

Abstract
4-Hydroxynonenal (4-HNE), formed as a consequence of oxidative stress, exists at increased concentrations in Alzheimer's disease (AD) patients and is found in amyloid β peptide (Aβ) plaques associated with AD. While it remains an open question whether oxidative stress is a causative factor or a consequence of AD, we show here that 4-HNE, putatively resulting from the peroxidation of lipids, covalently modifies Aβ, triggering its aggregation. These Aβ modifications result from 1,4 conjugate addition and/or Schiff base formation, they occur at multiple locations on a single Aβ peptide, and they result in covalent crosslinking of Aβ peptides. The consequence of these reactions is that 4-HNE accelerates formation of Aβ protofibrils while inhibiting the production of straight, mature fibrils. Recent studies implicating Aβ oligomers and protofibrils in the neurotoxic process that ultimately leads to AD suggests that the Aβ aggregates induced by 4-HNE may be important in the pathogenesis of AD. These results provide further incentive to understand the role of oxidative stress and small-molecule Aβ modifications in sporadic AD.
Although compelling genetic and biochemical evidence indicates that the process of amyloid β peptide (Aβ)1 amyloidogenesis causes Alzheimer's disease (AD) (1, 2), the amount of fibrillar amyloid found in the brain does not correlate well with disease severity (3). However, the amyloidogenesis of Aβ and other amyloidogenic proteins also affords spherical aggregates, annular structures, protofibrils, and other soluble oligomeric species both in vitro and in vivo (4, 5), the concentration of which better correlates with the severity of AD (6, 7). Furthermore, the neurotoxicity of these diffusible aggregates is now established (8-13). Herein we explore the possibility that the covalent modification of Aβ by the oxidative metabolite 4-hydroxynonenal (4-HNE) could be the trigger for Aβ misassembly into toxic oligomers leading to sporadic AD cases.
Oxidative metabolites of cholesterol containing an aldehyde functional group (14) form Schiff bases with Aβ, accelerating the in vitro aggregation of Aβ 1-40 (15, 16). These metabolites trigger Aβ 1-40 to form kinetically stable spherical aggregates under quiescent conditions, and fibrils upon agitation via a two-step mechanism (15, 16). These metabolites also hasten α-synuclein amyloid or Lewy body formation thought to cause Parkinson's disease, through what appears to be a non-covalent mechanism (17). 4-HNE (1), long associated with AD, is one of the most common products and toxic markers of oxidative stress (18, 19). It putatively arises from the peroxidation of ω-6 polyunsaturated fatty acids (Figure 1A) (20). Since 4-HNE contains a highly reactive α,β-unsaturated aldehyde, it can react with nucleophiles by 1,2 addition (Schiff base formation) or 1,4 addition (21) or by combinations thereof (Figure 1B) (22). When a nucleophilic side chain of a protein or peptide such as Aβ reacts with 4-HNE, protein conjugates form, substantially altering the structural and physical properties of the side chain it modifies, and because of the small size of Aβ, the entire peptide. Amino groups on lysine, histidine, and the N-terminus can react with 4-HNE by 1,2 addition affording Schiff bases, whereas the nucleophilic residues cysteine, histidine, and lysine and the N-terminus can add to 4-HNE by 1,4 conjugate addition (23, 24). 4-HNE can covalently crosslink proteins by reacting sequentially by 1,4 and 1,2 addition with residues on the same or on two different polypeptide chains (23) (Figure 1B).
Figure 1.

(A) 4-hydroxynonenal (1) is produced from the peroxidation of ω-6-polyunsaturated fatty acids in a mechanism involving reactive oxygen species (ROS) in the presence of Fe2+. (B) 4-HNE reacts with various nucleophilic protein residues to form hemi-acetals, Schiff bases by 1,2 addition, and 1,4-conjugates. Combining these reactivities, one molecule of 4-HNE can covalently crosslink polypeptides (PP) together. Crosslinked peptides are shown in the box.
Pathophysiological links have been made between 4-HNE and neurodegenerative diseases including Parkinson's disease, amyotrophic lateral sclerosis, and diffuse Lewy body disease, as well as AD (25). 4-HNE is elevated in the brain (26, 27) and plasma (28) of Alzheimer's patients (∼20 μM) in comparison to age matched controls (0.1 – 10 μM) (29). 4-HNE is therefore in large excess in vivo relative to the Aβ 1-40 concentration (1 – 10 nM) under physiological or pathophysiological conditions (30, 31). 4-HNE also co-localizes with Aβ amyloid deposits immunohistochemically (32). Moreover, 4-HNE has been shown to react with Aβ (1-42) in vitro (33), but its effect, if any, on the aggregation of Aβ has not been studied. Herein we show that 4-HNE enhances the misassembly of Aβ into small protofibrillar aggregates, but actually inhibits the conversion of Aβ aggregates into straight fibrils.
Materials and Methods
Preparation of seed-free Aβ 1-40
Aβ 1-40 purchased from SynPep (Dublin, CA) was pretreated using a method previously reported (15) to ensure that monomeric Aβ was used in the following experiments. Briefly, lyophilized Aβ 1-40 was dissolved at 2.5 mg/mL in 2 mM NaOH. The pH was adjusted to 10.5 by addition of 100 mM NaOH. The sample was sonicated in a water bath for 20 min, and filtered through a 0.2 μm syringe filter followed by a 10 kDa molecular weight cutoff Centricon filter (Millipore, Billerica, MA). The concentration of the resultant solution was checked by absorbance at 280 nm (ε = 1280 M-1cm-1) and diluted to 200 μM with double distilled H2O previously brought to pH 10.5 with NaOH.
Quiescent aggregation of Aβ 1-40
Stock solutions of 4-HNE (Cayman Chemical, Ann Arbor, MI) in ethanol were added to phosphate buffer (100 mM sodium phosphate, 600 mM NaCl, pH 7.2) to produce solutions twice the desired final concentration of 4-HNE (1% EtOH by volume). Seed-free Aβ 1-40 (200 μM, pH 10.5) was diluted 1:1 with the buffer solution to produce final solutions of 100 μM Aβ 1-40, 50 mM NaPi, 300 mM NaCl, 0.5% EtOH by volume, 0 – 100 μM 4-HNE, pH 7.4. The mixture was vortexed, aliquotted into separate tubes for each timepoint, and incubated quiescently at 37 °C. At desired time points, aliquots were briefly vortexed, 20 μL were added to 480 μL of a thioflavin T (TfT) solution (20 μM in 50 mM NaPi, pH 7.2), and the fluorescence was measured (excitation at 440 nm, emission at 485 nm, Aviv ATF-105 Spectrofluorometer, Aviv Biomedical, Lakewood, NJ, or Varian Cary Eclipse fluorometer, Varian, Inc. Palo Alto, CA).
Atomic force microscopy (AFM)
From an aliquot of the aggregation solution above, 20 μL was adsorbed to a surface of freshly cleaved mica (5 × 5 mm) for 1 min. The liquid was absorbed into filter paper. Salt and unbound material was removed through three washes by adding 30 μL of water to the mica and immediately absorbing it into filter paper. AFM images were recorded in tapping mode with a Digital Instruments multimode scanning probe microscope with FESP tips and a Nanoscope IIIa controller (Veeco, Woodbury, NY).
Size exclusion chromatography and light scattering
Aβ 1-40 (100 μM) was incubated with or without 4-HNE (100 μM) under quiescent conditions as described above. After a given amount of time, 75 μL of the aggregating Aβ solution was combined with 75 μL of 50 mM NaPi, filtered through a 0.22 μm syringe filter (Millipore), and injected onto an AKTA FPLC (GE Healthcare, Piscataway, NJ) using a 100 μL injection loop. The mixture was separated by size exclusion chromatography employing a Superdex 75 HR 10/30 column (GE Healthcare, Piscataway, NJ) eluted with 50 mM NaPi, 100 mM NaCl, and 0.03% NaN3, pH 7.2. Detection was accomplished by absorbance at 280 nm and by static light scattering (Dawn EOS, Wyatt Technology Corporation, Santa Barbara, CA). There is a 0.1 μm filter after the FPLC injection loop that precludes large aggregates from reaching the column.
MALDI-TOF
Low salt solutions of Aβ 1-40 (100 μM) and 4-HNE (100 μM) in phosphate buffer (10 mM NaPi, 50 mM NaCl, pH 7.8) were incubated for 5 h on a rocking platform at 37 °C. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) was performed on the sample by the Scripps Center for Mass Spectrometry (The Scripps Research Institute, La Jolla, CA).
Dot blots
Solutions containing Aβ 1-40 (100 μM) and 4-HNE (500 μM, 0.78% final volume of EtOH) in phosphate buffer (50 mM NaPi, 300 mM NaCl, pH 7.4) were incubated overnight at 37 °C on a rocking platform. NaBH4 (280 mM stock for a final concentration of 8.2 mM) was added, the mixture was vortexed and incubated quiescently for 30 min at 37 °C. Sodium dodecyl sulfate (SDS, 10 μL of 20% solution) and dithiothreitol (DTT, 10 μL of 18.6% solution) were added to 100 μL of the reduced Aβ solution and boiled for 10 min. The resulting mixture was extracted 4 times with 120 μL of ethyl acetate. This was dialyzed into 5 mM NaPi, pH 7.2 through a 1000 molecular weight cut off dialysis cassette (Spectra/Por CE Irradiated DispoDialyzer, Spectrum Laboratories, Inc. Rancho Dominguez, CA). Application of samples (20 μL for samples not dialyzed, 40 μL for dialyzed samples) onto PVDF membranes was followed by blocking the membrane with 5% milk, washing with TBST, and visualization using a primary antibody of rabbit anti-4-HNE (Alpha Diagnostic International, San Antonio, TX) and a goat anti-rabbit secondary antibody (Pierce, Rockford, IL)
SDS-PAGE analysis
Solutions prepared as in the previous section containing Aβ 1-40 (100 μM) and 4-HNE (0, 100, or 500 μM) in phosphate buffer (50 mM NaPi, 300 mM NaCl, pH 7.4, 220 μL total) were incubated at 37 °C on a rocking platform for 4 days. The resultant mixture was reduced with NaBH4 followed by dissolution in 70% HFIP. This solution was sonicated for 1 h, lyophilized, dissolved in 450 μL water, and sonicated again for 1 h. 15 μL of each sample were then run in a reducing, denaturing gel (10-20% tris/tricene, Bio-Rad Laboratories, Inc. Hercules, CA) followed by a western blot, probing the system with 6E10 antibody (Signet Laboratories, Inc. Dedham, MA), a monoclonal antibody specific for Aβ, to analyze the crosslinking of Aβ.
Results
Pretreated monomeric Aβ 1-40 (100 μM) was incubated with 4-HNE as a function of concentration (1 – 100 μM) under quiescent conditions (37 °C). The 75 h Aβ aggregation time course was followed by thioflavin T (TfT) fluorescence. TfT binding to amyloid fibrils, protofibrils, and spherical aggregates results in a substantial increase in the TfT quantum yield, so monitoring TfT fluorescence in the presence of Aβ reports on the amount of Aβ present as aggregates (15, 16, 34). Representative Aβ 1-40 aggregation time courses for a range of 4-HNE concentrations are depicted in Figure 2A. The TfT fluorescence of Aβ incubated in the absence of 4-HNE remains low over the 75 h time course. In contrast, the TfT fluorescence of Aβ incubated with 4-HNE increases as the incubation proceeds in a manner proportional to the 4-HNE concentration. This effect was reproducible over 4 experiments, for which the time to reach half maximal Aβ 1-40 TfT fluorescence (t50) varied by only 9% at a high 4-HNE concentration (100 μM). The t50 for lower concentrations of 4-HNE varied within 30%; the error for the latter samples was larger because less amyloid formed, so there was more noise in the TfT time courses. The TfT fluorescence of each experiment was normalized by setting the endpoint (75 h) value of the sample containing 100 μM Aβ 1–40 and 100 μM 4-HNE to 1 (arbitrary units), enabling direct time course comparisons across experiments. Combining the 75 h normalized fluorescence values from 5 experiments showed that the endpoint TfT fluorescence increased linearly with the amount of 4-HNE present (p < 0.0001) (Figure 2B). Thus, 4-HNE increases the amyloidogenicity of Aβ 1-40 in a dose dependent manner. The initial rate of Aβ amyloidogenesis for samples with varying 4-HNE concentrations was found to be proportional to the concentration of 4-HNE present (see figure S1, Supporting Information), however the rate does not double as the concentration of 4-HNE is doubled, which means the aggregation reaction is not first order in 4-HNE concentration. This indicates that the rate of amyloidogenesis is dependent on 4-HNE, but that the mechanism is likely complicated, depending on many factors including the rate of reaction between 4-HNE and Aβ 1-40, the location and type of modification, and the assembly interactions between modified and unmodified Aβ 1-40.
Figure 2.

(A) Quiescent aggregation of Aβ 1-40 (100 μM; 37 °C) in the presence or absence of 4-HNE, monitored by TfT fluorescence. (B) Normalized endpoint TfT fluorescence (t = 75 h) of Aβ 1-40 (100 μM) aggregated with 4-HNE (0 – 100 μM) for multiple experiments. Linear regression has a statistically significant (p < 0.0001) positive slope, indicating a dose dependence of Aβ aggregation on 4-HNE. AFM images show protofibrillar Aβ 1-40 (100 μM) after 8 h of incubation alone (C), and after 8 h of incubation with 4-HNE (100 μM) (D). (E) and (F) are magnifications of (C) and (D), respectively (images are representative of 12 images from 4 experiments). Height scale shown on right.
The morphology of Aβ 1-40 aggregates present in each sample was evaluated by AFM after 8 h of reaction. Although the sample with equimolar 4-HNE and Aβ 1-40 (100 μM) exhibited a higher TfT fluorescence than Aβ 1-40 samples lacking 4-HNE, protofibrillar species were detected by AFM analysis in both samples. All protofibrillar aggregates formed, whether from Aβ 1-40 alone (Figure 2C, zoom-in Figure 2E) or from Aβ in the presence of 4-HNE (Figure 2D, zoom-in Figure 2F), measured 2-4 nm in height. These protofibrillar species were up to several hundred nanometers in length. However, the samples containing 4-HNE had a consistently higher abundance of protofibrils adhering to the mica than those lacking 4-HNE, indicating that 4-HNE increases the propensity of Aβ to form protofibrils. These protofibrils presumably bind TfT, which explains the higher TfT signal of Aβ incubated with 4-HNE (Figure 2A).
To further study the differences between the aggregation of Aβ 1-40 in the presence and absence of 4-HNE, samples were examined by size exclusion chromatography with in-line static light scattering, an approach used previously to evaluate Aβ aggregation (35). Initially, the absorbance chromatograms show a single peak eluting at the monomer volume. As the incubation time of Aβ 1-40 alone (Figure 3A) or Aβ 1-40 with 4-HNE (Figure 3B) increases, the Aβ monomer peak shrinks and a peak corresponding to oligomeric Aβ that elutes in the void volume of the column grows. These changes in peak areas are much larger in samples containing 4-HNE, indicating significant aggregation only in the presence of 4-HNE (Figures 3A – 3D). The oligomer peak contains quaternary structures ranging from 300 – 3,500 kDa in molecular weight according to the static light scattering data, which reflect a composition of 70 – 800 Aβ 1-40 monomers, respectively. While the decrease in the monomer peak monitored by absorbance signifying aggregation is barely noticeable in the absence of 4-HNE, changes are apparent in the appearance of the oligomer peak monitored by light scattering both in the absence (Figure 3E) and in the presence of 4-HNE (Figure 3F). The signal intensity of light scattering is greater for the Aβ 1-40 samples incubated with 4-HNE, indicating that more oligomers are present in those samples at any given time. This supports the notion that 4-HNE increases the conversion of Aβ 1-40 monomers into oligomers, which correlates with the TfT and AFM data that also show more protofibril formation with 4-HNE.
Figure 3.
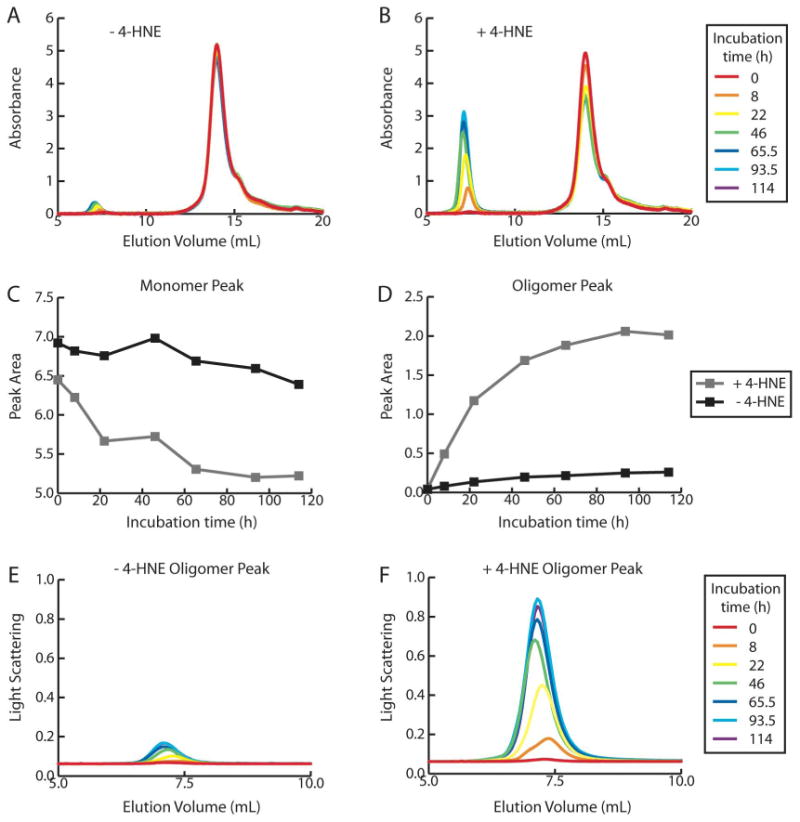
Size exclusion chromatograms of Aβ 1-40 (100 μM) incubated quiescently (37 °C) in the absence (A) and presence (B) of 4-HNE (100 μM), monitored by absorbance at 280 nm. The decrease in monomeric peak area (C) and increase in oligomeric peak area (D) is more pronounced in the presence of 4-HNE. The 90° light scattering signal of the oligomeric peak reaches a higher intensity in the presence (F) of 4-HNE compared to Aβ 1-40 incubated alone (E).
Since 4-HNE accelerates Aβ 1-40 protofibril formation over a time course of 75 h, a period that is not sufficient for Aβ 1-40 to form long straight fibrils (Figure 2E), a longer time course was utilized to discern whether 4-HNE could trigger Aβ 1-40 amyloid fibril formation. Although the time required for Aβ to form fibrils in the absence of 4-HNE varied between experiments, the overall finding discussed below was consistent over 4 experiments. As shown in Figure 4A (blue line), Aβ 1-40 incubated quiescently exhibited a lag phase in its TfT fluorescence followed by a growth phase. In contrast the Aβ samples incubated with 4-HNE displayed no lag phase and an immediate increase in TfT fluorescence, but did not undergo a further increase in TfT fluorescence beyond that seen in the first 75 h, a phase shown above to represent protofibril formation (Figure 2C-F). An influence of 4-HNE is also observed with Aβ 1-42 aggregation (Supporting Figure S2). After aggregating quiescently for 15 days, Aβ 1-42 (10 μM) incubated with stoichiometric or higher concentrations of 4-HNE exhibits inhibition of TfT fluorescence relative to Aβ 1-42 incubated without 4-HNE. This is consistent with the lower final TfT fluorescence exhibited by Aβ 1-40 in the presence of 4-HNE compared to Aβ 1-40 aggregated alone.
Figure 4.
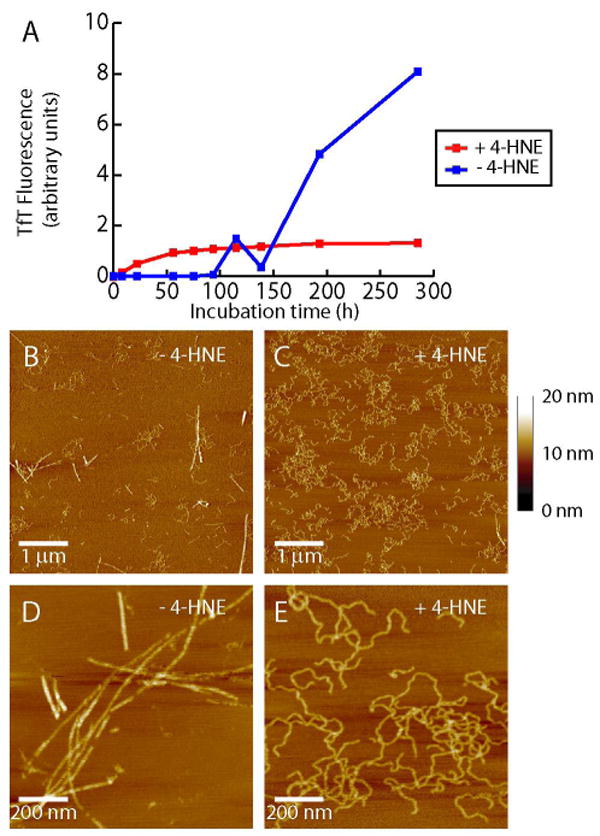
(A) Quiescent aggregation of Aβ 1-40 (100 μM) in the presence (100 μM) and absence of 4-HNE, monitored by TfT fluorescence. AFM images of Aβ 1-40 aggregated alone for 285 h (B, magnified in D) show long straight fibrils. Only curved fibrils are observed when Aβ 1-40 is incubated for 285 h in the presence of 4-HNE (C, magnified in E) (images are representative of 12 images from 4 experiments). Height scale shown on right.
AFM analyses of Aβ 1-40 aggregation samples lacking 4-HNE reveal mainly long, straight amyloid fibrils 2-7 nm in height. Only a few curved fibrillar species 2-4 nm high are observed after 285 h of incubation in the absence of 4-HNE (Figure 4B, zoom-in Figure 4D). In contrast, Aβ 1-40 samples containing 4-HNE and incubated for 285 h reveal predominantly curved fibrillar species 2-4 nm in height (Figure 4C, zoom-in Figure 4E) by AFM and lack detectable straight fibrils. Both the long straight fibrils and curved fibrillar aggregates appear to have similar lengths, varying from hundreds of nanometers to several microns in length. 4-HNE seems to prohibit the formation of long, straight amyloid fibrils thought to arise from lateral protofibril assembly, likely as a consequence of the side chain modifications discussed above. Although they appear different morphologically, both the long straight fibrils formed in the absence of 4-HNE and the curved fibrillar aggregates formed in the presence of 4-HNE (short curved protofibrils early in the time course, followed by longer curved fibrils) appear to be rich in β-sheet structure based on their circular dichroism spectra (Supporting Figure S3).
To better understand the mechanism of action of 4-HNE, its interaction with the Aβ 1-40 peptide was further studied. A solution of Aβ 1-40 (100 μM) was incubated with 4-HNE (100 μM) for 5 h in a low salt buffer at 37 °C. The resultant solution was examined by MALDI-TOF MS revealing peaks at molecular weights of 4335, 4491, 4648, and 4804 (± 4) Da (Figure 5A). Within the error of the measurement (0.1% accuracy, or ± 4 Da), these masses are consistent with the molecular weight of unmodified Aβ 1-40 and Aβ molecules attached covalently to 1, 2, and 3 substructures of molecular weight 156 Da, respectively. 4-HNE, when attached to a peptide by 1,4-conjugate addition raises the molecular weight of the peptide by 156 Da, whereas 1,2 addition requires loss of H2O from 4-HNE resulting in a molecular weight increase of 138 Da, which was not observed even if the samples were reduced with NaBH4 prior to MALDI-TOF MS analysis (after reduction a 140 Da increase is expected). These results are consistent with a previous mass spectrometry study reporting that 4-HNE has the ability to form adducts with Aβ 1-42 by 1,4-conjugate addition (33). This same type of adduct is likely forming here on up to 3 different Aβ 1-40 sites, possibly at His6, His13, His14, Lys16, Lys28, or at the N-terminus.
Figure 5.
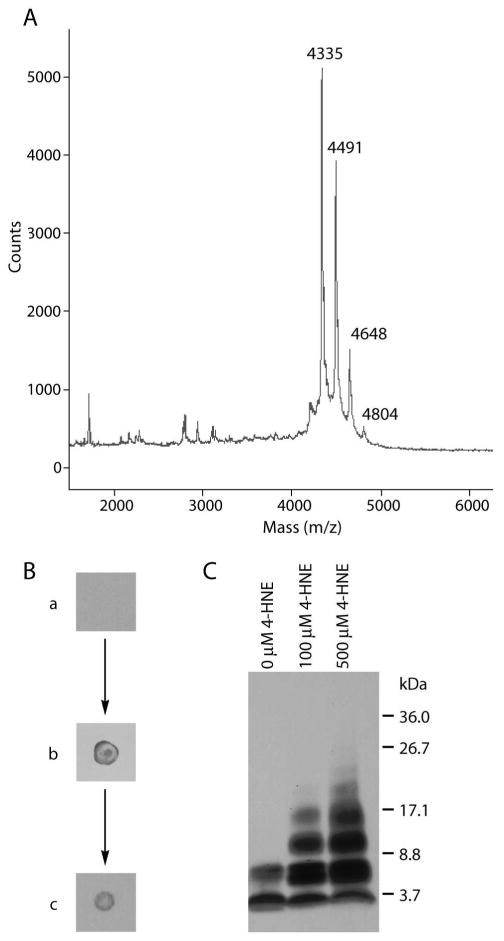
(A) MALDI-TOF MS of Aβ 1-40 (100 μM) incubated with 4-HNE (100 μM) for 5 h reveals multiple peaks corresponding to the molecular mass of Aβ 1-40 plus 0, 1, 2, or 3 adducts of 4-HNE. (B) Dot blots using an anti-HNE antibody detected adducts of HNE to Aβ 1-40. (a) Aβ 1-40 (100 μM) was (b) incubated with 4-HNE (500 μM). The solution was reduced with NaBH4, boiled with SDS and DTT, extracted with ethyl acetate, and dialyzed through a 1 kDa membrane to yield (c), a sample in which 4-HNE was still detectable. (C) Aβ 1-40 (100 μM) incubated with 4-HNE (0, 100, 500 μM) for 4 days was reduced with NaBH4 and treated with HFIP. Crosslinked oligomers as large as hexamers were separated by SDS-PAGE and detected by a western blot procedure.
If 4-HNE were to bind noncovalently with high affinity to Aβ, the complex would also exhibit the observed molecular weight increase of 156 Da. Therefore, to scrutinize the covalent nature of this interaction, Aβ 1-40 (100 μM) was incubated with 4-HNE (500 μM). The resultant sample was detectable by an antibody specific for 4-HNE-protein adducts using a dot blot (Figure 5B). This antibody binds to 4-HNE-protein conjugates, but not 4-HNE or Aβ 1-40 alone. The Aβ-4-HNE complex and remaining 4-HNE were reacted with NaBH4, which reduces any aldehydes or Schiff bases to alcohols and amines, respectively, preventing Schiff base hydrolysis and further reactions between free 4-HNE and Aβ 1-40. The reduced Aβ-4-HNE solution was then boiled in the presence of SDS and DTT and subsequently extracted with ethyl acetate to remove any non-covalently bound 4-HNE. The peptide was isolated by dialysis through a 1 kDa cutoff membrane and probed with the anti-4-HNE-protein antibody using a dot blot (Figure 5B). The 4-HNE-Aβ adduct was still detectable after rigorous treatments to remove non-specifically bound 4-HNE, indicating that the attachment of 4-HNE to Aβ 1-40 was covalent.
4-HNE can also covalently crosslink Aβ peptides. 4-HNE can react via 1,4-conjugate addition with one Aβ chain and Schiff base formation with a second to afford a covalent dimer. To test whether this was occurring, solutions of Aβ 1-40 were incubated with increasing concentrations of 4-HNE and reduced with NaBH4. The resulting aggregates were broken apart with HFIP (Supporting Figure S4) and analyzed under denaturing conditions by SDS-PAGE (Figure 5C). In the absence of 4-HNE, Aβ runs on the gel as a monomer and dimer, whereas in the presence of equimolar concentrations of 4-HNE, oligomers up to tetramers can be clearly observed. When a concentration of 4-HNE corresponding to a 5-fold molar excess over Aβ 1-40 is employed, oligomers as large as hexamers can be detected. That higher order oligomers are detected as the stoichiometry of 4-HNE increases strongly supports the hypothesis that covalent crosslinking of Aβ 1-40 by 4-HNE occurs. Only shorter oligomers are detected if the reduction step is omitted (Supporting Figure S4) which indicates that part of the interaction between 4-HNE and Aβ 1-40 involves Schiff base formation, which is reversible without reduction owing to hydrolysis under the denaturing conditions of SDS-PAGE. Therefore, 4-HNE most likely crosslinks Aβ 1-40 by 1,4-conjugate addition with one chain and Schiff base formation with another. Crosslinked conjugates of Aβ 1-40 (100 μM) with 4-HNE (100 μM) are detectable by SDS-PAGE in the quiescent aggregation reaction after 18.5 h of incubation, are more abundant after 46.5 h, and reach maximal abundance at the same time the TfT fluorescence curve plateaus (Supporting Figure S4). In other words, the time scales for the appearance of crosslinks and protofibrils are indistinguishable. While this does not prove that the crosslinks cause the protofibrils to form, these results are consistent with the hypothesis that crosslinking is important in preventing the formation of long straight fibrils.
Discussion
The majority of elderly Alzheimer's disease patients suffer from sporadic AD, not familial AD caused by known predisposing mutations (1, 36). The reasons why some elderly people get AD, while others do not, are not understood. It has been hypothesized that oxidative stress may be a risk factor for AD, as oxidative stress markers are found in higher concentrations in AD patients than in age matched controls (26-28). Whether oxidative stress is a cause or consequence of AD remains an unanswered question (25). Some groups have shown that Aβ can induce an oxidative stress response in the presence of neurons, synaptosomes, or even whole organisms such as C. elegans, and that the products formed from the oxidation of lipids are the cause of neuronal toxicity (37). However, others argue that the appearance of oxidative stress markers precedes that of plaques and other pathological markers of AD (38).
Here we have shown that 4-HNE, an oxidative stress marker, hastens Aβ protofibril and curved fibril formation, but precludes the formation of long straight fibrils formed in the absence of 4-HNE. 4-HNE covalently modifies Aβ 1-40 via 1,4-conjugate addition, and can crosslink Aβ peptides to each other, putatively by subsequent Schiff base formation. Collectively, these activities induce the formation of short Aβ oligomers (2 to 6 monomers in size) that are stable to SDS-PAGE and detectable by western blotting. These small oligomers likely serve as a nucleus or template to recruit modified and unmodified monomeric Aβ into larger oligomers, or they may bind other oligomers to afford protofibrils. Regardless of the exact mechanism, which is under further investigation, the net effect of 4-HNE on Aβ 1-40 aggregation is clear. It hastens the formation of Aβ 1-40 protofibrils, cf. Figures 2E and 2F, producing more aggregates as seen by TfT fluorescence, size exclusion chromatography, static light scattering, and AFM. Additionally, it inhibits the conversion of these protofibrils to long, straight amyloid fibrils, even upon extended incubation. Instead the protofibrils are converted into curved fibrils exhibiting lowered fluorescence in the presence of 4-HNE. This is relevant given that it is now thought that oligomers and protofibrillar structures, not long mature amyloid fibrils, are the species of Aβ responsible for neurodegeneration (8-13). Therefore, 4-HNE, formed by oxidative stress as a consequence of infection, injury, or Aβ deposition may induce Aβ to form toxic oligomers more rapidly than it would in the absence of oxidative stress metabolites. 4-HNE may extend the lifetime of these toxic protofibrils and curved fibrils preventing their conversion into less toxic long straight fibrils, sustaining toxicity, exacerbating neuronal death, ultimately leading to AD.
Our results suggest a model in which oxidative stress leads to the production of 4-HNE and related small molecule oxidation products (15, 16) that hasten Aβ aggregation and toxicity, likely by altering the oligomers formed, which in turn causes oxidative stress that leads to the formation of even more lipid peroxidation products, such as 4-HNE and more toxic Aβ oligomers. In this manner, a vicious cycle of events ensues wherein oxidative stress affords oxidized molecules that trigger toxic Aβ oligomerization which begets more oxidative stress and aggregation which could result in the onset of memory loss and cognitive dysfunction, hallmarks of Alzheimer's disease.
Supplementary Material
The initial rate of Aβ 1-40 amyloidogenesis was proportional to the concentration of 4-HNE. 4-HNE decreases the endpoint TfT fluorescence of Aβ 1-42. Aβ 1-40 forms β-sheet rich aggregates in the presence and absence of 4-HNE. The formation of SDS-PAGE stable higher order oligomers is reduced when the NaBH4 step is omitted. The timecourses for curved fibril formation monitored by TfT fluorescence and higher-order oligomer formation are coincident. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank M. R. Ghadiri for use of his AFM.
Footnotes
We thank the Skaggs Institute of Chemical Biology, the Lita Annenberg Hazen Foundation, the NIH (NS 50636), and the Bundy Foundation for financial support.
Abbreviations: AD, Alzheimer's disease; Aβ, amyloid beta peptide; AFM, atomic force microscopy; DTT, dithiothreitol; FPLC, fast performance liquid chromatography; 4-HNE, 4-hydroxynonenal; MALDI-TOF MS, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; TfT, thioflavin T.
References
- 1.Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 2.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 3.Berg L, McKeel DW, Jr, Miller JP, Storandt M, Rubin EH, Morris JC, Baty J, Coats M, Norton J, Goate AM, Price JL, Gearing M, Mirra SS, Saunders AM. Clinicopathologic studies in cognitively healthy aging and Alzheimer's disease: relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch Neurol. 1998;55:326–335. doi: 10.1001/archneur.55.3.326. [DOI] [PubMed] [Google Scholar]
- 4.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 5.Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing Activities Protect Against Age Onset Proteotoxicity. Science. 2006 doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- 6.Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 8.Dahlgren KN, Manelli AM, Stine WB, Jr, Baker LK, Krafft GA, LaDu MJ. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem. 2002;277:32046–32053. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- 9.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 11.Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 13.Baglioni S, Casamenti F, Bucciantini M, Luheshi LM, Taddei N, Chiti F, Dobson CM, Stefani M. Prefibrillar amyloid aggregates could be generic toxins in higher organisms. J Neurosci. 2006;26:8160–8167. doi: 10.1523/JNEUROSCI.4809-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wentworth P, Jr, Nieva J, Takeuchi C, Galve R, Wentworth AD, Dilley RB, DeLaria GA, Saven A, Babior BM, Janda KD, Eschenmoser A, Lerner RA. Evidence for ozone formation in human atherosclerotic arteries. Science. 2003;302:1053–1056. doi: 10.1126/science.1089525. [DOI] [PubMed] [Google Scholar]
- 15.Bieschke J, Zhang Q, Powers ET, Lerner RA, Kelly JW. Oxidative metabolites accelerate Alzheimer's amyloidogenesis by a two-step mechanism, eliminating the requirement for nucleation. Biochemistry. 2005;44:4977–4983. doi: 10.1021/bi0501030. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Q, Powers ET, Nieva J, Huff ME, Dendle MA, Bieschke J, Glabe CG, Eschenmoser A, Wentworth P, Jr, Lerner RA, Kelly JW. Metabolite-initiated protein misfolding may trigger Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:4752–4757. doi: 10.1073/pnas.0400924101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bosco DA, Fowler DM, Zhang Q, Nieva J, Powers ET, Wentworth P, Jr, Lerner RA, Kelly JW. Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate alpha-synuclein fibrilization. Nat Chem Biol. 2006;2:249–253. doi: 10.1038/nchembio782. [DOI] [PubMed] [Google Scholar]
- 18.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 19.Ji C, Amarnath V, Pietenpol JA, Marnett LJ. 4-hydroxynonenal induces apoptosis via caspase-3 activation and cytochrome c release. Chem Res Toxicol. 2001;14:1090–1096. doi: 10.1021/tx000186f. [DOI] [PubMed] [Google Scholar]
- 20.Pryor WA, Porter NA. Suggested mechanisms for the production of 4-hydroxy-2-nonenal from the autoxidation of polyunsaturated fatty acids. Free Radic Biol Med. 1990;8:541–543. doi: 10.1016/0891-5849(90)90153-a. [DOI] [PubMed] [Google Scholar]
- 21.Uchida K, Stadtman ER. Modification of histidine residues in proteins by reaction with 4-hydroxynonenal. Proc Natl Acad Sci U S A. 1992;89:4544–4548. doi: 10.1073/pnas.89.10.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nadkarni DV, Sayre LM. Structural definition of early lysine and histidine adduction chemistry of 4-hydroxynonenal. Chem Res Toxicol. 1995;8:284–291. doi: 10.1021/tx00044a014. [DOI] [PubMed] [Google Scholar]
- 23.Carini M, Aldini G, Facino RM. Mass spectrometry for detection of 4-hydroxy-trans-2-nonenal (HNE) adducts with peptides and proteins. Mass Spectrom Rev. 2004;23:281–305. doi: 10.1002/mas.10076. [DOI] [PubMed] [Google Scholar]
- 24.Szapacs ME, Riggins JN, Zimmerman LJ, Liebler DC. Covalent adduction of human serum albumin by 4-hydroxy-2-nonenal: kinetic analysis of competing alkylation reactions. Biochemistry. 2006;45:10521–10528. doi: 10.1021/bi060535q. [DOI] [PubMed] [Google Scholar]
- 25.Zarkovic K. 4-hydroxynonenal and neurodegenerative diseases. Mol Aspects Med. 2003;24:293–303. doi: 10.1016/s0098-2997(03)00024-4. [DOI] [PubMed] [Google Scholar]
- 26.Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer's disease. J Neurochem. 1997;68:2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- 27.Markesbery WR, Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer's disease. Neurobiol Aging. 1998;19:33–36. doi: 10.1016/s0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- 28.McGrath LT, McGleenon BM, Brennan S, McColl D, Mc IS, Passmore AP. Increased oxidative stress in Alzheimer's disease as assessed with 4-hydroxynonenal but not malondialdehyde. QJM. 2001;94:485–490. doi: 10.1093/qjmed/94.9.485. [DOI] [PubMed] [Google Scholar]
- 29.Dianzani MU. 4-hydroxynonenal from pathology to physiology. Mol Aspects Med. 2003;24:263–272. doi: 10.1016/s0098-2997(03)00021-9. [DOI] [PubMed] [Google Scholar]
- 30.Mehta PD, Pirttila T, Patrick BA, Barshatzky M, Mehta SP. Amyloid beta protein 1-40 and 1-42 levels in matched cerebrospinal fluid and plasma from patients with Alzheimer disease. Neurosci Lett. 2001;304:102–106. doi: 10.1016/s0304-3940(01)01754-2. [DOI] [PubMed] [Google Scholar]
- 31.Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C, et al. Isolation and quantification of soluble Alzheimer's beta-peptide from biological fluids. Nature. 1992;359:325–327. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- 32.Ando Y, Brannstrom T, Uchida K, Nyhlin N, Nasman B, Suhr O, Yamashita T, Olsson T, El Salhy M, Uchino M, Ando M. Histochemical detection of 4-hydroxynonenal protein in Alzheimer amyloid. J Neurol Sci. 1998;156:172–176. doi: 10.1016/s0022-510x(98)00042-2. [DOI] [PubMed] [Google Scholar]
- 33.Magni F, Galbusera C, Tremolada L, Ferrarese C, Kienle MG. Characterisation of adducts of the lipid peroxidation product 4-hydroxy-2-nonenal and amyloid beta-peptides by liquid chromatography/electrospray ionisation mass spectrometry. Rapid Commun Mass Spectrom. 2002;16:1485–1493. doi: 10.1002/rcm.743. [DOI] [PubMed] [Google Scholar]
- 34.LeVine H., 3rd Thioflavine T interaction with synthetic Alzheimer's disease beta-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 1993;2:404–410. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. Amyloid beta-protein fibrillogenesis. Detection of a protofibrillar intermediate. J Biol Chem. 1997;272:22364–22372. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- 36.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 37.Butterfield DA. Amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer's disease brain. Free Radic Res. 2002;36:1307–1313. doi: 10.1080/1071576021000049890. A review. [DOI] [PubMed] [Google Scholar]
- 38.Behl C. Oxidative stress in Alzheimer's disease: implications for prevention and therapy. Subcell Biochem. 2005;38:65–78. doi: 10.1007/0-387-23226-5_3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The initial rate of Aβ 1-40 amyloidogenesis was proportional to the concentration of 4-HNE. 4-HNE decreases the endpoint TfT fluorescence of Aβ 1-42. Aβ 1-40 forms β-sheet rich aggregates in the presence and absence of 4-HNE. The formation of SDS-PAGE stable higher order oligomers is reduced when the NaBH4 step is omitted. The timecourses for curved fibril formation monitored by TfT fluorescence and higher-order oligomer formation are coincident. This material is available free of charge via the Internet at http://pubs.acs.org.