

Abstract
Phosphomevalonate kinase (PMK) catalyzes the cation dependent reaction of mevalonate 5-phosphate with ATP to form mevalonate 5-diphosphate and ADP, a key step in the mevalonate pathway for isoprenoid/sterol biosynthesis. Animal PMK proteins belong to the nucleoside monophosphate (NMP) kinase family. For many NMP kinases, multiple basic residues contribute to the neutralization of the negatively charged pentacoordinate phosphate reaction intermediate. Loss of basicity can result in catalytically impaired enzymes. Based on this precedent, conserved basic residues of human PMK have been mutated and purified forms of the mutated proteins have been kinetically and biophysically characterized. K48M and R73M mutants exhibit diminished Vmax values in both reaction directions (>1000-fold) with only slight Km perturbations (<10-fold). In both forward and reverse reactions, R110M exhibits a large (>10,000-fold) specific activity diminution. R111M exhibits substantially inflated Km values for mevalonate 5-phosphate and mevalonate 5-diphosphate (60 and 30-fold, respectively) as well as decreases (50-fold (forward) and 85-fold (reverse)) in Vmax. R84M also exhibits inflated Km values (50 and 33-fold for mevalonate 5-phosphate and mevalonate 5-diphosphate, respectively). The Ki values for R111M and R84M product inhibition by mevalonate 5-diphosphate are inflated by 45- and 63-fold; effects are comparable to the 30- and 38-fold inflations in Km for mevalonate 5-diphosphate. R141M exhibits little perturbation in Vmax (14-fold (forward) and 10-fold (reverse)) but has inflated Km values for ATP and ADP (48 and 136-fold, respectively). The Kd of ATP for R141M, determined by changes in tryptophan fluorescence, is inflated 27-fold compared to wt PMK. These data suggest that R110 is important to PMK catalysis, which is also influenced by K48 and R73. R111 and R84 contribute to binding of mevalonate 5-phosphate and R141 to binding of ATP.
Phosphomevalonate kinase (PMK1; EC 2.7.4.2) catalyzes the reversible ATP dependent phosphorylation of mevalonate 5-phosphate to produce mevalonate 5-diphosphate and ADP.
The reaction represents a key step in the mevalonic acid mediated biosynthesis of isopentenyl diphosphate and other polyisoprenoid metabolites. Disruption of the PMK encoding gene in yeast (1) has demonstrated the essentiality of this enzyme. Enzyme activity was documented in pig liver (2) and initial characterization work employed the purified porcine enzyme (3, 4). The sequence of human PMK has been deduced (5) and expression of a recombinant GST-human PMK fusion construct (6) produced protein that was used to test the site of feedback inhibition of the cholesterogenic pathway.
Analysis of the rapidly expanding genomic database indicates that animal PMKs and low homology invertebrate PMKs are encoded by genes that are nonorthologous to plant, fungal, and bacterial PMK genes (7). The recent discovery that the mevalonate pathway for isoprenoid biosynthesis is functionally critical in gram positive cocci (8) has been a prelude to expression of prokaryotic forms of the enzyme. For example, the enzymes from both E. faecalis (9) and S. pneumoniae (10) have been expressed, isolated, and partially characterized; the latter enzyme has been used for crystallization and elucidation of a protein structure (10). The fold of the nonorthologous bacterial protein confirms it to be a member of the GHMP kinase (Galactokinase/Homoserine kinase/Mevalonate kinase/Phosphomevalonate kinase) family.
No empirical structural information is available for the divergent animal PMK proteins. Animal/invertebrate PMKs have been predicted to be a member of the nucleoside monophosphate kinase family of phosphotransferases based on a large scale analysis of phosphotransferase protein sequences (7, 11, 12). A recombinant human protein has been successfully overexpressed as an N-terminal His6 tagged enzyme (13). The purified human enzyme, as well as several mutant proteins containing substitutions in the predicted Walker A P-loop, has been characterized. The results support the assignment of human PMK to the nucleoside monophosphate (NMP) kinase family. This hypothesis is also compatible with the results of a recently generated homology model of human PMK (13).
Basic residues have been shown to play an important role in substrate binding and catalysis for a variety of NMP kinases (14–17). Considering this precedent and the charged nature of PMK’s two phosphorylated substrates, basic residues appear to be attractive targets for any investigation into PMK function. This report describes the identification of invariant basic residues in known and putative animal/invertebrate PMK proteins. These basic residues have been conservatively mutated; the mutant proteins have been expressed and purified. The mutant PMKs have been characterized by kinetic and biophysical methods and compared to the wild-type enzyme. This account presents the results of those experiments, which provide insight into active site function of several of these conserved basic residues.
Materials and Methods
Materials
Deoxynucleotides and Pfu DNA polymerase used for mutagenesis were purchased from Stratagene. Primers used for mutagenesis were obtained from Integrated DNA Technologies. Plasmid DNA was propagated in E. coli JM109 cells (Promega). Reagents for plasmid DNA purification were purchased from Eppendorf (miniprep) and Qiagen (midiprep). DNA fragments were purified by agarose gel electrophoresis and isolated using a Qiaquick gel extraction kit (Qiagen). DNA sequencing was performed at the DNA Core Facility (University of Missouri - Columbia). For protein expression, E. coli BL21(DE3) cells were obtained from Novagen. Isopropyl-β-D-thiogalactopyranoside (IPTG) was purchased from Research Products International Corporation, Ni-Sepharose from GE Healthcare, and imidazole from Lancaster Synthesis Incorporated. 2′ (3′)-O-(2,4,6-Trinitrophenyl)adenosine-5′-triphosphate (TNP-ATP) was obtained from Molecular Probes. Chemicals, buffers, media components and antibiotics were purchased from Fisher Scientific. All other biochemical reagents and coupling enzymes were purchased from Sigma Chemical Company.
Syntheses of Mevalonate 5-Phosphate and Mevalonate 5-Diphosphate
The syntheses of mevalonate 5-phosphate and mevalonate 5-diphosphate have been previously reported (18, 19) and are briefly summarized. Methyl 3-hydroxy-3-methyl-5-iodopentanoate was synthesized by reacting mevalonolactone with trimethylsilyl iodide, followed by diazomethane derivatization to form the methyl ester (18). The product was subsequently purified by silica gel chromatography. Methyl 5-phosphomevalonate was synthesized by reacting the purified methyl 3-hydroxy-3-methyl-5-iodopentanoate with an excess of tetrabutylammonium phosphate (19). The methyl 5-phosphomevalonate was converted to the lithium salt by passage over a Dowex 50 column (lithium form). Deesterification was accomplished by alkaline hydrolysis in 0.5 N LiOH for 20 hours at 4°C. The resulting mevalonate 5-phosphate was purified by anion exchange chromatography using a DEAE-Sephadex A25 column. The chromatographically purified product was then analyzed and the concentration of the physiologically active R isomer was determined by enzymatic end point assay. Mevalonate 5-diphosphate was synthesized in a similar manner but tetrabutylammonium pyrophosphate was used as the phosphorylation reagent and deesterification was performed after DEAE chromatography(18).
Mutagenesis
A full circle PCR method, which employed a Stratagene QuikChange site-directed mutagenesis protocol, was used to generate the desired mutations. The presence of the mutation and the integrity of the remaining coding sequence were verified by DNA sequencing. Primer sequences used in the mutagenic reactions are:
| K48M forward: | 5′-CTCTCTGGTCCACTCATGGAACAGTATGCTCAG-3′, |
| K48M reverse: | 5′-CTGAGCATACTGTTCCATGAGTGGACCAGAGAG-3′; |
| K69M forward: | 5′-CAGCACCTACATGGAGGCCTTTC-3′, |
| K69M reverse: | 5′-GAAAGGCCTCCATGTAGGTGCTG-3′; |
| R73M forward: | 5′-TACAAGGAGGCCTTTATGAAGGACATGATCCGC-3′, |
| R73M reverse: | 5′-GCGGATCATGTCCTTCATAAAGGCCTCCTTGTA-3′; |
| R84M forward: | 5′-GAGAGGAGAAAATGCAGGCTGACCCAG-3′, |
| R84M reverse: | 5′-CTGGGTCAGCCTGCATTTTCTCCTCTC-3′; |
| R93M forward: | 5′-CCAGGCTTCTTTTGCATGAAGATTGTGGAGGGC-3′, |
| R93M reverse: | 5′-GCCCTCCACAATCTTCATGCAAAAGAAGCCTGG-3′; |
| R110M forward: | 5′ - CTGGTGAGTGACACAATGAGAGTGTCTGACATC-3′, |
| R110M reverse: | 5′ - GATGTCAGACACTCTCATTGTGTCACTCACCAG-3′; |
| R111M forward: | 5′ - GTGAGTGACACACGGATGGTGTCTGACATCCAG-3′, |
| R111M reverse: | 5′ - CTGGATGTCAGACACCATCCGTGTGTCACTCAC-3′; |
| R130M forward: | 5′ - GTGACGCAGACGGTCATGGTTGTAGCGTTGGAG-3′, |
| R130M reverse: | 5′ - CTCCAACGCTACAACCATGACCGTCTGCGTCAC-3′; |
| R138M forward: | 5′ - GCGTTGGAGCAGAGCATGCAGCAGCGGGGCTGG-3′, |
| R138M reverse: | 5′ - CCAGCCCCGCTGCTGCATGCTCTGCTCCAACGC-3′; |
| R141M forward: | 5′ - CAGAGCCGACAGCAGATGGGCTGGGTGTTCACG-3′, |
| R141M reverse: | 5′ - CGTGAACACCCAGCCCATCTGCTGTCGGCTCTG-3′. |
Protein Expression
Chemically competent E. coli BL21(DE3) Rosetta cells were transformed with the pET15b(+) plasmid that encodes either human WT or mutant phosphomevalonate kinase containing an N-terminal His6 affinity tag. In our initial report on this recombinant enzyme (13), we indicated that a C-terminal his tagged form of PMK was also constructed, expressed, and characterized. It is comparable in activity and Km values to the N-tagged enzyme, so no influence of the N-tag is apparent. The transformed cells were plated onto LB agar containing ampicillin (amp) and chloramphenicol (cam). These plates were incubated overnight at 37°C. 2 ml of LB-amp-cam was inoculated with a single colony and allowed to grow to moderate turbidity (A600 ~0.3). This culture was used to inoculate 10 LB-amp-cam plates (100μl/plate). The plates were incubated overnight at 37°C. The resulting lawns were used to inoculate 500 ml LB-amp-cam (A600 ~1.0). The liquid culture was incubated at 30°C for 1 hr prior to induction with 1 mM IPTG. The culture was harvested 4 hours post induction (A600 ~2.0) by centrifugation.
Enzyme Purification
Bacterial pellets were resuspended in 100 ml of buffer containing 50 mM KPi, 100 mM KCl, 5 mM imidazole, 0.5 mM DTT at pH 7.8. Lysis was accomplished by passage thru a microfluidizer at ~17 kpsi. The lysate was clarified by centrifugation at ~100,000 × g and the supernatant loaded onto ~0.5 – 1.0 ml of Ni-Sepharose Fast Flow resin. The column was washed with the lysis buffer until A280 < 0.005 and the protein was eluted using the lysis buffer supplemented with 300 mM imidazole. The fractions containing PMK were pooled and the concentration determined spectrophotometrically using an extinction coefficient (ε280 = 32 290 M−1cm−1) calculated from the deduced protein composition.
Steady-State Kinetics
For measurement of enzyme activity, a coupled enzyme assay was employed (20, 21). In the forward reaction, initial velocities were determined by coupling the production of ADP to the oxidation of NADH with pyruvate kinase/lactate dehydrogenase (6U/assay) and rate of decrease in absorbance at 340 nm monitored. In the reverse reaction, activity was determined by coupling the production of ATP to the reduction of NADP+ with hexokinase/glucose 6-phosphate dehydrogenase (3U/assay) and the rate of increase in absorbance at 340 nm was monitored. All assays were done at 30°C in the presence of 100 mM MOPS, 200 mM KCl, 1 mM DTT, 10 mM MgCl2 at pH 7.0. Spectrophotometric measurements were performed using a Lambda 35 spectrophotometer (Perkin Elmer). Catalytically impaired mutants were assayed fluorometrically using an excitation wavelength of 340 nm and monitoring the rate of change in NADH or NADPH fluorescence (emission wavelength of 460 nm) using a Photon Technologies International spectrofluorometer. The activity of WT PMK was also determined fluorometrically and was in good agreement with activity measured spectrophotometrically. All assays were started with the addition of mevalonate 5-phosphate in the forward reaction and mevalonate 5-diphosphate in the reverse reaction. For estimates of maximum velocity (Vmax) and Michaelis constant (Km), the reaction velocities at various substrate concentrations were fit to the Michaelis-Menten equation using the Kaleidagraph Graphing and Data Analysis Software (Synergy Software, Reading, PA). One unit of activity corresponds to one μmol of substrate converted to product in one minute.
TNP-ATP Binding to Wild-Type and Mutant PMK Proteins
The recombinant wild-type and mutant enzymes were tested for active site structural integrity using TNP-ATP, a fluorescent analogue of ATP. TNP-ATP was titrated into buffer alone or into buffer containing a fixed concentration of enzyme. The buffer used for these fluorescence measurements was 100 mM MOPS, 100 mM KCl, 1 mM DTT at pH 7.0. Excitation wavelength used in these experiments was 409 nm. Emission spectra were scanned from 500 – 600 nm, using a 5 nm slit width. TNP-ATP concentration was determined by absorbance at 409 nm using the extinction coefficient of 26 400 M−1 cm−1 (22). For data analysis, values measured at the fluorescent emission peak (533 to 535 nm) for bound probe were corrected for free TNP-ATP/buffer. Thus, these corrected fluorescence enhancement data were plotted against TNP-ATP concentrations and analyzed by nonlinear regression to yield the ligand concentration required for half saturation (S½) and an extrapolated maximum binding (Bmax) using the Kaleidagraph Graphing and Data Analysis Software (Synergy Software, Reading, PA). Binding stoichiometries were determined by Scatchard analysis.
Product Inhibition of WT and Mutant PMKs
For inhibition by mevalonate 5-diphosphate, initial velocities were determined using the spectrophotometric assay. The concentration of the variable substrate, mevalonate 5-phosphate, used in these assays ranged from 42 to 105 μM for WT, 204 to 1021 μM for R84M, and 95 to 1235 μM for R111M. The ATP concentration was kept constant and saturating. The concentration of the inhibitor, mevalonate 5-diphosphate, ranged from 70 to 150 μM for WT, 1270 to 3505 μM for R84M, and 731 to 2194 μM for R111M. These data were fit to a competitive inhibition model using Sigmaplot 10.0/Enzyme Kinetics Module 1.3 (Systat Software, Inc.).
Determination of an Equilibrium Dissociation Constant for ATP by Intrinsic Tryptophan Fluorescence
All fluorescence measurements were performed using a Photon Technologies International spectrofluorometer. Intrinsic tryptophan fluorescence of WT (0.20 – 0.33 μM) and R141M (0.28 – 0.66 μM) was measured at 25°C in 1.7 mL 100 mM Tris-Cl, 100 mM KCl, 1 mM DTT at pH 7.5. The concentration of MgCl2 in these assays was 20 and 31 mM for WT and R141M, respectively. Samples were excited at 295 nm and the emission was monitored between 310 – 450 nm. For data analysis, values measured at the fluorescent emission peak of ~333 nm were corrected for buffer background fluorescence and for dilution. Thus, these corrected fluorescence data were plotted against ATP concentrations, analyzed by nonlinear regression and fit to a 2-site model using GraphPad Prism version 4.00 for Windows (GraphPad Software, San Diego California USA, (www.graphpad.com). The Kd estimates reflect the average of three separate titrations, done at three different protein concentrations.
Results
Rationale for Functional Investigation of Conserved Basic Phosphomevalonate Kinase Residues that Contribute to Active Site Function
No empirical structural information for animal or invertebrate PMK is available. The crystal structure of the non-orthologous S. pneumoniae phosphomevalonate kinase has been determined (10) but offers little insight into animal/invertebrate PMKs. Several different groups have suggested that the animal/invertebrate enzyme belongs to the nucleoside monophosphate kinase family of enzymes(7, 11–13). Given this information and also these constraints, 13 protein sequences of known/putative animal/invertebrate PMKs were obtained from public databases. A sequence alignment was generated using the BioEdit program (23).
Based on this alignment, 14 basic residues (K17, R18, K19, K22, K48, K69, R73, R84, R93, R110, R111, R130, R138, and R141) were determined to be absolutely conserved (Figure 1.). Residues 17 – 19, and 22 are part of an atypical basic rich “Walker A” P-loop and have been previously mutated and functionally evaluated (13). For NMP kinase fold enzymes, there is strong precedent for important functional contributions from basic residues outside of the P-loop sequence (14, 16, 24–26) These observations suggested the utility of functional tests of the remaining invariant basic residues. The basic side chains of these residues have been replaced by conservative mutations to methionine; the mutant proteins have been expressed, and isolated in highly purified forms (Figure 2.)
Figure 1. Phosphomevalonate kinase sequences.

(A) The amino acid sequence of H. sapiens (human) phosphomevalonate kinase is shown and the residues numbered to correspond to the text. Conserved basic residues mutated in this study are shown in bold, blue letters. Conserved basic residues previously mutated and evaluated (17–19, 22) are shown in bold, red letters (13). (B) Sequence alignment of animal/invertebrate phosphomevalonate kinases. Conserved basic residues mutated in this study are shown in bold, blue letters. Conserved basic residues previously mutated and evaluated (17–19, 22) are shown in bold, red letters (13). All sequences were obtained from public databases. Alignment was generated using the BioEdit program (23). Sequences correspond to the following organisms and accession numbers: H. sapiens (human), Q15126; B. taurus (bovine), Q2KIU2; M. musculus (mouse), Q9D1G2; R. norvegicus (rat), NP_001008353; P. troglodytes (chimpanzee), XP_513842; C. familiaris (dog), XP_855082; M. mulatta (rhesus monkey), XP_001114509; D. rerio (zebra fish), XM_680509.1; X. laevis (African clawed frog), NP_001089752; C. elegans (flatworm), CAD66220; B. mori (domestic silkworm), BAF62110; D. melanogaster (fruit fly), AAF53833; A. gambiae (African malaria mosquito), XP_310779.
Figure 2. SDS-PAGE of wild-type and mutant human PMKs.
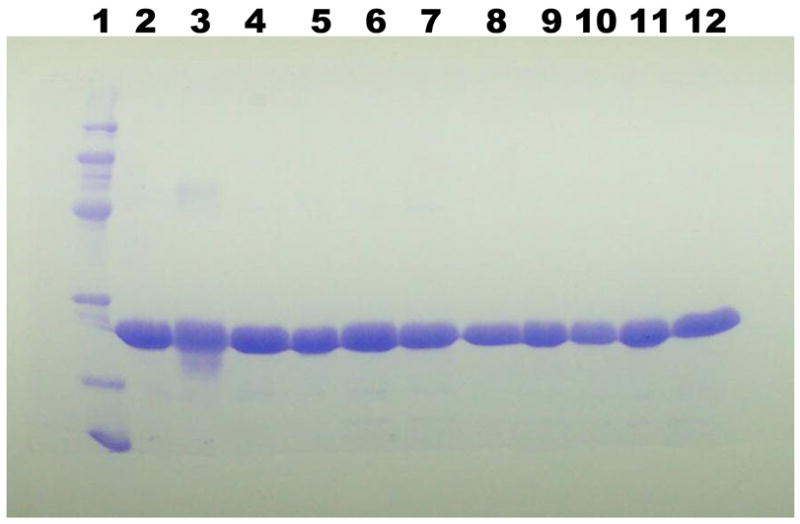
Lane 1: molecular mass markers (phosphorylase b, 97.4 kDa; bovine serum albumin, 66.2 kDa; ovalbumin, 45 kDa; carbonic anhydrase, 31 kDa; trypsin inhibitor, 21.5 kDa; lysozyme, 14.4 kDa. Lanes 2 – 12 contain 10 μg of human PMK proteins corresponding to wt, K48M, K69M, R73M, R84M, R93M, R110M, R111M, R130M, R138M, R141M, respectively.
Kinetic Characterization of Human Phosphomevalonate Kinase Mutants
Three of the ten mutant proteins (R93M, R130M, R138M) displayed no major change in either apparent Km (< 5-fold) or Vmax (< 10-fold) values for either the forward (Table 1) or reverse (Table 2) reactions. Substitution to eliminate the positively charged side chainresulted in a moderate to large decrease in catalysis (Vmax) for three of the mutants (K48M (forward: ~1300-fold; reverse: ~1200-fold), K69M (forward: ~450-fold; reverse: ~1100-fold), and R73M (forward: ~2600-fold; reverse: ~2300-fold). The perturbation in the observed Km for substrates in either forward or reverse reactions was minimal for the K48M, K69M, and R73M mutants (< 6-fold).
Table 1.
Steady-State Kinetic Constants of Wild-Type and Mutant PMKs Determined for the Forward Reaction. a
| Enzyme | Km(R-MVP),app (μM) | Vmax(R-MVP) (U/mg) | Km(ATP),app (μM) | Vmax(ATP) (U/mg) |
|---|---|---|---|---|
| WTb | 34±3 | 46.4±1.0 | 107±14 | 52.0±1.1 |
| K48M | 131±7 | 0.036±0.001 | 236±16 | 0.035±0.001 |
| K69M | 110±8 | 0.11±0.01 | 268±22 | 0.09±0.01 |
| R73M | 100±8 | 0.018±0.001 | 94±16 | 0.015±0.001 |
| R84M | 1710±49 | 5.89±0.04 | 303±14 | 3.91±0.08 |
| R93M | 48±3 | 33.1±0.8 | 101±6 | 32.4±0.5 |
| R110Mc | n.d | n.d | n.d | 0.0026±0.0007 |
| R111M | 2040±225 | 0.95±0.04 | 792±102 | 0.69±0.03 |
| R130M | 102±9 | 14.8±0.4 | 229±16 | 13.2±0.3 |
| R138M | 47±7 | 17.4±0.6 | 518±61 | 19.9±0.8 |
| R141Md | 225±23 | 3.2±0.1 | 5200±474 | 3.6±0.1 |
Spectrophotometric assays were performed in 100 mM MOPS, 200 mM KCl, 1 mM DTT (pH 7.0) in the presence of 10 mM MgCl2 at 30°C. Values were determined by fitting data to a Michaelis-Menten equation. Errors represent the standard error of the fit.
Herdendorf and Miziorko, Biochemistry 45 (2006)
Specific activity determination under standard conditions using a fluorescent assay using the same buffer conditions. Error represents the standard deviation of 5 measurements.
Assay contained 20 mM MgCl2. nd indicates not determined.
Table 2.
Steady-State Kinetic Constants of Wild-Type and Mutant PMKs Determined for the Reverse Reaction. a
| Enzyme | Km(R-MVPP),app (μM) | Vmax(R-MVPP) (U/mg) | Km(ADP),app (μM) | Vmax(ADP) (U/mg) |
|---|---|---|---|---|
| WTb | 41±3 | 11.3±0.2 | 47±5 | 12.0±0.2 |
| K48M | 7±1 | 0.0099±0.0001 | 60±6 | 0.0079±0.0002 |
| K69M | 44±3 | 0.0104±0.0002 | 106±10 | 0.009±0.001 |
| R73M | 41±7 | 0.0052±0.0003 | 118±20 | 0.0041±0.0002 |
| R84M | 1550±112 | 0.58±0.02 | 238±10 | 0.52±0.01 |
| R93M | 45±2 | 4.8±0.5 | 91±4 | 4.83±0.06 |
| R110Mc | nd | nd | nd | 0.00019±0.00005 |
| R111M | 1250±91 | 0.14±0.01 | 93±11 | 0.062±0.002 |
| R130M | 102±6 | 3.60±0.06 | 164±12 | 3.58±0.06 |
| R138M | 57±4 | 6.8±0.2 | 451±41 | 6.9±0.2 |
| R141Md | 139±7 | 0.95±0.02 | 5620±1260 | 1.2±0.1 |
Spectrophotometric assays were performed in 100 mM MOPS, 200 mM KCl, 1 mM DTT (pH 7.0) in the presence of 10 mM MgCl2 at 30°C. Values were determined by fitting data to a Michaelis-Menten equation. Errors represent the standard error of the fit.
Herdendorf and Miziorko, Biochemistry 45 (2006)
Specific activity determination under standard conditions using a fluorescent assay using the same buffer conditions. Error represents the standard deviation of 6 measurements.
Assay contained 20 mM MgCl2. nd indicates not determined.
Two of the PMK mutants (R84M and R111M) are clearly characterized by a substantial inflation in the apparent Km for the non-nucleotide substrates (mevalonate 5-phosphate:50-fold and 60-fold, respectively (Table 1); mevalonate 5-diphosphate: 38-fold and 30-fold, respectively (Table 2)). While the R84M protein exhibited only modest effects (decreases) in Vmax (~ 8-fold (forward reaction); ~ 21-fold (reverse reaction)), R111M was more catalytically impaired in both reaction directions (forward: ~53-fold; reverse: ~ 86-fold).
In contrast with those Km effects on mevalonate 5-phosphate and mevalonate 5-diphosphate, R141M is most clearly characterized by a significantly inflated apparent Kmfor the nucleotide substrates (forward (ATP): ~50-fold (Table 1); reverse (ADP): ~120-fold (Table 2)). There was little effect on Vmax for the R141M mutation (forward reaction: ~ 10-fold; reverse reaction: ~ 14-fold) or on Km (<7-fold) for non-nucleotide substrates. Estimates of Km values for the R110M mutant PMK were precluded by the diminished activity of this mutant. Use of a more sensitive spectrofluorometric assay provided aspecific activity estimate for R110M in forward and reverse reactions but not accurate measurements at sub-saturating substrate concentrations. A major diminution in the specific activity is observed in both forward (1.9 × 104-fold; Table 1) and reverse (6.3 × 104-fold; Table 2) reactions.
Evaluation of Active Site Integrity
In view of the large catalytic effect on R110M and the substantial Km,ATP effect on R141M, it seemed important to ensure that these introduced mutations have not significantly perturbed the overall structure of the enzymes. For this reason, the interaction of the fluorescent ATP analog, TNP-ATP, with WT, R110M and R141M proteins was compared. It has been previously shown that the TNP-ATP, an alternative substrate for PMK, binds efficiently to the WT protein, producing an enhanced fluorescence with a concomitant “blue shift” in the emission maximum (13). The TNP-ATP probe also binds to the most catalytically impaired mutant enzyme (R110M) and to the mutant PMK (R141M) that has the most significant perturbation in Km for the nucleotide substrates. Analysis of the data from titration of these proteins with TNP-ATP suggests that there are no major differences in the binding stoichiometries (0.8 – 1.2 per site) or in equilibrium binding constants (2.3 – 3.9 μM) between WT and these mutant proteins (Figure 3; Table 3). For these mutants that exhibit the largest contrast with wild type PMK in terms of kinetic parameters, the TNP-ATP data suggest that the ATP binding sites remain largely intact. Thus, these proteins exhibit no major structural perturbations that might complicate straightforward interpretation of kinetic characterization results.
Figure 3. Scatchard analysis of TNP-ATP binding to wild-type and mutant human PMKs.
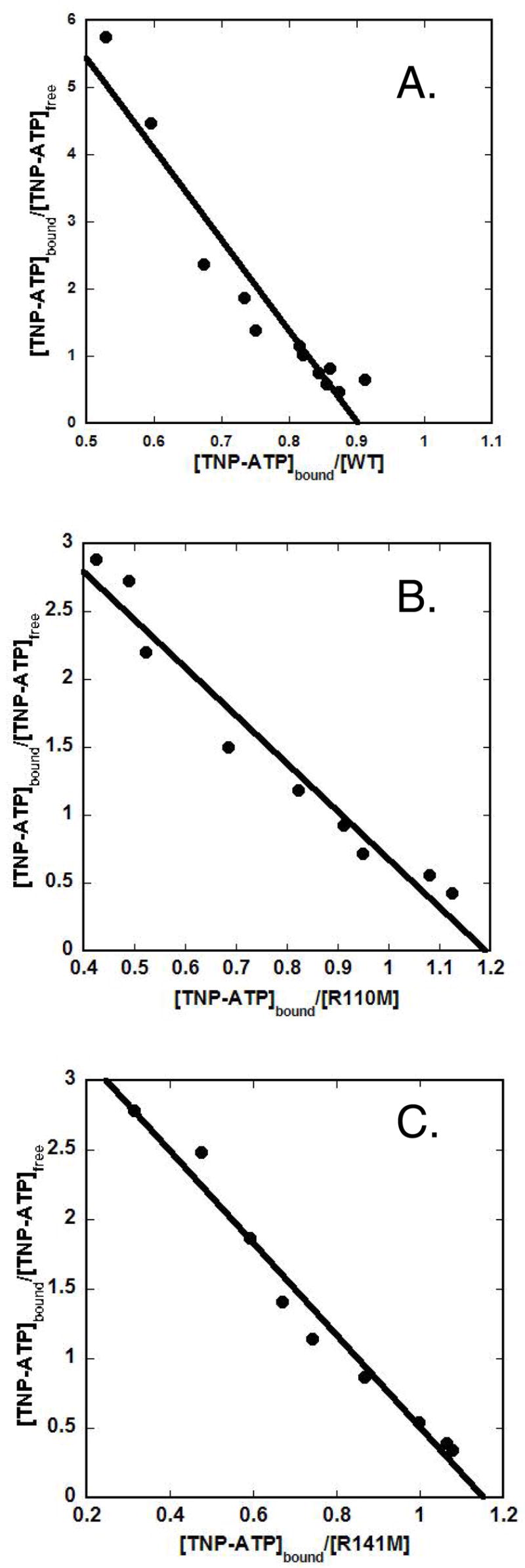
Panel (A) displays the Scatchard plot for wild-type PMK. Panel (B) displays the Scatchard plot for R110M. Panel (C) displays the Scatchard plot for R141M. Binding measurements were performed under the conditions described in the Methods and in Table 3.
Table 3.
Comparison of TNP-ATP Binding to Wild-Type and Mutant PMKs a
Fluorescent titrations were performed in 100 mM MOPS, 100 mM KCl, 1 mM DTT (pH 7.0) at 25°C.
S½ ([ligand] at half maximal binding) values were estimated by fitting the titration data to the nonlinear regression equation y = Bmax[X]/(S½ + [X]). Errors represent the standard error of the fit.
The n value (binding stoichiometry) was estimated by Scatchard analysis.
Product inhibition of Wild Type and Mutant PMKs
To provide a test of whether the inflation in the apparent Km values for mevalonate 5-diphosphate (Table 2) measured using R84M (38-fold) or R111M (30-fold) mutant proteins is primarily due to weakened binding of the non-nucleotide substrates, mevalonate 5-diphosphate inhibition constants were determined for the wild type enzyme and these mutant PMKs. The data obtained from inhibition of the forward reaction by the product, mevalonate 5-diphosphate, werefit to a competitive inhibition pattern (Figure 4), so Ki comparisons should primarily reflect differences in binding affinity. The inhibition constants for WT, R84M and R111M were estimated to be 18 μM, 1140 μM, and 806 μM, respectively (Table 4). The inflation in zthe inhibition constants for R84M and R111M, when compared to wild type PMK, is 63-fold and 45-fold, respectively. These data correlate reasonably well with the changes in Km for mevalonate 5-diphosphate. The combined observations stronglysuggest that R84M and R111M significantly contribute to the binding of PMK’s non-nucleotide substrates.
Figure 4. Product inhibition of wild-type and mutant human PMKs.
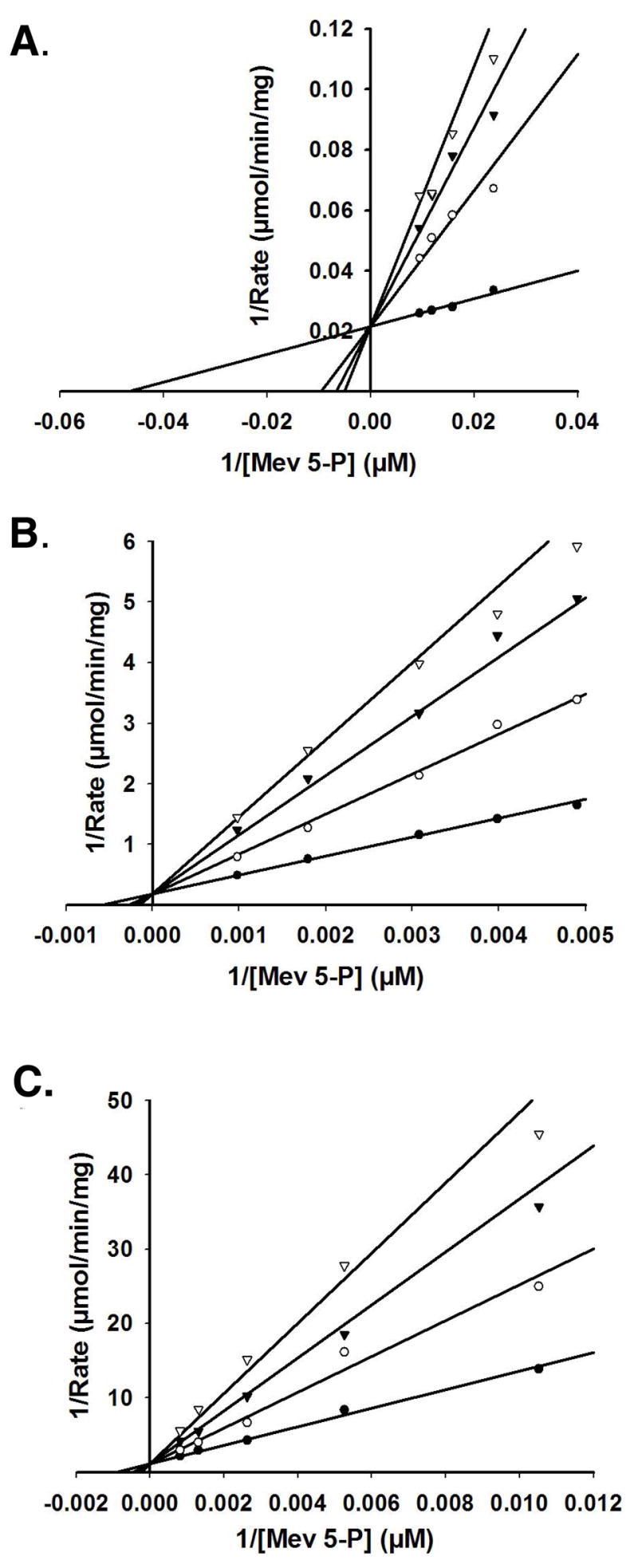
Double-reciprocal plots of the initial velocity for PMK formation of ADP as a function of mevalonate 5-phosphate concentration, measured at different levels of the product inhibitor mevalonate 5-diphosphate. Data were fit to a competitive inhibition model using SigmaPlot 10.0/Enzyme Kinetics 1.3. (Systat Software, Inc.). Panel (A) displays the product inhibition for wild-type PMK. Mevalonate 5-diphosphate concentrations utilized were (●) 0.0 mM, (○) 0.07 mM, (▼) 0.11 mM, (▽) 0.15 mM. Panel (B) displays the product inhibition for R84M PMK. Mevalonate 5-diphosphate concentrations utilized were (●) 0.0 mM, (○) 1.27 mM, (▼) 2.44 mM, (▽) 3.50 mM. Panel (C) displays the product inhibition for R111M PMK. Mevalonate 5-diphosphate concentrations utilized were (●) 0.0 mM, (○) 0.73 mM, (▼) 1.46 mM, (▽) 2.19 mM.
Table 4.
Comparison of Michaelis Constants and Product Inhibition Constants of Mevalonate diphosphate for Wild-Type and Mutant PMKs. a
| Enzyme | Km(R-MVPP) (μM) | Fold Inflation | Ki(R-MVPP)b (μM) | Fold Inflation |
|---|---|---|---|---|
| WT | 41±3 | - | 18±1 | - |
| R84M | 1550±112 | 38 | 1140±51 | 63 |
| R111M | 1250±91 | 30 | 806±85 | 45 |
Spectrophotometric assays were performed in 100 mM MOPS, 200 mM KCl, 1 mM DTT (pH 7.0) in the presence of 10 mM MgCl2 at 30°C. [ATP] was kept saturating at 5 mM. Errors represent the standard error of the fit.
[R-Mev 5-PP] ranged from 1 – 5 times Ki values. Ki values were estimated by fitting the data to a competitive inhibition model using the equation velocity = Vmax[X]/(Km(1+[I]/Ki) + [X]).
Determination of an Equilibrium Dissociation Constant for ATP by Intrinsic Tryptophan Fluorescence
For the R141M mutant, apparent Km values for the nucleotide substrates are inflated by ~ 50-fold (ATP) and ~ 120-fold (ADP) (Table 1,2). To test whether thisinflation in the apparent Km values reflects weakened binding of the nucleotide substrate, an independent estimate of nucleotide binding was pursued. Human PMKcontains 5 tryptophan residues (W79, W104, W117, W143, W166) (Figure 1). Two of these tryptophan residues, based on an extended protein sequence alignment of many phosphotransferases (12), lie in regions expected to interact with the nucleotide substrate. W104 is four residues upstream of the predicted Walker B carboxylate (D108) that would be expected to interact with the divalent cation of the metal·ATP complex. W143 is two residues downstream from the mutated residue (R141). R141 aligns with residues from several NMP kinases known to be in the “lid” domain. A conserved arginine in this region of the NMP kinase “lid” has been implicated in formation of a critical interface with the adenine moiety of ATP in thymidylate, shikimate, gluconate, and dephospho-coenzyme A kinases(27–30). On this basis, it seemed reasonable to test whether ATP influences PMK tryptophan fluorescence. For both wild type PMK and R141M PMK (which exhibits a substantial inflation of Km for nucleotide substrates, tryptophan fluorescence data were measured as a function of ATP concentration (Fig. 5). The data were best fit to a two-site binding equation to estimate the ATP equilibrium dissociation constants for WT and R141M PMKs. The dissociation constants for the first (tight) site for WT and R141M were estimated to be 30 μM and 816 μM, respectively (Table 5). When compared to corresponding parameters for wild type PMK, the inflation in Kd1 (~27-fold) correlates reasonably well the inflation in the apparent Km,ATP (~49-fold). These observations suggest that R141 plays a role in the binding of the nucleotide substrates. Second (weak) site dissociation constants for WT and R141M PMK proteins were estimated to be 14 mM and 41 mM, respectively. These second site binding constants are well above the physiological concentration of ATP and were not considered for further analysis.
Figure 5. Tryptophan fluorescence quenching of WT and R141M PMK by ATP.
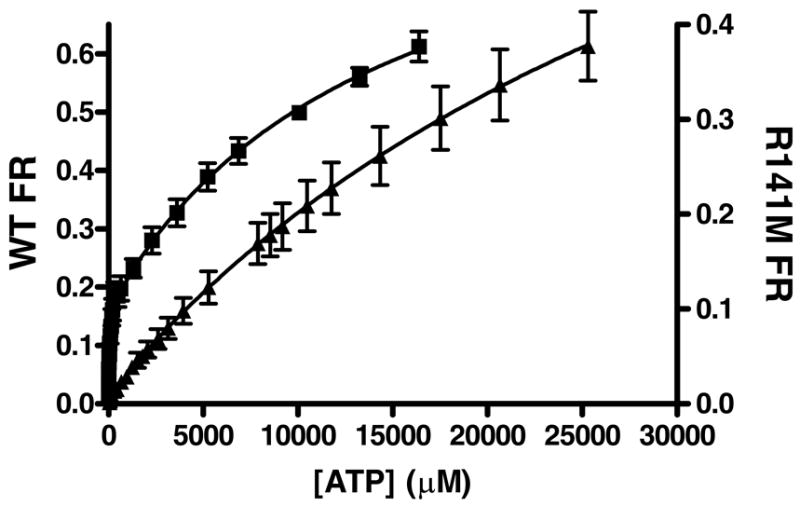
The average fractional response of 3 WT(■) concentrations (0.20 – 0.33 μM) and 3 R141M (▲) concentrations (0.28 – 0.66 μM) is plotted as a function of ATP concentration. All measurements were done at 25°C in 1.7 mL 100 mM Tris-Cl, 100 mM KCl, 1 mM DTT at pH 7.5. The concentration of MgCl2 in these assays was 20 and 31 mM for WT and R141M, respectively. Samples were excited at 295 nm and the emission was monitored between 310 – 450 nm. For data analysis, values measured at the fluorescent emission peak of ~333 nm were corrected for buffer background fluorescence and for dilution. These data were analyzed by nonlinear regression and fit to a 2-site model [y = Bmax1[X]/(Kd1 + [X]) + Bmax2[X]/(Kd2 + [X])] using GraphPad Prism version 4.00 for Windows (GraphPad Software, San Diego California USA, (www.graphpad.com). Error bars represent the standard deviation of the three different protein concentrations.
Table 5.
Comparison of Michaelis Constants and Equilibrium Binding Constants for ATP to Wild-Type and Mutant PMKs. a
| Enzyme | Km(ATP) (μM) | Fold Inflation | Kd(ATP) (μM) c | Fold Inflation |
|---|---|---|---|---|
| WT | 107±14 | - | 30±3 | - |
| R141M | 5200±474b | 49 | 816±160 | 27 |
Spectrophotometric assays were performed in 100 mM MOPS, 200 mM KCl, 1 mM DTT (pH 7.0) in the presence of 10 mM MgCl2 at 30°C. [ATP] was kept saturating (5 mM). Errors represent the standard error of the fit.
Assays contained 20 mM MgCl2.
Determined by tryptophan fluorescence. Averaged data points from 3 [protein] were fit to a two site model using the equation y = Bmax1[X]/(Kd1 + [X]) + Bmax2[X]/(Kd2 + [X]).
Kd2 values (WT: 14 mM, R141M: 41 mM) are too weak to be physiologically relevant and are not considered in the comparison. Errors represent the standard error of the fit.
Discussion
Nucleoside monophosphate kinase family proteins include numerous examples of phosphoryl transfer between two nucleotides (e.g. dexoynucleotide kinase (31), adenylate kinase (14–17), thymidylate kinase (30), etc.) or between a nucleotide and the alcohol group of a non-nucleotide acceptor metabolite (e.g., fructose 6-phosphate 2-kinase (24), phosphoribulokinase (25), shikimate kinase (28), gluconate kinase (27)). Phoshomevalonate kinase is unusual in this family of proteins in that it catalyzes a phosphoryl transfer from a nucleotide donor (ATP) to the phosphate of a non-nucleotide metabolite acceptor (mevalonate 5-phosphate). Several limitations to analysis of the protein include the lack of a structure of the animal/invertebrate enzyme as well as the divergence of yeast and lower PMK proteins from the NMP kinase family fold. Nevertheless, the reported sequences of animal and invertebrate PMK proteins have recently grown to an appreciable number and it has been possible to identify a manageable number of conserved basic residues and to test their potential function. Fortunately, when charged side chains of basic residues were eliminated by a methionine substitution, the resulting mutant proteins could be expressed and isolated in a fashion comparable to wild type enzyme, allowing pursuit of the functional analyses reported in Results. Of six mutants (K48M, R73M, R84M, R110M, R111M, R141M) which exhibit substantial diminution in kcat or inflation in substrate Km (Tables 1, 2), reasonable functional explanations are possible for most of these and are summarized below.
In the case of PMK’s R110, this residue is only two residues downstream of the putative “Walker B” carboxyl (IWLVSD108; (12)) that typically binds cation of the M2+-ATP substrate. Deoxynocleotide kinase (31) also exhibits such a motif but the functional test of the comparable arginines has only been reported in this work on PMK. Ostermann et al. (30) have proposed that, for thymidylate kinase, a basic residue near a Walker B motif bridges phosphoryl donor and acceptor substrates. In such a case, the basic side chain (e.g. PMK R110’s guanidinium group) could be critical in affecting the proximity and alignment of substrates, so the large kcat effect (~104-fold decrease in specific activity) would be compatible with such a role in catalysis.
R111 in PMK is also close to the “Walker B” motif and, since phosphoryl acceptor must be situated in close proximity to allow efficient in-line phosphoryl transfer from the donor to occur, it is reasonable that mevalonate 5-phosphate’s Km (Table 1) or mevalonate 5-diphosphate’s Km (Table 2) would be affected if R111’s guanidinium group is substituted. Similar inflations of Km for the phosphoryl acceptor in the NMP kinase family enzymes phosphoribulokinase (32) and fructose 6-phosphate-2-kinase (26) occur upon arginine mutagenesis. Thus, the prediction that R111 supports binding of PMK’s non-nucleotide substrate seems reasonable, especially since the product inhibition data (Table 4) for R111M confirm the inflation observed in Km for mevalonate 5-phosphate and mevalonate 5-diphosphate.
In considering residues which are not so easily associated with established sequence motifs and reinforced by some defined regions in a PMK homology model (13), reliance on large scale phosphotransferase sequence alignments can provide some insight. For example, PMK R141 falls into the RX(2–3)R sequence commonly observed in the “lid” region of NMP kinases. Basic residues in this sequence have been proposed to interact with the substrate adenine moiety (12). PMK’s R141 could function in such a capacity. This would explain R141M’s dominant effects on apparent nucleotide affinity, with contrasting observations of large Km effects for ATP and ADP (~50–100-fold) but moremodest effects on Km for non-nucleotide substrate.
Finally, in considering PMK’s K48M, which exhibits a significant (>103 -fold) effect on kcat but little effect on Km of any substrate, it may be useful to note the homology to CMP kinase’s R41 (12). R41 in CMK kinase (33) hydrogen bonds to the alpha phosphoryl of CMP, which is the acceptor substrate in the CMP kinase reaction. If PMK’s K48 should support a similar function, substitution of its charged side chain (e.g. K48M) could disrupt PMK’s ability to precisely orient the acceptor with respect to the donor gamma phosphoryl of ATP. For such a function, a kcat effect of the magnitude observed for K48M would be quite reasonable.
Footnotes
This work was supported in part by NIDDK
Abbreviations used are: PMK, phosphomevalonate kinase; MVP, mevalonate 5-phosphate; MVPP, mevalonate 5-diphosphate; TNP-ATP, 2′ (3′)-O-(2,4,6-Trinitrophenyl)adenosine-5′-triphosphate; NMP, nucleoside monophosphate.
References
- 1.Tsay YH, Robinson GW. Cloning and characterization of ERG8, an essential gene of Saccharomyces cerevisiae that encodes phosphomevalonate kinase. Mol Cell Biol. 1991;11:620–631. doi: 10.1128/mcb.11.2.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hellig H, Popjak G. Studies on the biosynthesis of cholesterol: XIII. phosphomevalonic kinase from liver. J Lipid Res. 1961;2:235–243. [Google Scholar]
- 3.Bazaes S, Beytia E, Jabalquinto AM, Solis de Ovando F, Gomez I, Eyzaguirre J. Pig liver phosphomevalone kinase. 1. Purification and properties. Biochemistry. 1980;19:2300–2304. doi: 10.1021/bi00552a003. [DOI] [PubMed] [Google Scholar]
- 4.Lee CS, O’Sullivan WJ. Improved procedures for the synthesis of phosphomevalonate and for the assay and purification of pig liver phosphomevalonate kinase. Biochim Biophys Acta. 1985;839:83–89. doi: 10.1016/0304-4165(85)90184-9. [DOI] [PubMed] [Google Scholar]
- 5.Chambliss KL, Slaughter CA, Schreiner R, Hoffmann GF, Gibson KM. Molecular cloning of human phosphomevalonate kinase and identification of a consensus peroxisomal targeting sequence. J Biol Chem. 1996;271:17330–17334. doi: 10.1074/jbc.271.29.17330. [DOI] [PubMed] [Google Scholar]
- 6.Hinson DD, Chambliss KL, Toth MJ, Tanaka RD, Gibson KM. Post-translational regulation of mevalonate kinase by intermediates of the cholesterol and nonsterol isoprene biosynthetic pathways. J Lipid Res. 1997;38:2216–2223. [PubMed] [Google Scholar]
- 7.Smit A, Mushegian A. Biosynthesis of isoprenoids via mevalonate in Archaea: the lost pathway. Genome Res. 2000;10:1468–1484. doi: 10.1101/gr.145600. [DOI] [PubMed] [Google Scholar]
- 8.Wilding EI, Brown JR, Bryant AP, Chalker AF, Holmes DJ, Ingraham KA, Iordanescu S, So CY, Rosenberg M, Gwynn MN. Identification, evolution, and essentiality of the mevalonate pathway for isopentenyl diphosphate biosynthesis in gram-positive cocci. J Bacteriol. 2000;182:4319–4327. doi: 10.1128/jb.182.15.4319-4327.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doun SS, Burgner JW, 2nd, Briggs SD, Rodwell VW. Enterococcus faecalis phosphomevalonate kinase. Protein Sci. 2005;14:1134–1139. doi: 10.1110/ps.041210405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romanowski MJ, Bonanno JB, Burley SK. Crystal structure of the Streptococcus pneumoniae phosphomevalonate kinase, a member of the GHMP kinase superfamily. Proteins. 2002;47:568–571. doi: 10.1002/prot.10118. [DOI] [PubMed] [Google Scholar]
- 11.Houten SM, Waterham HR. Nonorthologous gene displacement of phosphomevalonate kinase. Mol Genet Metab. 2001;72:273–276. doi: 10.1006/mgme.2000.3133. [DOI] [PubMed] [Google Scholar]
- 12.Leipe DD, Koonin EV, Aravind L. Evolution and classification of P-loop kinases and related proteins. J Mol Biol. 2003;333:781–815. doi: 10.1016/j.jmb.2003.08.040. [DOI] [PubMed] [Google Scholar]
- 13.Herdendorf TJ, Miziorko HM. Phosphomevalonate kinase: functional investigation of the recombinant human enzyme. Biochemistry. 2006;45:3235–3242. doi: 10.1021/bi052231u. [DOI] [PubMed] [Google Scholar]
- 14.Dahnke T, Shi Z, Yan H, Jiang RT, Tsai MD. Mechanism of adenylate kinase. Structural and functional roles of the conserved arginine-97 and arginine-132. Biochemistry. 1992;31:6318–6328. doi: 10.1021/bi00142a022. [DOI] [PubMed] [Google Scholar]
- 15.Tian GC, Yan HG, Jiang RT, Kishi F, Nakazawa A, Tsai MD. Mechanism of adenylate kinase. Are the essential lysines essential? Biochemistry. 1990;29:4296–4304. doi: 10.1021/bi00470a006. [DOI] [PubMed] [Google Scholar]
- 16.Yan HG, Dahnke T, Zhou BB, Nakazawa A, Tsai MD. Mechanism of adenylate kinase. Critical evaluation of the X-ray model and assignment of the AMP site. Biochemistry. 1990;29:10956–10964. doi: 10.1021/bi00501a013. [DOI] [PubMed] [Google Scholar]
- 17.Yan HG, Shi ZT, Tsai MD. Mechanism of adenylate kinase. Structural and functional demonstration of arginine-138 as a key catalytic residue that cannot be replaced by lysine. Biochemistry. 1990;29:6385–6392. doi: 10.1021/bi00479a007. [DOI] [PubMed] [Google Scholar]
- 18.Reardon JE, Abeles RH. Inhibition of cholesterol biosynthesis by fluorinated mevalonate analogues. Biochemistry. 1987;26:4717–4722. doi: 10.1021/bi00389a018. [DOI] [PubMed] [Google Scholar]
- 19.Wang CZ, Miziorko HM. Methodology for synthesis and isolation of 5-phosphomevalonic acid. Anal Biochem. 2003;321:272–275. doi: 10.1016/s0003-2697(03)00435-4. [DOI] [PubMed] [Google Scholar]
- 20.Liu F, Dong Q, Myers AM, Fromm HJ. Expression of human brain hexokinase in Escherichia coli: purification and characterization of the expressed enzyme. Biochem Biophys Res Commun. 1991;177:305–311. doi: 10.1016/0006-291x(91)91983-j. [DOI] [PubMed] [Google Scholar]
- 21.Tchen TT. Enzymes in Sterol Biogenesis. Methods Enzymol. 1962;5:489–499. [Google Scholar]
- 22.Hiratsuka T, Uchida K. Preparation and properties of 2′ (or 3′)-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate, an analog of adenosine triphosphate. Biochim Biophys Acta. 1973;320:635–647. doi: 10.1016/0304-4165(73)90143-8. [DOI] [PubMed] [Google Scholar]
- 23.Hall TA. BioEdit: A user-friendly biological sequence alignment editor anda analysis program for Window 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- 24.Li L, Lin K, Pilkis J, Correia JJ, Pilkis SJ. Hepatic 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase. The role of surface loop basic residues in substrate binding to the fructose-2,6-bisphosphatase domain. J Biol Chem. 1992;267:21588–21594. [PubMed] [Google Scholar]
- 25.Runquist JA, Harrison DH, Miziorko HM. Rhodobacter sphaeroides phosphoribulokinase: identification of lysine-165 as a catalytic residue and evaluation of the contributions of invariant basic amino acids to ribulose 5-phosphate binding. Biochemistry. 1999;38:13999–14005. doi: 10.1021/bi9910326. [DOI] [PubMed] [Google Scholar]
- 26.Tsujikawa T, Watanabe F, Uyeda K. Hexose phosphate binding sites of fructose 6-phosphate,2-kinase:fructose 2,6-bisphosphatase. Biochemistry. 1995;34:6389–6393. doi: 10.1021/bi00019a018. [DOI] [PubMed] [Google Scholar]
- 27.Kraft L, Sprenger GA, Lindqvist Y. Conformational changes during the catalytic cycle of gluconate kinase as revealed by X-ray crystallography. J Mol Biol. 2002;318:1057–1069. doi: 10.1016/S0022-2836(02)00215-2. [DOI] [PubMed] [Google Scholar]
- 28.Krell T, Coggins JR, Lapthorn AJ. The three-dimensional structure of shikimate kinase. J Mol Biol. 1998;278:983–997. doi: 10.1006/jmbi.1998.1755. [DOI] [PubMed] [Google Scholar]
- 29.Obmolova G, Teplyakov A, Bonander N, Eisenstein E, Howard AJ, Gilliland GL. Crystal structure of dephospho-coenzyme A kinase from Haemophilus influenzae. J Struct Biol. 2001;136:119–125. doi: 10.1006/jsbi.2001.4428. [DOI] [PubMed] [Google Scholar]
- 30.Ostermann N, Schlichting I, Brundiers R, Konrad M, Reinstein J, Veit T, Goody RS, Lavie A. Insights into the phosphoryltransfer mechanism of human thymidylate kinase gained from crystal structures of enzyme complexes along the reaction coordinate. Structure. 2000;8:629–642. doi: 10.1016/s0969-2126(00)00149-0. [DOI] [PubMed] [Google Scholar]
- 31.Teplyakov A, Sebastiao P, Obmolova G, Perrakis A, Brush GS, Bessman MJ, Wilson KS. Crystal structure of bacteriophage T4 deoxynucleotide kinase with its substrates dGMP and ATP. Embo J. 1996;15:3487–3497. [PMC free article] [PubMed] [Google Scholar]
- 32.Sandbaken MG, Runquist JA, Barbieri JT, Miziorko HM. Identification of the phosphoribulokinase sugar phosphate binding domain. Biochemistry. 1992;31:3715–3719. doi: 10.1021/bi00129a022. [DOI] [PubMed] [Google Scholar]
- 33.Bertrand T, Briozzo P, Assairi L, Ofiteru A, Bucurenci N, Munier-Lehmann H, Golinelli-Pimpaneau B, Barzu O, Gilles AM. Sugar specificity of bacterial CMP kinases as revealed by crystal structures and mutagenesis of Escherichia coli enzyme. J Mol Biol. 2002;315:1099–1110. doi: 10.1006/jmbi.2001.5286. [DOI] [PubMed] [Google Scholar]