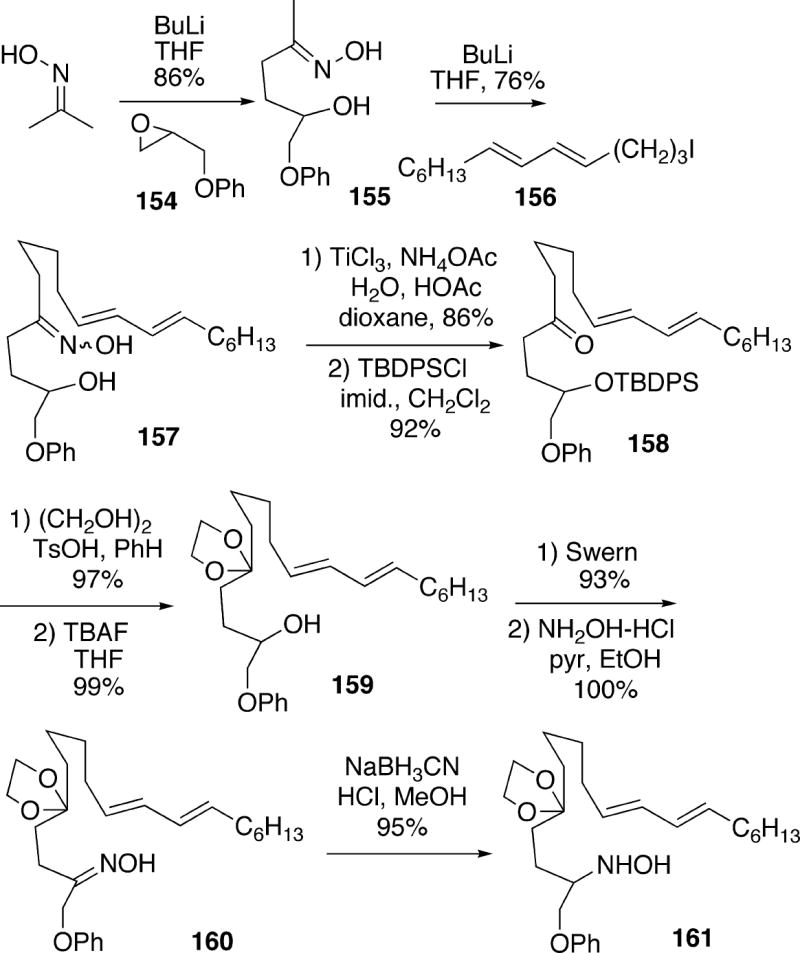
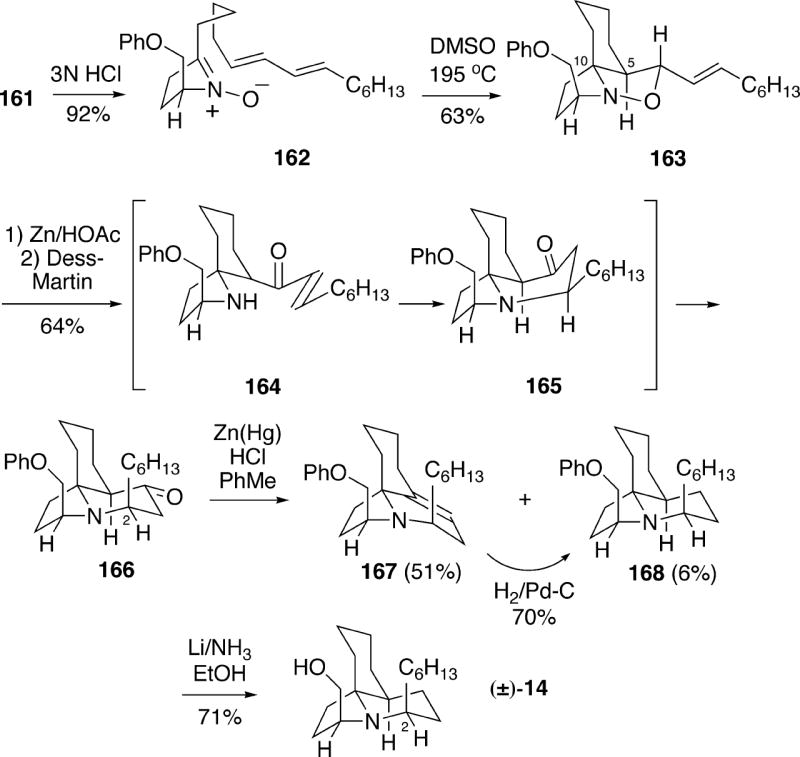
Treatment of hydroxylamino ketal 161 with aqueous HCl led to the desired cyclic nitrone diene 162 in 92% yield (Scheme 30). Thermolysis of 162 in DMSO solution overnight at 195 °C produced cycloadduct 163 in 63% yield as a single stereo- and regioisomer. Cleavage of the N-O bond of this isoxazolidine could be effected with Zn/HOAc to give the amino alcohol, and oxidation with the Dess-Martin reagent led to tricycle 166 via in situ cyclization of amino enone 164. The formation of the C(2) axial epimer occurs for stereoelectronic reasons since conjugate addition of the amino group to the enone functionality in 164 must occur through a transition state initially leading to the boat B-ring 165, which ring flips to the observed all-chair product 166. It should be noted that ketone 166 could be protected as its ethylene ketal, and the phenyl protecting group removed by Birch reduction, followed by acid hydrolysis, to afford unnatural (±)-2- epicylindricine C (Cf. Scheme 16).
To complete the synthesis of 14, tricyclic ketone 166 was reduced under Clemmensen reduction conditions, surprisingly leading to olefin 167 as the major product, along with a small amount of the desired compound 168. It was possible, however, to stereoselectively reduce alkene 167 by catalytic hydrogenation to produce tricycle 168. X-ray analysis of the picrate salt of amine 168 showed that the compound exists in the conformation shown, rather than in the flip form 14a originally suggested by Biard for lepadiformine (see Figure 3). Finally, removal of the phenyl protecting group from 168 afforded racemic 14, which was found to be different from authentic lepadiformine by a comparison of proton and carbon NMR spectra, and moreover the synthetic compound showed no indication of being a zwitterion.
5.1.2 Oppolzer Nitrone-Based Approach
In 2000, Oppolzer and coworkers described intramolecular nitrone/olefin dipolar cycloaddition methodology for synthesis of enantiopure spirocyclic systems like those found in the cylindricines.32 Their approach is closely analogous to that of the Weinreb group outlined above for preparation of racemic systems. The Oppolzer strategy utilized chiral auxiliary 169 to access enantiomerically pure material for this cycloaddition process (Scheme 31). Thus, readily prepared ester 170 was first converted to the acylsultam 171.32a This compound was deprotonated with sodium hexamethyldisilazide and hydroxyaminated with 1-nitroso-1-chlorocyclohexane, followed by acid-promoted cyclization to produce nitrone 172 with complete diastereoselectivity. Thermolysis of this nitrone olefin in toluene produced a mixture of regioisomeric cycloadducts 173 and 175. The minor desired isoxazolidine 173 arises via dipolar cycloaddition through the conformation shown in structure 172, whereas the major cycloadduct is produced from conformation 174. It should be noted that the Weinreb group had found similar regiochemical problems in a very closely related intramolecular nitrone/olefin cycloaddition.4b
Scheme 31.
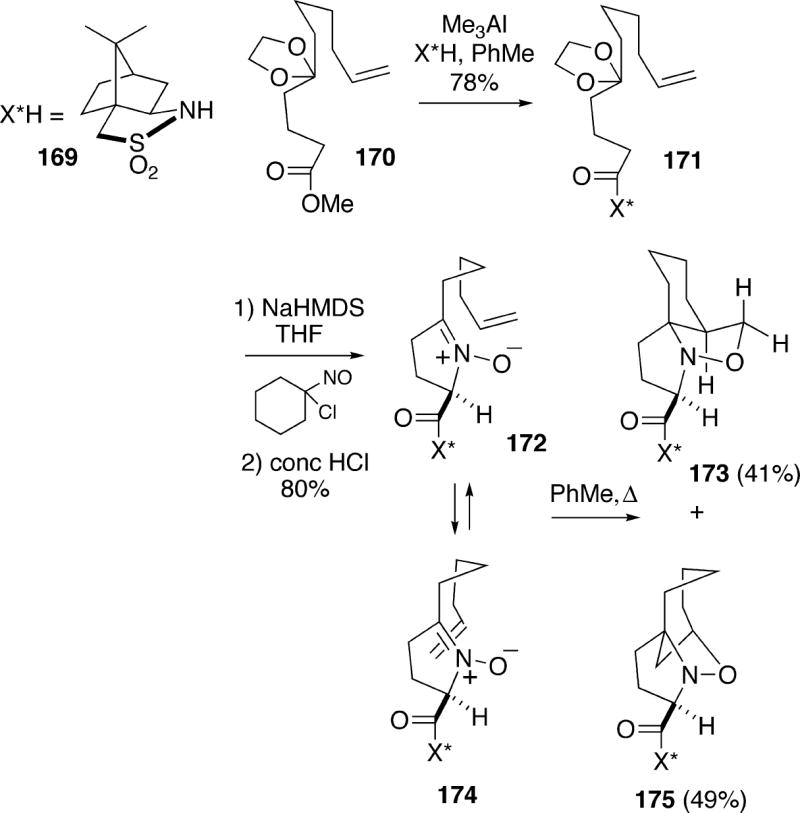
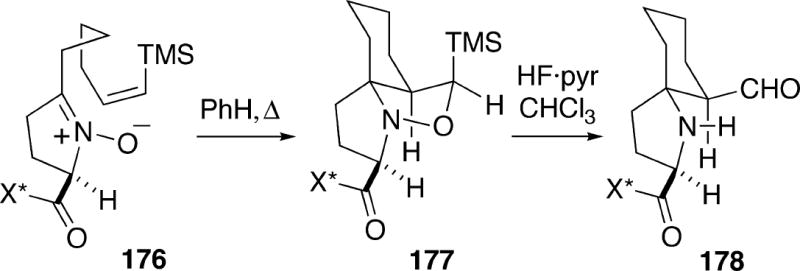
In order to improve regioselectivity in the dipolar cycloaddition, a vinylsilane was investigated as the dipolarophile.32b Thus, nitrone Z-vinylsilane176 was prepared using chemistry similar to that shown above (Scheme 32). Thermolysis of this nitrone in benzene for several days provided only cycloadduct 177, although the yield was not reported. This cycloadduct could be cleaved to aldehyde 178, once again in unspecified yield.
Scheme 32.
5.1.3 Pearson Synthesis of C(2) and C(13) Epimers of 14
Pearson, et al. synthesized the three diastereomers of structure 14 at C(2) and C(13) utilizing azaallyl anion 1,3-dipolar cycloaddition methodology.5 In this approach, stannyl amine 180 was first condensed with cyclohexanone 179 to produce imine 181. Without isolation, this compound was converted to the azaallyl anion with butyllithium, and combined with phenyl vinyl sulfide to stereo- and regioselectively form spirocyclic adduct 182 (Scheme 33). This compound could then be converted in three steps to alcohol 183. Reductive removal of the thiophenyl group was followed by hydrolytic cleavage of the Cbz and ketal moieties, and the resulting amino ketone underwent reductive amination using sodium borohydride to afford tricycle 184. This compound was compared with natural lepadiformine and was found to be different.
Scheme 33.
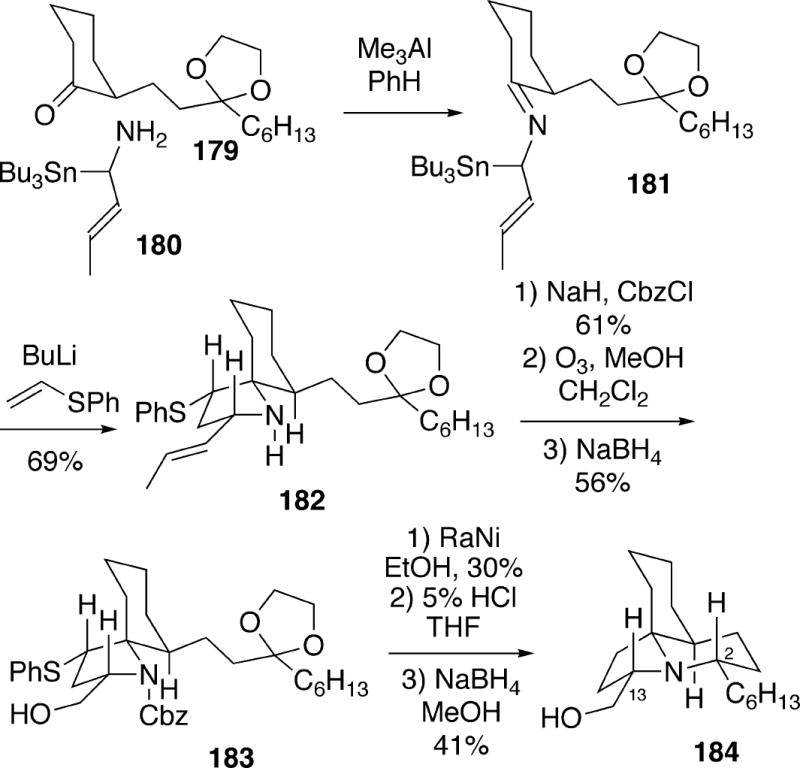
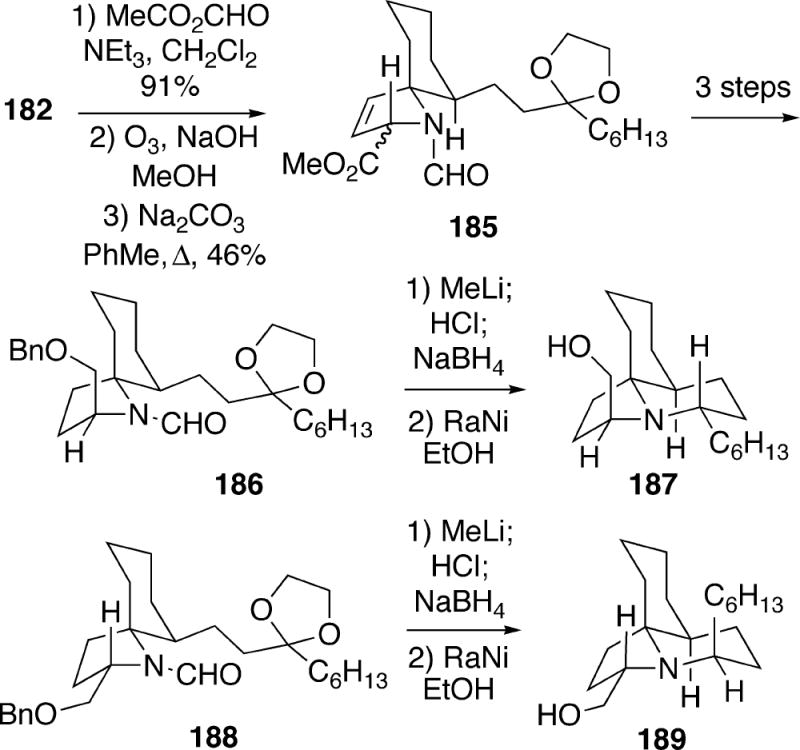
To prepare two other stereoisomers of 14, cycloadduct 182 was transformed in three steps to an epimeric mixture of methyl esters 185 (Scheme 34). Three additional steps served to provide pure samples of isomeric compounds 186 and 188. A deprotection/reductive amination sequence then led to tricycles 187 and 189, which had spectra that did not match those of lepadiformine.
Scheme 34.
5.1.4 Kibayashi Synthesis of Racemic Structure 14
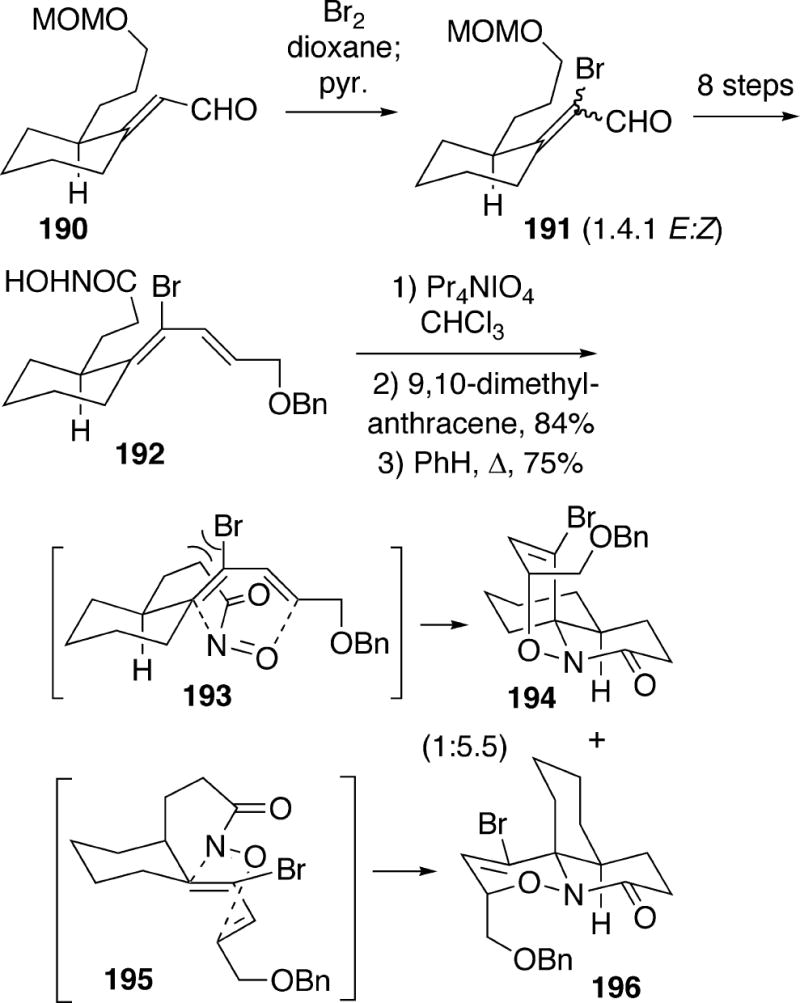
Kibayashi and coworkers used an intramolecular nitroso Diels-Alder-based strategy for preparation of (±)-14 similar to that previously discussed for synthesis of racemic cylindricine C (Cf. Schemes 20 and 21).6a,b In this approach, it was possible to reverse the stereoselectivity of the key [4+2]-cycloaddition step relative to that in Scheme 21 by introducing a bromine substituent into the diene. Thus, unsaturated aldehyde 190 was first brominated to afford a 1.4:1 mixture of E:Z isomers 191 (Scheme 35). This compound was processed via an eight-step sequence, including a separation of the geometric isomers, to eventually produce the bromodiene hydroxamic acid 192 required for the cycloaddition. The best way to conduct the Diels-Alder reaction was to first oxidize hydroxamic acid 192 to the corresponding acylnitroso compound with tetrapropylammonium periodate, and then trap it as a [4+2]-cycloadduct with 9,10-dimethylanthracene. Thermal reversion of this reaction, followed by intramolecular cycoaddition led to a 1:5.5 mixture of adducts 194 and 196. It was suggested that transition state conformation 193, leading to the minor isomer 194, is destabilized relative to the alternative conformation 195 by a severe steric interaction between the bromine on the diene and the tether. Adduct 196 has the requisite A/B-ring cis-1-azadecalin stereochemistry for cylindricine-type structures.
Scheme 35.
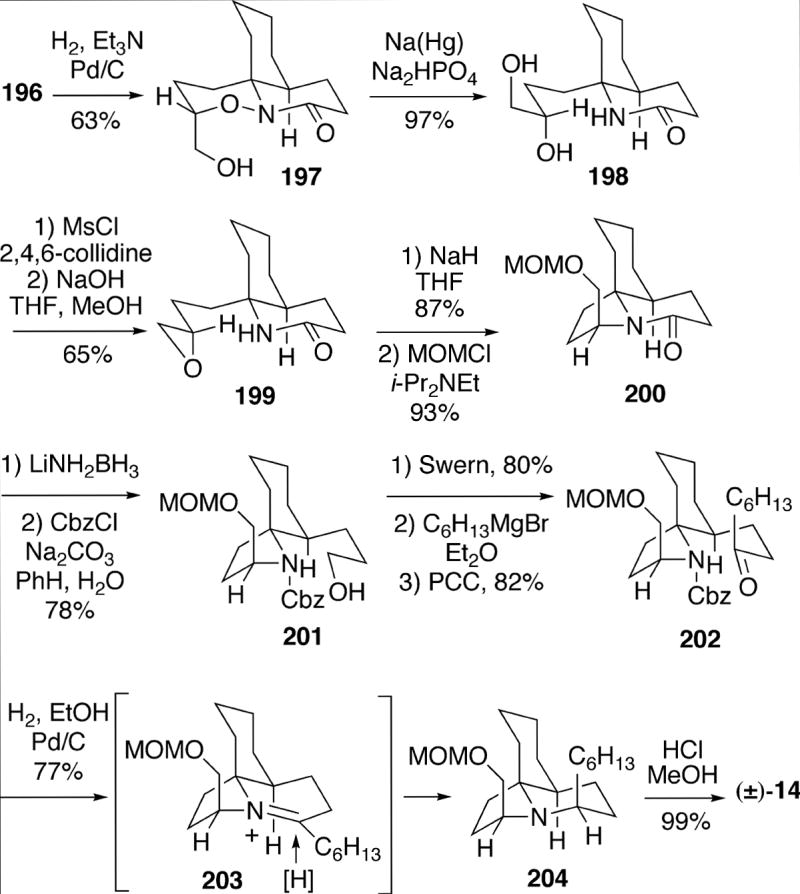
Adduct 196 was utilized for the synthesis of 14, and was first hydrogenated to afford tricyclic alcohol 197 (Scheme 36). The N-O bond of this intermediate was cleaved with sodium amalgam to generate diol lactam 198, which was next converted to epoxide 199. Treatment of intermediate 199 with sodium hydride led to intramolecular 5-exo attack of the lactam nitrogen on the epoxide to form the C-ring, and after alcohol protection tricyclic lactam 200 was generateded in good overall yield. In order to further elaborate the B-ring, this lactam was reduced to the ring-opened amino alcohol, which was protected as Cbz derivative 201 and transformed in three straightforward steps into ketone 202. Removal of the Cbz protecting group and reductive amination then provided the requisite tricycle 204. This process occurs via reduction of intermediate iminium salt 203 from the least hindered face. Finally, hydrolytic cleavage of the MOM protecting group afforded racemic compound 14.
Scheme 36.
5.2 Synthesis of Authentic Lepadiformine 15a
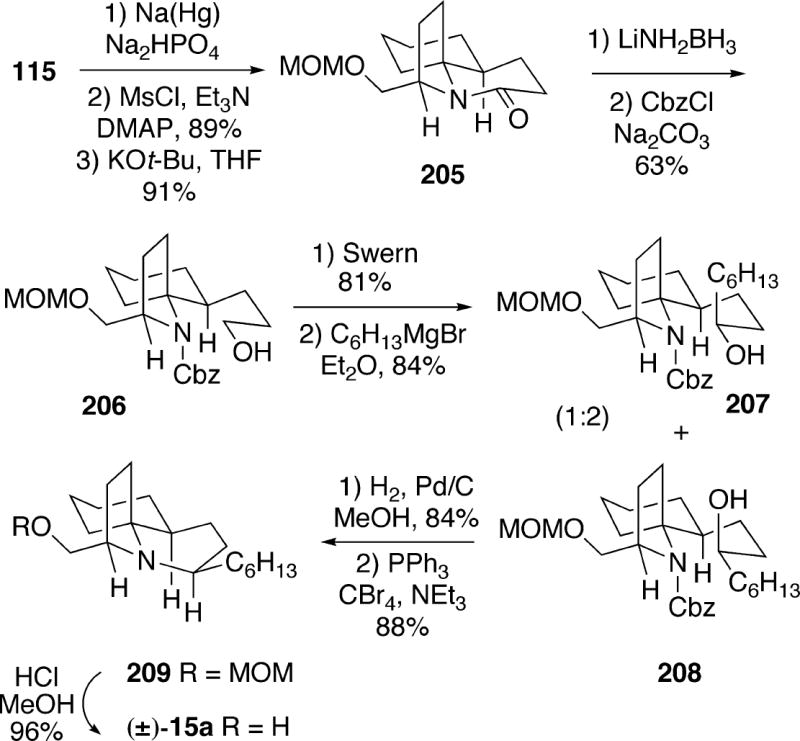
5.2.1 Kibayashi Synthesis of (±)-Lepadiformine
Using the key nitroso Diels-Alder-derived intermediate 115 which had been used in their synthesis of fasicularin (Cf. Scheme 22), the Kibayashi group prepared structure 15a and found that its hydrochloride salt was in fact identical to lepadiformine, thereby unambiguously establishing the structure of this alkaloid.6b Oxazine 115 was reduced with sodium amalgam and the resulting alcohol was converted to the mesylate, which upon treatment with potassium t-butoxide cyclized with inversion to provide tricyclic lactam 205 (Scheme 37). Reductive ring opening of this lactam, and protection of the resulting amine as its Cbz derivative led to alchol 206. The primary alcohol 206 was oxidized to the aldehyde and addition of hexylmagnesium bromide afforded alcohols 207 and 208 in a 1:2 ratio. The minor adduct 207 could be inverted to 208 by a Mitsunobu procedure. Removal of the Cbz group from 208, followed by cyclization of the resulting amino alcohol with inversion afforded tricycle 209. Finally, removal of the MOM group with methanolic HCl led to racemic 15a. The hydrochloride salt of the free base 15a was prepared and was found to be identical with natural lepadiformine. It should be noted that natural lepadiformine was originally isolated by evaporation of an aqueous HCl solution. In addition, X-ray analysis of the synthetic lepadiformine showed that the B-ring exists as a twist boat.
Scheme 37.
5.2.2 Funk Synthesis of (±)-Lepadiformine
Greshock and Funk applied their 2-amidoacrolein-based methodology to a total synthesis of the lepadiformine structure 15a.33 Thus, thermolysis of dioxin 210 produced amidoacrolein 211 via a retro Diels-Alder process (Scheme 38). This compound underwent a [4+2]-cycloaddition reaction with diene 212 under high pressure to afford endo adduct 213 as a single regio- and stereoisomer. The aldehyde functionality of 213 was reduced, and catalytic hydrogenation served to both reduce the alkene and remove the N-benzyl group. The resulting alcohol sulfonamide was converted to the N-tosylaziridine 214 by a Mitsunobu-type process. This compound could then be coupled with excess allylmagnesium bromide, followed by acetal hydrolysis, to generate alkene aldehyde 215 in high overall yield.
Scheme 38.
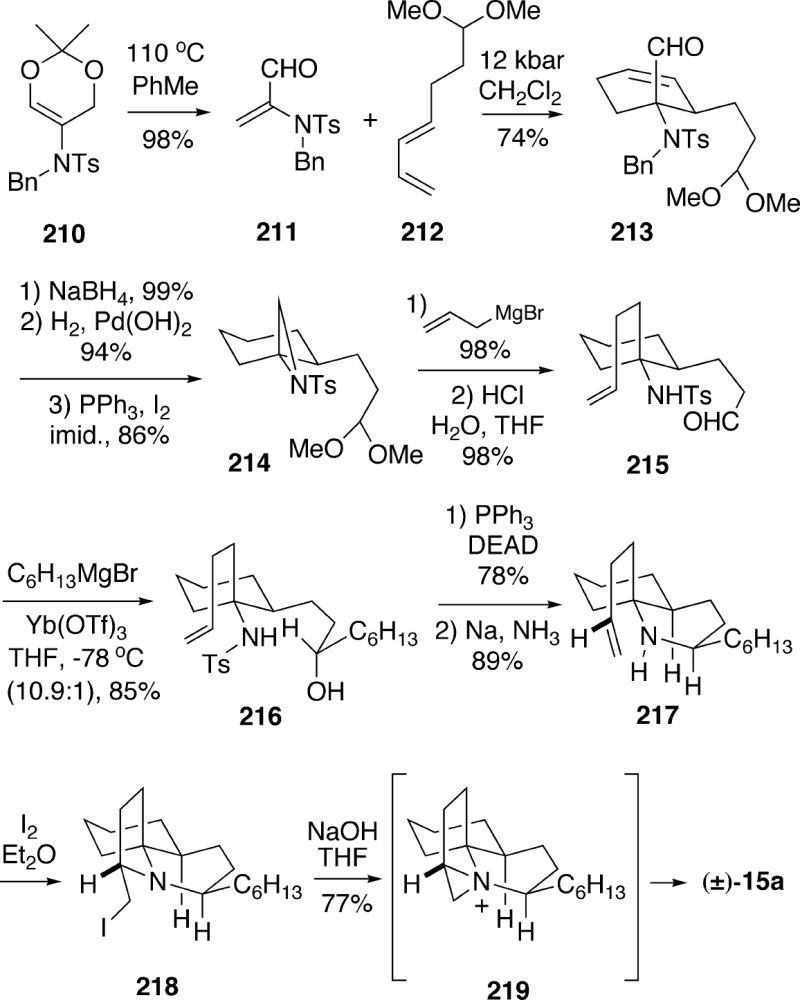
The hexyl group was introduced into this aldehyde using hexylmagnesium bromide in the presence of ytterbium triflate to afford 216 as the major product with 10.9:1 stereoselectivity. In the absence of the ytterbium catalyst the stereoselectivity was significantly lower. It was suggested that metal chelation between the aldehyde carbonyl and the sulfonamide was responsible for the good selectivity in this addition. To complete the synthesis, alcohol 216 was first cyclized with inversion, and the tosyl group was removed to lead to bicyclic amino olefin 217. Finally, treatment of 217 with iodine led to iodomethyl tricycle 218, which upon treatment with sodium hydroxide afforded racemic lepadiformine (15a). This final step probably proceeds via the aziridinium ion 219.
5.2.3 Weinreb Synthesis of (±)-Lepadiformine
In 2001 Weinreb et al. disclosed an approach to the structure 15a of lepadiformine which featured a stereoselective intramolecular spirocyclization of an N-acyliminium ion with an allylic silane.34 The substrate 222 for this reaction was easily prepared from cyclic imine 220 by metallation followed by alkylation with allylsilane iodide 221, then acylation with o-nitrobenzoyl chloride (Scheme 39). Exposure of enamides 222 to trifluoroacetic acid led to a single spirocycle 224 in 57% overal yield based upon imine 220. This cyclization most likely occurs via an N-acyliminium ion which cyclizes through the preferred conformation 223 having both the allylsilane and N-acylimine groups pseudo-equatorial. The alternative conformation 225, which would lead to diastereomer 226, is probably destabilized relative to 223 by a steric interaction between the bulky pseudo-axial N-acyl group and the axial hydrogens of the newly forming six-membered ring.
Scheme 39.

Using the radical transposition methodology previously developed by the Weinreb group,35 functionalization of compound 224 was next investigated in order to provide a handle for introduction of the hydroxymethyl group of the alkaloid. To effect this transformation, the nitro functionality of 224 was first reduced to the o-aminobenzamide 227 which was then subjected to the conditions for conversion of such functionality to an α-methoxybenzamide, thereby cleanly producing 228 (Scheme 40). It was then possible to alkylate the acyliminium ion 230 derived from α-methoxybenzamide 228 with the cuprate from (allyldimethylsilyl)methylmagnesium bromide (229) to afford an 87% yield of a 7:1 mixture of epimeric products, with the desired silane 231 being the major stereoisomer. This transformation presumably occurs via preferred attack of the cuprate from the least congested face of N-acyliminium ion 230. A Tamao oxidation then allowed conversion of silane 231 to the hydroxymethyl compound 232. The alkene 232 was hydroformylated, the resulting aldehyde was transformed to the acetal, and the N-benzoyl group was removed by basic hydrolysis to give amino alcohol 233.
Scheme 40.
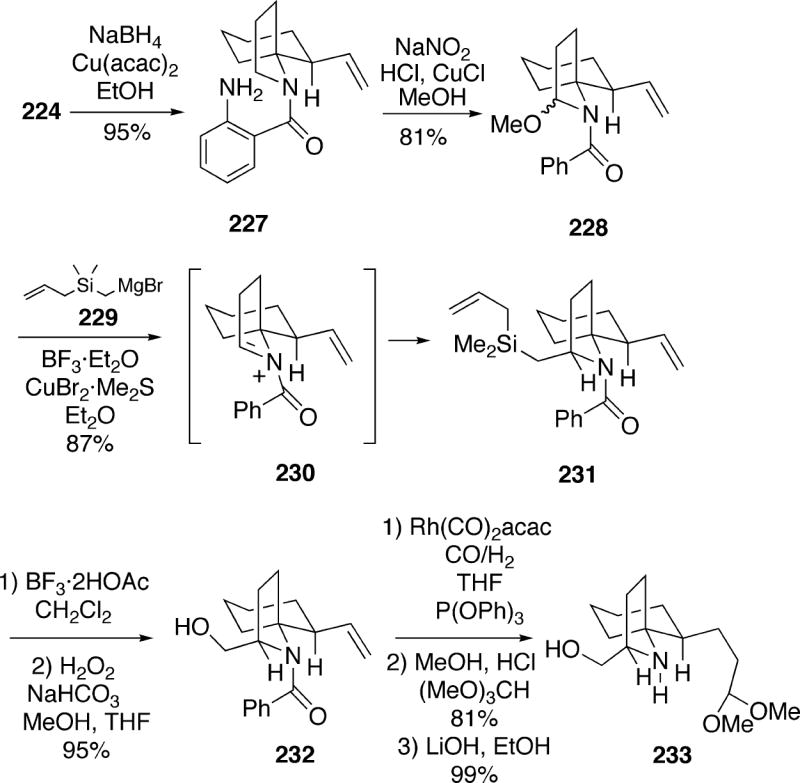
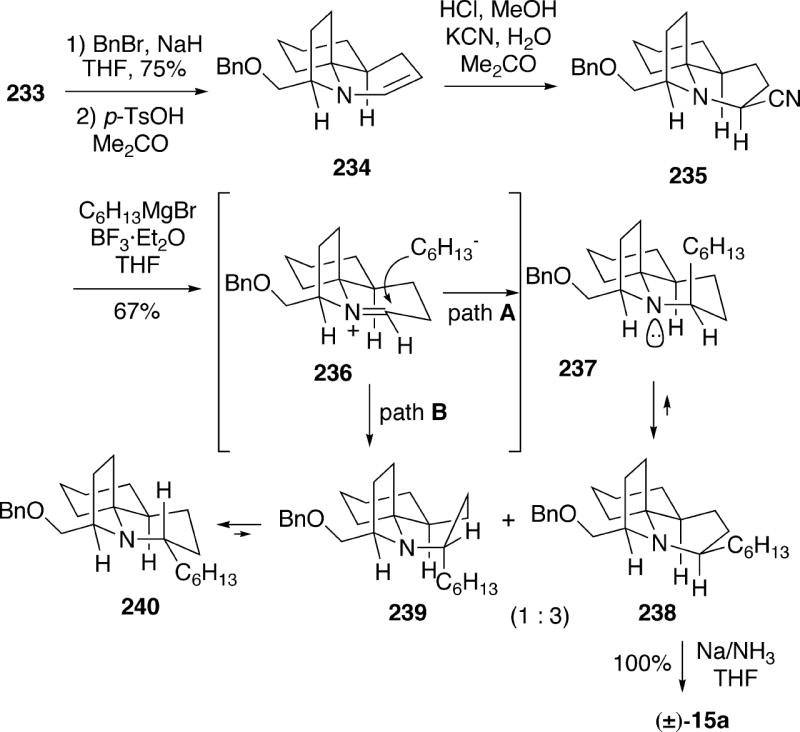
The remaining phase of the synthesis involved construction of the lepadiformine B-ring. Thus, amino alcohol 233 was converted to the corresponding amine benzyl ether and then treated with acid to afford the unstable tricyclic enamine 234 (Scheme 41). This enamine could be converted to the more stable α-amino nitrile 235 which appears to contain a twist boat B-ring. The hexyl chain could be introduced by treating this amino nitrile with hexylmagnesium bromide in the presence of boron trifluoride etherate in THF producing a 3:1 mixture of the desired alkylation product 238 along with its C(2) epimer 240. The stereoelectronic principles summarized by Stevens were used to rationalize the results of this reaction.36 Thus, anti-periplanar addition of the Grignard reagent to the iminium salt 236 from the preferred “axial” direction (path A) will generate an initial chair B-ring as in 237, and this compound would then undergo conformational inversion to the more stable lepadiformine twist boat conformation 238. Nucleophilic attack on the iminium ion 236 from the opposite face (path B), however, will initially produce an unfavorable B-ring boat 239, which then flipped to the more stable chair conformer 240 now with an equatorial hexyl group. To complete the synthesis, reductive cleavage of the benzyl ether group from tricycle 238 afforded racemic lepadiformine (15a).
Scheme 41.
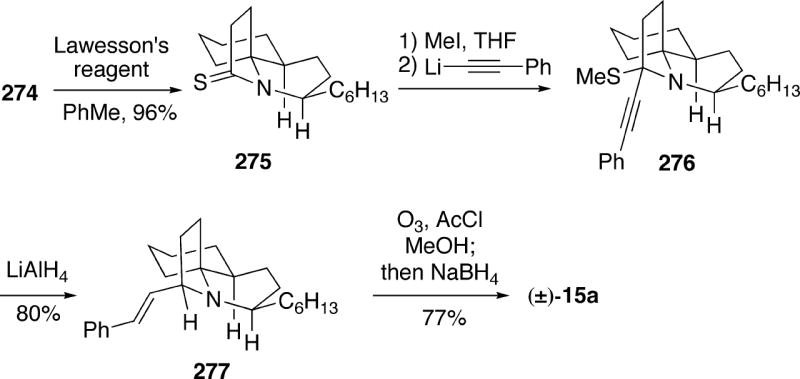
5.2.4 Weinreb Synthesis of (-)-Lepadiformine
The basic strategy outlined above for synthesis of racemic lepadiformine was extended to achieve an enantioselective total synthesis of the alkaloid, which also established its absolute configuration.34b Therefore, readily prepared (S)-Boc-lactam 241, derived from (S)-pyroglutamic acid, was coupled with lithio (Z)-allylsilane 242 to give adduct 243 (Scheme 41). Without isolation, this intermediate was then exposed to boron trifluoride acetic acid complex in methylene chloride to provide spirocycle 245 as a single stereoisomer in 52% overall yield via selective attack by the allylsilane onto the less encumbered face of N-acyliminium ion 244. Spirocycle 245 was converted into (-)-lepadiformine (15a) as had been done for the racemic compound (Cf. Schemes 40 and 41). Comparison of the optical rotation of the synthetic material with that of the natural product, along with comparison of the proton NMR spectra of their Mosher esters, proved the absolute configuration of the alkaloid to be as shown in structure 15a.
5.2.5 Kibayashi Synthesis of (-)-Lepadiformine
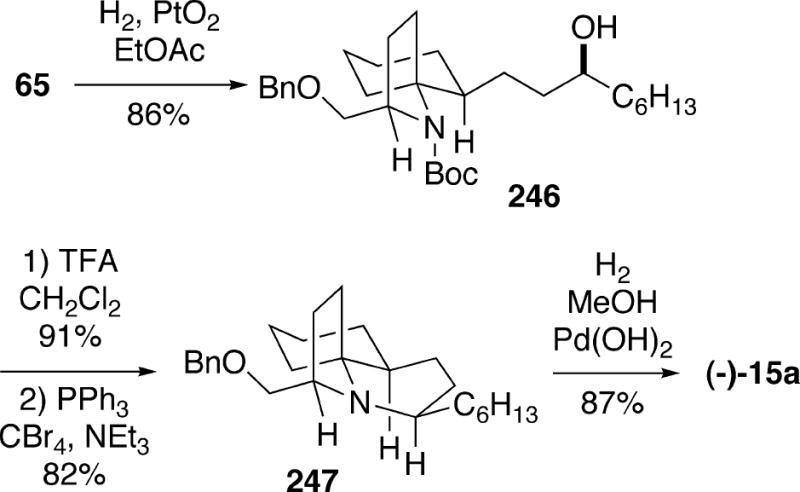
Kibayashi et al. described a total synthesis of (-)-lepadiformine which made use of a common intermediate also utilized in the syntheses of (+)-cylindricine C and (-)-fasicularin described above.19,37 Thus, enantiopure allylic alcohol 65 was hydrogenated to produce alcohol 246 (Scheme 43). Removal of the Boc group of 246 and cyclization of the resulting amino alcohol with inversion led to tricycle 247. Finally, hydrogenolysis of the benzyl group yielded (-)-lepadiformine (15a). This material was found to be indentical to the natural alkaloid by chiral HPLC.
Scheme 43.
5.2.6 Hsung Synthesis of (-)-Lepadiformine
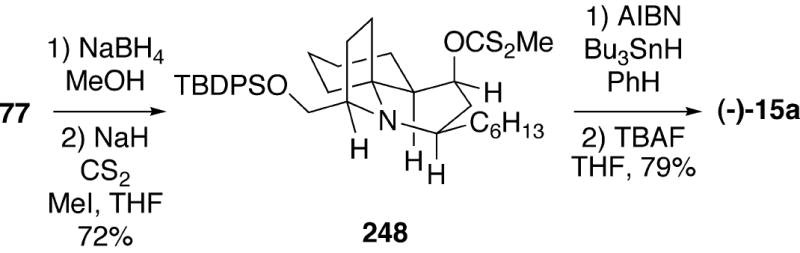
The Hsung group has used intermediate 77, prepared during their enantioselective total synthesis of (+)-cylindricine C, to complete a synthesis of (-)-lepadiformine.20 Sodium borohydride reduction of ketone 77 provided the alcohol which was converted into the corresponding xanthate 248 (Scheme 44). Barton-McCombie deoxygenation of 248, followed by removal of the silyl group then afforded (-)-lepadiformine (15a) in good yield.
Scheme 44.
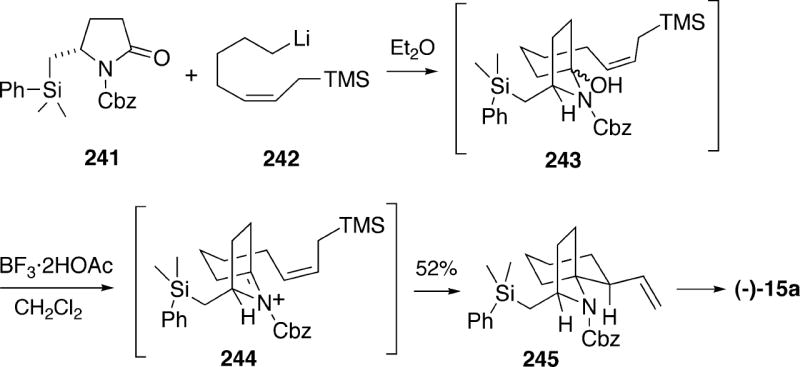
5.2.7 Kim Formal Synthesis of (-)-Lepadiformine
In 2006, Kim et al. reported a new approach to (-)-lepadiformine (15a) which uses an amino acid ester-enolate Claisen rearrangement and a ring closing metathesis as key steps.38 The synthesis made use of enantiomerically pure acid 249, which can be obtained in eight steps from (S)-pyroglutamic acid (Scheme 47). This acid was first converted to (E)-allylic ester 250, which exposed to lithium hexamethyldisilazide and t-butyldimethylsilyl chloride to generate the silylketene acetal 251. This intermediate undergoes a Claisen rearrangement predominantly through the conformation shown to produce an 8:1 mixture of the desired olefin acid 252 along with its diastereomer at C(5, 10). Alkene acid 252 was then esterified, followed by hydroboration with 9-BBN, and the resuting borane was coupled with vinyl bromide to afforded the homologated alkene 253. The ester functionality of 253 could be transformed into alkene 254 in three straightforward steps.
Scheme 47.
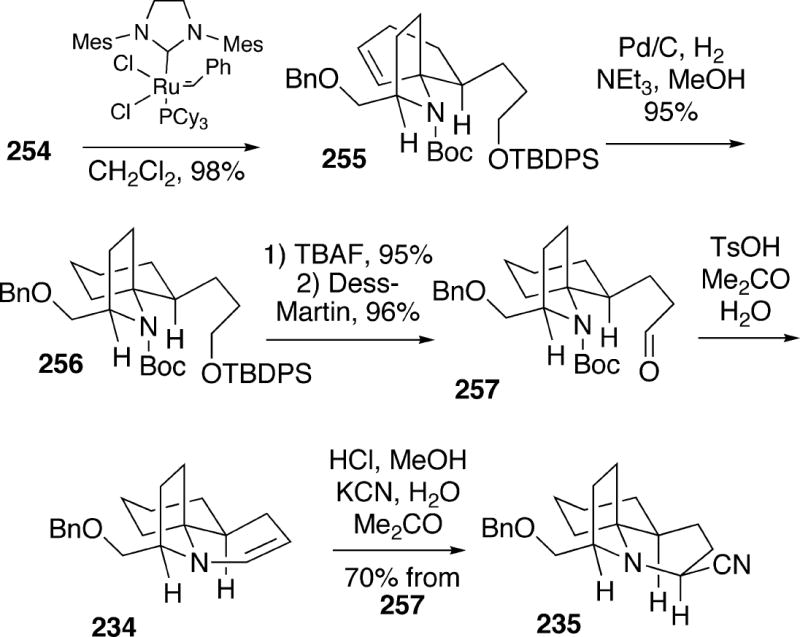
The carbocyclic A-ring of the alkaloid was then formed in high yield by a ring closing metathesis reaction of diene 254 using the second generation Grubbs catalyst, leading to azaspirocyclohexene 255 (Scheme 46). Catalytic hydrogenation of the double bond of 255 produced 256, and following desilylation, the resulting primary alcohol was oxidized to the corresponding aldehyde 257 using the Dess-Martin periodinane. Finally, treatment of 257 with p-toluenesulfonic acid in aqueous acetone led to removal of the Boc protecting group, and subsequent cyclization to the unstable tricyclic enamine 234, previously prepared by the Weinreb group in their lepadiformine synthesis (Cf. Scheme 41). This enamine was then converted in situ to the more stable amino nitrile 235, which has been converted to (-)-lepadiformine (15a).
Scheme 46.
6. Addendum
6.1 Hsung Synthesis of (-)-Cylindricine C
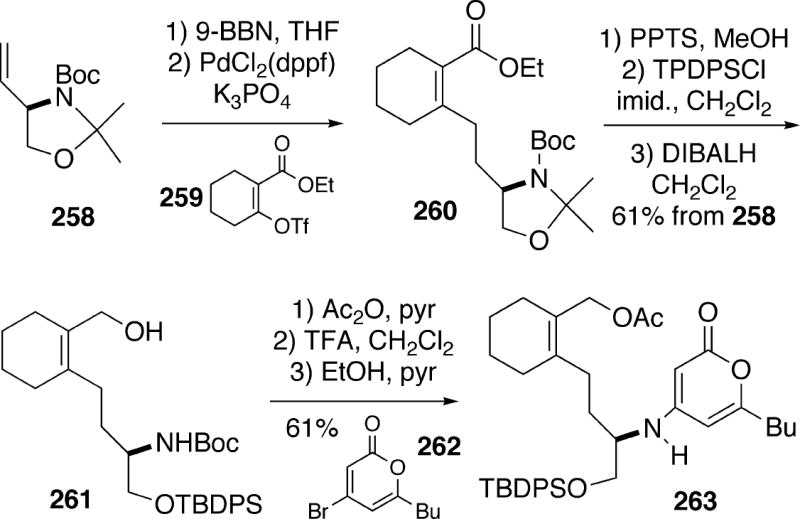
A recent paper from the Hsung group has described a new apporach to (-)-cylindricine C which utilizes a pivotal aza-[3+3]-cycloaddition strategy to access the B-ring of the alkaloid.39 This strategy is different from the one previously used for (+)-cylindricine C (Cf. Schemes 13,14). The source of chirality in this work was (R)-vinyl oxazoline 258, prepared in five steps from L-serine (Scheme 47). Hydroboration of 258 with 9-BBN, followed by a Suzuki-Miyaura coupling of the resulting borane with vinyl triflate 259 gave unsaturated ester 260. This intermediate was then converted in three straightforward operations into alcohol 261. The hydroxyl group of 261 was protected as the corresponding acetate, and the Boc group was subsequently removed. Coupling of the amine product with bromo-α-pyrone 262 led to amino pyrone 263.
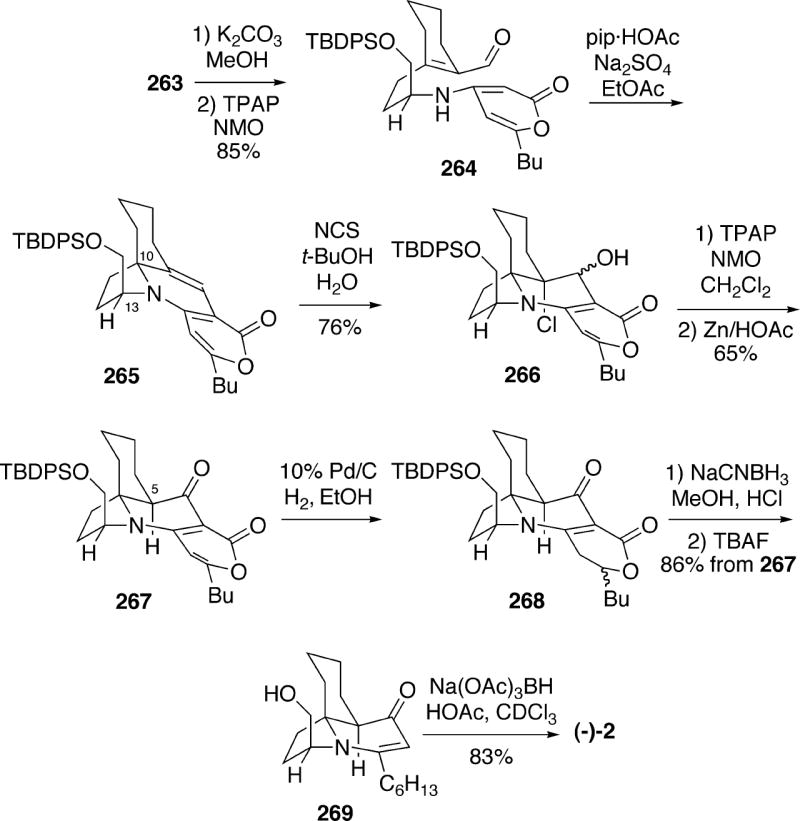
Compound 263 was then converted to aldehyde 264 (Scheme 48). The key step in the synthesis was the intramolecular cycloaddition of amino pyrone aldehyde 264, which could be effected by heating with piperidinium acetate as catalyst to afford tetracycle 265. This product was formed in 68% yield as a 9:1 mixture of stereoisomers, with the major isomer having the required configuration at C(10, 13) for the alkaloid. To continue the synthesis, pyrone 265 was treated with NCS in aqueous t-butanol to produce chlorohydrin 266 as a single stereoisomer (configuration not determined). Oxidation of alcohol 266 to the corresponding ketone, followed by reductive dechlorination led to ketone 267 having the necessary stereochemistry at C(5) for the cylindricines.. α-Pyrone ketone 267 could subsequently be partially reduced to afford dihydropyrone 268 as a 2:1 mixture of diastereomers. In an interesting transformation, reduction of intermediate 268 with sodium cyanoborohydride in the presence of either HCl or acetic acid led to a reductive decarboxylation product, which after removal of the silyl protecting group yielded vinylogous amide alcohol 269. Finally, it was found that reduction of this vinylogous amide with sodium triacetoxyborohydride produced (-)-cylindricine C (2) stereoselectively in good yield. It might be noted that catalytic hydrogenation of 269 with the Crabtree catalyst afforded C(2)-epicylindricine C.
Scheme 48.
6.2 Renaud Synthesis of (±)-Lepadiformine
Schar and Renaud have recently reported a new approach to racemic lepadiformine which makes use of an interesting free radical carboazidation strategy developed by this group.40 The synthesis began with cyclohexanone which was converted in a short, straightforward sequence into exo-methylene compound 270 (Scheme 49). Carboazidation of olefin 270 using the conditions shown in the scheme led to a 3:2 mixture of diastereomeric azido esters 271 and 272. These isomers could be separated here, or used as the mixture for the ensuing reactions. Catalytic hydrogenation of the desired stereoisomeric azide 272 produced the corresponding amino ketone, which under the reaction conditions underwent stereoselective intramolecular reductive amination yielding bicyclic amino ester 273. This intermediate could then be cyclized to tricyclic lactam 274 in 72% yield starting from the pure azide 272 (43% overall yield from the mixture of 271/272).
Scheme 49.
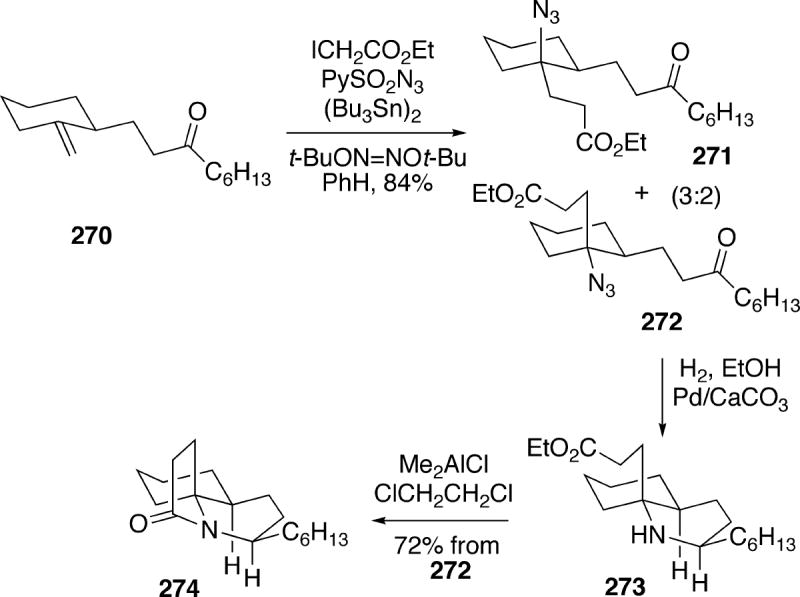
To complete the synthesis it was necessary to introduce C(14) and its attendant stereochemistry into the system. Therefore, the tricyclic lactam 274 was first converted to thiolactam 275 (Scheme 50). This compound was then S-methylated, followed by addition of lithium 2-phenylacetylide leading to adduct 276. Without isolation, this intermediate was exposed to lithium aluminum hydride, affording alkene 277 with high diastereoselectivity in 80% overall yield from thiolactam 275. Finally, ozonolysis of 277 under acidic comditions, and subsequent reduction of the resulting aldehyde led to racemic lepadiformine (15a). This synthesis requires 10 steps starting from cyclohexanone and proceeds in 15% overall yield.
Scheme 50.
Scheme 42.
Scheme 45.
Acknowledgments
I am grateful to the National Institutes of Health (CA-034303) for financial support of the research from the Weinreb lab described in this review.
Biography

References
- 1.(a) Blackman AJ, Li C, Hockless DCR, Skelton BW, White AH. Tetrahedron. 1993;49:8645. [Google Scholar]; (b) Li C, Blackman AJ. Aust J Chem. 1994;47:1355–1361. [Google Scholar]; (c) Li C, Blackman AJ. Aust J Chem. 1995;48:955. [Google Scholar]
- 2.Patil AD, Freyer AJ, Reichwein R, Carte B, Killmer LB, Faucette L, Johnson RK, Faulkner DJ. Tetrahedron Lett. 1997;38:363. [Google Scholar]
- 3.(a) Biard JF, Guyot S, Roussakis C, Verbist JF, Vercauteren J, Weber JF, Boukef K. Tetrahedron Lett. 1994;35:2691. [Google Scholar]; (b) Juge M, Grimaud N, Biard JF, Sauviat MP, Nabil M, Verbist JF, Petit JY. Toxicon. 2001;39:1231. doi: 10.1016/s0041-0101(01)00079-4. [DOI] [PubMed] [Google Scholar]; (c) Sauviat MP, Vercauteren J, Grimaud N, Juge M, Nabil M, Petit JY, Biard JF. J Nat Prod. 2006;69:000. doi: 10.1021/np050215s. [DOI] [PubMed] [Google Scholar]
- 4.(a) Werner KM, de los Santos JM, Weinreb SM, Shang M. J Org Chem. 1999;64:686. doi: 10.1021/jo981897+. [DOI] [PubMed] [Google Scholar]; (b) Werner KM, de los Santos JM, Weinreb SM, Shang M. J Org Chem. 1999;64:4865. doi: 10.1021/jo990266s. [DOI] [PubMed] [Google Scholar]
- 5.(a) Pearson WH, Barta NS, Kampf JW. Tetrahedron Lett. 1997;38:3369–3372. [Google Scholar]; (b) Pearson WH, Ren Y. J Org Chem. 1999;64:688. doi: 10.1021/jo982120j. [DOI] [PubMed] [Google Scholar]
- 6.(a) Abe H, Aoyagi S, Kibayashi C. Tetrahedron Lett. 2000;41:1205. [Google Scholar]; (b) Abe H, Aoyagi S, Kibayashi C. J Am Chem Soc. 2000;122:4583. doi: 10.1021/ja040213e. [DOI] [PubMed] [Google Scholar]; (c) Kibayashi C, Aoyagi S, Abe H. Bull Chem Soc Jpn. 2003;76:2059. [Google Scholar]
- 7.For an overview of this synthetic work see: Weinreb SM. Acc Chem Res. 2003;36:59. doi: 10.1021/ar0200403.
- 8.Dutta S, Abe H, Aoyagi S, Kibayashi C, Gates KS. J Am Chem Soc. 2005;127:15004. doi: 10.1021/ja053735i. [DOI] [PubMed] [Google Scholar]
- 9.Snider BB, Liu T. J Org Chem. 1997;62:5630. [Google Scholar]
- 10.Stella L. Angew Chem Int Ed. 1983;22:337. [Google Scholar]
- 11.Liu JF, Heathcock CH. J Org Chem. 1999;64:8263. doi: 10.1021/jo991020q. [DOI] [PubMed] [Google Scholar]
- 12.Brown JD, Foley MA, Comins DL. J Am Chem Soc. 1988;110:7445. [Google Scholar]; Comins DL, Dehghani A. J Org Chem. 1995;60:794. [Google Scholar]
- 13.Molander GA, Ronn M. J Org Chem. 1999;64:5183. doi: 10.1021/jo990363l. [DOI] [PubMed] [Google Scholar]
- 14.Borjesson L, Csoregh I, Welch CJ. J Org Chem. 1995;60:2989. [Google Scholar]
- 15.Trost BM, Rudd MT. Org Lett. 2003;5:4599. doi: 10.1021/ol035752n. [DOI] [PubMed] [Google Scholar]
- 16.Yeh MCP, Knochel P. Tetrahedron Lett. 1989;30:4799. [Google Scholar]
- 17.Trost BM, Rudd MT. J Am Chem Soc. 2003;125:11516. doi: 10.1021/ja036410f. [DOI] [PubMed] [Google Scholar]
- 18.Arai T, Abe H, Aoyagi S, Kibayashi C. Tetrahedron Lett. 2004;45:5921. [Google Scholar]
- 19.Abe H, Aoyagi S, Kibayashi C. J Am Chem Soc. 2005;127:1473. doi: 10.1021/ja040213e. [DOI] [PubMed] [Google Scholar]
- 20.(a) Liu J, Hsung RP, Peters SD. Org Lett. 2004;6:3989. doi: 10.1021/ol048353g. [DOI] [PubMed] [Google Scholar]; (b) Liu J, Swidorski JJ, Peters SD, Hsung RP. J Org Chem. 2005;70:3898–3902. doi: 10.1021/jo0501846. [DOI] [PubMed] [Google Scholar]
- 21.Wharton PS, Bohlen DH. J Org Chem. 1961;26:3615. [Google Scholar]
- 22.Canesi S, Bouchu D, Ciufolini MA. Angew Chem Int Ed. 2004;43:4336. doi: 10.1002/anie.200460178. [DOI] [PubMed] [Google Scholar]
- 23.Takahashi K, Ishiyama T, Miyaura N. J Organomet Chem. 2001;625:47. [Google Scholar]
- 24.Evans DA, Clark JS, Metternich R, Novack VJ, Sheppard GS. J Am Chem Soc. 1990;112:866. [Google Scholar]
- 25.Taniguchi T, Tamura O, Uchiyama M, Muraoka O, Tanabe G, Ishibashi H. Synlett. 2005:1179. doi: 10.1021/jo040264u. [DOI] [PubMed] [Google Scholar]
- 26.Hunter R, Richards P. Synlett. 2003:271. For a synthesis of a similar model fasicularin spirocycle see: Wardrop DJ, Zhang W, Landrie CL. Tetrahedron Lett. 2004;45:4229.
- 27.Maeng J-H, Funk RL. Org Lett. 2002;4:331. doi: 10.1021/ol016850g. For previous use of this methodology see: Maeng J-H, Funk RL. Org Lett. 2001;3:1125. doi: 10.1021/ol015506g.
- 28.(a) Fenster MDB, Dake GR. Org Lett. 2003;5:4313. doi: 10.1021/ol035566h. [DOI] [PubMed] [Google Scholar]; (b) Fenster MDB, Dake GR. Chem Eur J. 2005;11:639. doi: 10.1002/chem.200400749. [DOI] [PubMed] [Google Scholar]
- 29.(a) Dake GR, Fenster MDB, Hurley PB, Patrick BO. J Org Chem. 2004;69:5668. doi: 10.1021/jo0493572. [DOI] [PubMed] [Google Scholar]; (b) Dake GR, Fenster MDB, Fleury M, Patrick BO. J Org Chem. 2004;69:5676. doi: 10.1021/jo049356+. [DOI] [PubMed] [Google Scholar]
- 30.Herdeis C. Synthesis. 1986:233. [Google Scholar]
- 31.Haack K-J, Hashiguchi S, Fujii A, Ikariya T, Noyori R. Angew Chem Int Ed. 1997;36:285. [Google Scholar]; Matsumura K, Hashiguchi S, Ikariya T, Noyori R. J Am Chem Soc. 1997;119:8738. [Google Scholar]
- 32.(a) Bagley MC, Oppolzer W. Tetrahedron: Asymmetry. 2000;11:2625. [Google Scholar]; (b) Oppolzer W, Bochet CG. Tetrahedron: Asymmetry. 2000;11:4761. [Google Scholar]
- 33.Greshock TJ, Funk RL. Org Lett. 2001;3:3511. doi: 10.1021/ol0165903. [DOI] [PubMed] [Google Scholar]
- 34.(a) Sun P, Sun C, Weinreb SM. Org Lett. 2001;3:3507. doi: 10.1021/ol010179y. [DOI] [PubMed] [Google Scholar]; (b) Sun P, Sun C, Weinreb SM. J Org Chem. 2002;67:4337. doi: 10.1021/jo0201070. [DOI] [PubMed] [Google Scholar]
- 35.(a) Han G, McIntosh MC, Weinreb SM. Tetrahedron Lett. 1994;35:5813. [Google Scholar]; (b) Han G, LaPorte MG, McIntosh MC, Weinreb SM, Parvez M. J Org Chem. 1996;61:9483. [Google Scholar]; (c) Chao W, Weinreb SM. Tetrahedron Lett. 2000;41:9199. [Google Scholar]
- 36.Stevens RV. Acc Chem Res. 1984;17:289–296. [Google Scholar]; Polniaszek RP, Belmont SE. J Org Chem. 1990;55:4688. [Google Scholar]
- 37.Abe H, Aoyagi S, Kibayashi C. Angew Chem Int Ed. 2002;41:3017. doi: 10.1002/1521-3773(20020816)41:16<3017::AID-ANIE3017>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 38.Lee M, Lee T, Kim E-Y, Ko H, Kim D, Kim S. Org Lett. 2006;8:745. doi: 10.1021/ol053010j. [DOI] [PubMed] [Google Scholar]
- 39.Swidorski JJ, Wang J, Hsung RP. Org Lett. 2006;8:777. doi: 10.1021/ol053059p. [DOI] [PubMed] [Google Scholar]
- 40.Schar P, Renaud P. Org Lett. 2006;8 doi: 10.1021/ol060083+. ASAP. [DOI] [PubMed] [Google Scholar]