

Abstract
Coruscanone A, a plant derived cyclopentenedione derivative, showed potent in vitro antifungal activity against Candida albicans and Cryptococcus neoformans, comparable to amphotericin B and fluconazole. A series of analogs have been synthesized by modification of the cyclopentenedione ring, the enolic methoxy functionality, and the side chain styryl moiety of this natural product lead. A structurally close 1,4-benzoquinone analog was also prepared. All the compounds were examined for their in vitro activity against major opportunistic fungal pathogens including C. albicans, C. neoformans and Aspergillus fumigatus, and fluconazole-resistant C. albicans strains, with several analogs demonstrating potent antifungal activity. Structure activity relationship studies indicate that the 2-methoxymethylene-cyclopent-4-ene-1,3-dione structural moiety is the pharmacophore responsible for the antifungal activity of this class of compounds, while the side chain styryl-like moiety plays an important complementary role, presumably contributing to target binding.
Introduction
The limitations of current antifungal drugs, increased incidence of systemic fungal infections, and rapid development of drug resistance have highlighted the need for the discovery of new antifungal agents, preferably with novel mechanisms of action.1 While synthetic efforts remain the mainstream in antifungal drug discovery, as evidenced by the fact that 18 out of 23 antifungal drugs approved from 1980–2002 are synthetic (83% belong to the single azole class), unique antifungal natural products have demonstrated remarkable success in this regard.2 For example, two major drug classes currently in use, represented by amphotericin B, the ‘gold standard’ in the antifungal armamentarium, and the lipopeptide caspofungin, the most important antifungal drug approved in recent years, are derived from natural products.1
In the course of our search for prototype antifungal agents from natural sources, we discovered the antifungal lead, coruscanone A (1), from the ethanol extract of the piperaceous plant Piper coruscans.3 Coruscanone A possesses a 2-methoxymethylenecyclopent-4-ene-1,3-dione skeleton with a styryl moiety (designated as side chain below) attached to the methylene at C-2. This unique structural property facilitates the coexistence of two geometrical isomers along the C-2 exo-double bond in solution, via photoisomerization.3,4 It must be pointed out that all the compounds of this class described herein have two geometrical isomers in a ratio of almost 1:1 in solution; however, for convenience sake the structure of only one isomer is presented. Coruscanone A showed strong antifungal activity against two major opportunistic pathogens C. albicans and C. neoformans associated with immuno-compromised patients (AIDS, cancer, or organ transplant), comparable to the positive controls amphotericin B and fluconazole. The lack of antibacterial activity of 1 against Staphylococcus aureus indicated a good selectivity. Coruscanone A represents the most fungicidal plant metabolite that is active against C. albicans.3 In addition, 1 showed acceptable in vitro cytotoxicity against mammalian Vero cells, comparable to amphotericin B, and is readily accessible by total synthesis.3 Although the cyclopent-4-ene- and cyclopentane-1,3-dione structural moieties are present in a large number of synthetic compounds with therapeutic applications or potential, the functional groups in these compounds are di- or triketonic in nature,5 in contrast to the unique diketone enol ether form in 1. Also, little is known about the mechanisms of action of these compounds. Therefore, this drug-like, small molecule natural product is an ideal structural template for the synthesis of a series of analogs in order to explore their structure activity relationships (SAR), thus affording the information for further lead optimization of this class of compounds as potential new antimycotics.
Results and Discussion
Chemistry
To develop a robust pharmacophoric model and understand the basis for the antifungal activity of coruscanone A (1), its structure can be subdivided into three key structural moieties: cyclopentenedione ring (A), enolic methoxy functionality (S1), and the side chain styryl moiety (S2) (Fig. 1). Each structural moiety can be modified independently in order to facilitate the systematic refinement of the search for increasingly effective coruscanone A analogs.
Figure 1.
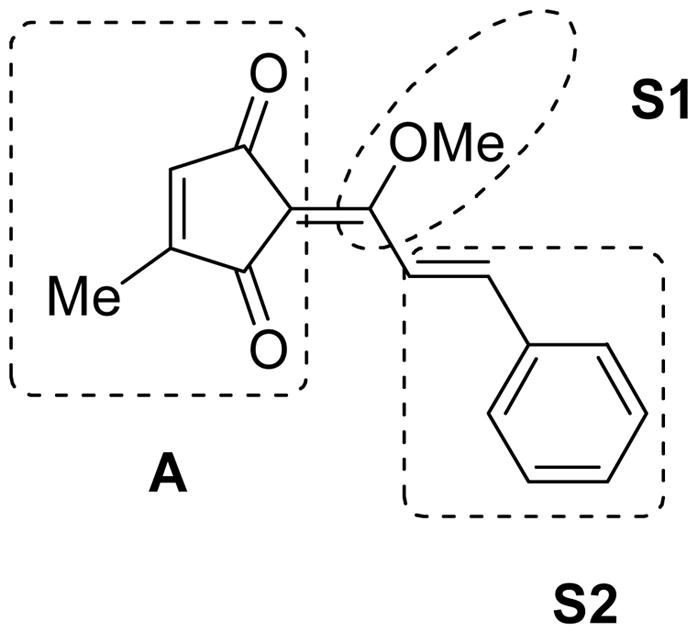
Key structural moieties of coruscanone A (1).
The general synthetic methodology for the preparation of coruscanone A analogs is a modified version of a method developed by Clemo et al.,6 and successful synthesis of coruscanone A has demonstrated its feasibility.3 As outlined in Scheme 1, the synthetic sequence starts with Wittig condensation between a maleic anhydride derivative, e.g., 2-methylmaleic anhydride (2), 2,3-dimethylmaleic anhydride (3) or 2-phenylmaleic anhydride (4) and a phosphorane (16–21, refer to Experimental section for their structures) prepared from the corresponding α-methyl ketone in a three step sequence, via brominated α-methyl ketone (5–9) and phosponium salt (10–15). The key step is the base-catalyzed rearrangement of the resulting 4-ylidenebutenolide (22–29), affording the corresponding cyclopent-4-ene-1,3-dione (30–37). Alkylation or acylation of the cyclopentenediones yields the enolic hydroxy substituted product (38–52).
1. Structural Modifications of the Cyclopentenedione Ring (A) of 1
Our first modification of the cyclopentenedione ring of coruscanone A involved the synthesis of 2-(1-methoxy-3-phenylpropenylidene)-4,5-dimethylcyclopent-4-ene-1,3-dione (38), since this compound would only have one isomer in solution due to the symmetric nature of the cyclopentenedione ring. It was thus prepared from 2,3-dimethylmaleic anhydride (3) and 4-phenyl-1-(triphenylphosphoranylidene)-(3E)-buten-2-one (16) that was available from styryl methyl ketone as shown in Scheme 1. The second compound, 2-[(E)-1-methoxy-3-phenylallylidene)-4-phenylcyclopent-4-ene-1,3-dione (39), represented replacement of the methyl substitutent on the cyclopentenedione ring of 1 with a phenyl group, which was prepared from 4-phenylmaleic anhydride (4) and phosphorane 16. It was postulated that introduction of the bulky phenyl group which extends conjugation might have a significant impact on the antifungal activity of the compound.
It should be pointed out that Wittig condensation of a monosubstituted maleic anhydride derivative, e.g. 2 or 4, with a phosphorane always results in a regional- and stereoselective formation of an E-butenolide as the predominant product, as observed in 22 and 24. In the case of disubstituted 2,3-dimethylmaleic anhydride (3), the intermediate butenolide 23 as the major product possesses Z-configuration at the C-4 double bond due to less steric hindrance between the olefinic proton and the cyclic methyl group. However, both types of butenolides can be readily converted to corresponding cyclopentenediones via a non-stereoselective base-catalyzed rearrangement.3,6
2. Structural Modification of the Enolic Methoxy Functionality of 1 by Alkylation and Acylation (S1)
Our previous study has indicated that O-methylation of the enolic hydroxy group plays a key role in the antifungal activity of coruscanone A (1) when compared to its demethylated precursor, coruscanone B (30), which shows only marginal antifungal activity.3 Thus, modification of the enolic methoxy functionality of 1 would have a significant impact on the antifungal activity of newly synthesized analogs. When 30 was alkylated with Et2SO4/K2CO3 in acetone and acetylated with acetic anhydride/pyridine, compounds 47 and 49 were obtained, respectively. Similarly, ethylation of cyclopentenedione 31 with Et2SO4/K2CO3 in acetone yielded 48. Acylation of 31 with acetic anhydride/pyridine, benzoyl chloride/THF/DMAP and cinnamoyl chloride/pyridine/DMAP afforded 50–52, respectively.
3. Structural Modification of the Styryl Side Chain (S2) of 1
Selection of α-methyl ketones to construct the side chain of synthetic coruscanone A analogs was based on minor modification of the styryl moiety of 1, with the aim of achieving improved antifungal potency and better selectivity, thus arriving at meaningful SAR information. With the retention of the structural moieties A and S1 of 1, six analogs (40–43, 45 and 46) were prepared according to the general synthetic approach illustrated in Scheme 1. While the preparation of 40–43 was fairly straightforward, the synthesis of 45 and 46 involved protection/deprotection of a phenolic hydroxy group at appropriate stages (see Experimental section). The side chains in these compounds vary from a phenyl group in 43, to heteroaromatic furanyl and thiophene moieties in 41 and 42, respectively, and additional substitution at the phenyl group of the styryl moiety in 40, 45 and 46.
The last, simplest synthetic entity in this series is 2-(1-hydroxyethylidene)-4-methylcyclopent-4-ene-1,3-dione (53) in which a methyl group replaces the styryl group in 1. It was prepared by an alternative method via reaction of 2-methylmaleic anhydride (2) and α-methylvinyl acetate in AlCl3/C2H4Cl2,7 followed by methylation with Me2SO4/K2CO3 in acetone.
4. Synthesis of 2-Methoxy-5-methyl-3-styryl-1,4-benzoquinone (60)
It is well-known that some 1,4-benzoquinones are reported to have good antifungal activity.8 Since coruscanone A (1) shows some similarity to 1,4-benzoquinone derivatives, a structurally close analog, 2-methoxy-5-methyl-3-styryl-1,4-benzoquinone (60), was synthesized for SAR studies. This would also indicate whether any correlation exists between the two classes of compounds regarding their biological activity. Thus, starting from commercially available methoxyhydroquinone (54), its methoxymethyl protected ether 55 was treated with pyridinium hydrobromide perbromide in MeOH to give the brominated product 56, which was subjected to bromine/lithium exchange in MeLi/THF. The lithio derivative was quenched with MeI, and the resultant C-methyl derivative 57 was converted to the lithium salt with BuLi/THF in a DoM-type reaction,9 and then coupled with phenylacetaldehyde to give benzyl alcohol 58. Dehydration and deprotection of 58 using mineral acid yielded stilbene 59, which was oxidized by DDQ to furnish 60 (Scheme 2). In view of the limited number of available methods for the synthesis of styryl benzoquinones,10 it is worthwhile mentioning that direct coupling of phenylacetaldehyde with di-ortho-oxgenated benzenes, followed by oxidation, is a facile method for preparation of 2-hydroxy (or methoxy)-3-styryl benzoquinone derivatives.
Biological Activity
Synthetic coruscanone A (1) and its analogs (30–43, and 45–53) as well as the 1,4-benzoquinone 60 were evaluated for their in vitro antifungal activity against three major opportunistic fungal pathogens C. albicans, C. neoformans, and Aspergillus fumigatus using the positive control drugs amphotericin B and fluconazole for comparison. Compounds showing 50% growth inhibition (IC50) at the concentration of less than 20 μg/ml were considered to be active and were further evaluated for their antifungal activity against clinical isolates of fluconazole-susceptible and resistant C. albicans (isolates #1 and #17, respectively) from AIDS patients during fluconazole therapy.11 The results indicated that compounds 1, 33, 39, 40–43, 45–47 and 49 showed varying degrees of antifungal activity (Table 1), while other compounds were inactive at 20 μg/mL. The in vitro cyctoxicity of the antifungal compounds against mammalian kidney cells (Vero and LLC-PK-1) was determined and compared with the anticancer drug doxorubicin. While coruscanone A remains the most potent antifungal compound among the synthetic derivatives, several analogs (40–42) demonstrated potent, but slightly weaker activity than 1. In addition, compounds that were active against C. albicans retained equivalent activity against fluconazole-resistant C. albicans isolate #17, which involves several resistance mechanisms, including overexpression of efflux pumps CDR1 (p-glycoprotein) and MDR1 (major facilitator) mRNA, as well as overexpression of fluconazole’s target gene, ERG11.11b Since overexpression of these genes leads to inefficacy of fluconazole via efflux pump activity and/or overproduction of ERG11’s product, 14α-lanosterol demethylase, a key enzyme in the biosynthesis of egosterol,11a coruscanone A and its analogs would target something other than ERG11, and/or are not a substrate for the CDR1 or MDR1 efflux pumps. Meaningful SAR information is summarized as follows:
Table 1.
In vitro Antifungal Activity and Cytotoxicity of Coruscanone A and Its Analogsa
| Antifungal Activity (IC50b/MICc/MFCd, μg/mL)
|
Cytotoxicity (IC50,b μg/mL)
|
||||||
|---|---|---|---|---|---|---|---|
| C. albicans ATCC 90028 | C. albicans #1e | C. albicans #17e | C. neoformans ATCC 90113 | A. fumigatus ATCC 90906 | Verof | LLC-PK-1g | |
| 1 | 0.80/2.08/2.08 | 0.47/1.25/1.25 | 0.68/2.08/2.08 | 2.04/8.33/8.33 | 19/>20/>20 | 4.9 | 3.4 |
| 33 | 13/>20/>20 | 10/>20/>20 | 13/>20/>20 | >20/>20/>20 | >20/>20/>20 | >10 | 6.0 |
| 39 | >20/>20/>20 | 20/>20/>20 | 18/>20/>20 | >20/>20/>20 | >20/>20/>20 | >10 | 5.1 |
| 40 | 1.39/4.17/4.17 | 0.64/2.08/2.92 | 0.86/2.92/2.92 | 2.23/12/12 | 20/>20/>20 | 5.5 | 3.1 |
| 41 | 1.40/4.17/4.17 | 0.93/2.50/2.50 | 2.40/4.17/6.67 | 3.40/10/13 | 8.98/20/20 | 3.3 | 3.3 |
| 42 | 1.21/3.33/3.33 | 0.74/2.50/2.50 | 1.39/4.17/4.17 | 2.49/12/12 | 9.81/20/>20 | 3.0 | 2.8 |
| 43 | 10.6/>20/>20 | 7.88/>20/>20 | 6.04/>20/>20 | >20/>20/>20 | >20/>20/>20 | >10 | >10 |
| 45 | 3.30/10/10 | 1.86/5.00/13 | 5.35/20/20 | 6.95/>20/>20 | >20/>20/>20 | 3.7 | 3.4 |
| 46 | 3.03/10/10 | 1.56/5.00/6.67 | 4.29/10/13 | 3.41/15/17 | >20/>20/>20 | 5.0 | 3.9 |
| 47 | 5.02/20/20 | 2.49/12/17 | 4.51/13/20 | 5.02/20/20 | >20/>20/>20 | 4.8 | 4.0 |
| 49 | 8.56/>20/>20 | 3.79/>20/>20 | 5.84/>20/>20 | 20/>20/>20 | >20/>20/>20 | 5.5 | 4.9 |
| AMBh | 0.15/0.63/1.04 | 0.12/0.52/1.25 | 0.17/0.63/0.83 | 0.41/1.25/1.67 | 0.73/1.25/2.5 | 7.0 | 2.4 |
| FLUj | 0.22/0.45/>200 | 0.24/0.45/>200 | 118/>200/>200 | 5.65/37/>200 | >200/>200/>200 | >10 | >10 |
| DOXk | nti | nti | nti | nti | nti | 7.5 | 0.65 |
Mean values based on three independent experiments.
50% growth inhibitory concentration.
Minimum Inhibitory Concentration.
Minimum Fungicidal Concentration.
Patient isolates during fluconazole therapy:11 #1, first isolate, azole-susceptible; #17, last isolate, azole-resistant.
African green monkey kidney fibroblast.
Pig kidney epithelial cells.
Amphotericin B.
Not tested.
Fluconazole.
Doxorobicin.
-
Modifications of the cyclopentenedione ring: Compound 38 with only one additional methyl group on the cyclopentenedione ring when compared to 1, and its derivatives 31, 48, and 50–52 surprisingly lack antifungal activity. This indicates that the antifungal mechanism of coruscanone A regarding its reactive site to the target would be on the cyclopentenedione ring that may be considered as the ‘warhead’ of the molecule. It is thus postulated that 1 may act like maleimide derivatives as a Michael acceptor,12 reacting at C-5 of the cyclopentendione ring with a nucleophilic site of the target (enzyme). Additional methyl substitution at C-5 such as in 38 would preclude such a reaction due to steric hindrance. When the methyl group in 1 is replaced by a large phenyl group in 39, the activity decreases significantly, presumably due to decreased affinity to the target.
The lack of antifungal activity of the benzoquinone derivative 60 further indicates that the cyclopentenedione structural moiety of coruscanone A analogs is selective to fungal pathogens, although a similar Michael addition reaction mechanism may also occur on the benzoquinone skeleton of 60.
Modifications to the enolic functionality: Different substitutions at the enolic hydroxy group affect the activity. Similar to coruscanone B (30), all the synthetic cyclopentenediones with a free enolic hydroxy group (31, 32, 34–36) are inactive except for the chlorine-containing compound 33 which shows moderate activity against C. albicans and fluconazole-resistant C. albicans strains. Compound 49 with O-acetyl substitution is only moderately active against C. albicans and marginally active against C. neoformans, and 47 with O-ethyl substitution is about 10-fold (MIC value) less active against C. albicans than 1. The weak activity of the two compounds, particularly 47, can not be simply explained by the reaction mechanism of Michael addition mentioned above. The binding affinity or selectivity of the structures of the compounds to the reactive site of the target (enzyme) must play an important role. At this point, the enolic O-methyl group remains the optimum functional group among all the synthetic analogs.
Modifications to the styryl side chain: The side chain plays an important role for the activity of coruscanone A analogs. Compounds 40 and 46 that have aromatic chloro- and methoxy substituents show a slightly weaker activity than 1. It is noteworthy that compounds 41 and 42 possessing five-membered heteroaromatic rings instead of a benzene ring, display significant activity against C. albicans and C. neoformans with enhanced activity against A. fumigatus compared to 1. However, when the styryl moiety is replaced by a phenyl group as in 43 or a methyl group as in 53, the activity is decreased drastically for 43 or completely lost for 53, implying the double bond of the styryl moiety or the intact styryl moiety (e.g., in 1) or styryl-like moiety (e.g., in 41 and 42) are necessary for the activity. This strongly suggests that the side chain is associated with binding affinity for the target.
Separation of antifungal activity from cytotoxicity: Although the above antifungal compounds show moderate in vitro cytotoxicity to the mammalian cells Vero (IC50s from 3.3 to >10 mg/mL) and LLC-PK-1 (IC50s from 3.1 to >10 μg/mL), their cytotoxicity does not seem to be necessarily correlated to their antifungal activity. For example, compound 47, which exhibits a cytotoxicity similar to 1 (IC50 of 4.8 v.s. 4.9 μg/mL against Vero cells, Table 1), is only moderately active against C. albicans (MIC of 20 v.s. 2.08 μg/mL). Compound 49, which is even less antifungal than 47, also shows cytotoxicity similar to 1 (IC50 of 5.5 v.s. 4.9 μg/mL against Vero cells, Table 1). In contrast, the most cytotoxic compounds, 41, 42 and 45, are not the most potent antifungal compounds. This indicates that these compounds may act via different mechanisms of action for their antifungal activity against C. albicans and cytotoxicity against mammalian cells, or that minor structural changes afford important alterations in fungal cell permeability/transport, etc. This would render an opportunity to improve the antifungal activity or therapeutic index of the analogs by further chemical modification of the lead structure without effecting cytotoxicity.
It was recently demonstrated that a synthetic cyclopentenedione derivative 61 showed a potent inhibition of human chymase, a potential drug target associated with cardiovascular diseases and chronic inflammation following fibrosis.13 A possible mechanism for inhibition of the enzyme by 61 was proposed as shown in Scheme 3, i.e., the enzyme interacts with the inhibitor at C-6 due to its strong electrophilicity, while the phenyl group of the inhibitor is enclosed by the residue of the S1 pocket.13 However, the reactive site of coruscanone A and its analogs to the target enzyme is apparently different from that of 61. Coruscanone A may act as a Michael acceptor reacting at C-5 of the cyclopentenedione ring with a nucleophile of the active site residue of the target (enzyme) to form an irreversible covalent adduct,14 while its styryl and methoxy groups are enclosed by other residues of the target (Scheme 4). The irreversible covalent nature of the adduct may also explain the fungicidal activity of coruscanone A. There is no doubt that Michael acceptors are associated with certain levels of toxicity towards mammalian cells, as observed in coruscanone A analogs (Table 1). However, numerous reports/patents have described the therapeutic potential of Michael acceptors from the perspective of drug discovery. For example, it is suggested that Michael acceptors can be structurally modified, so that they can react selectively with target nucleophiles or can be converted into selective Michael acceptors during metabolism for anticancer drug discovery. Representative natural cytotoxic compounds containing Michael acceptors include menadione and shikonin analogs, while the anticancer drug mitomycin is transformed by reductive activation into a conjugated dienone Michael acceptor.15 Another anticancer drug mitoxantrone acts via oxidative activation involving a Michael acceptor intermediate.16 Recent work has demonstrated that Michael acceptors are a new class of inhibitors specific for caspases and other Clan cysteine proteases. A large number of aza-peptides composed of a 2-pyridone-containing peptidomimetic binding determinant and an α,β-unsaturated Michael acceptor “warhead” have been synthesized for the development of orally bioavailable inhibitors for the 3C protease, which were tested against rhinoviruses.17 Moreover, extensive studies have demonstrated that Michael acceptors are an important class of compounds that can induce phase 2 detoxification enzymes [e.g., glutathione transferases, NAD(P)H:quinone reductase, glucoronosyl transferases, epoxide hydrolase] and thus possess anticarcinogenic properties.18 Moderately reactive Michael acceptors are receiving growing interest for the design of chemoprotective drugs.19 Therefore, there is great potential for the development of coruscanone A analogs carrying a Michael acceptor “warhead” as therapeutic agents.
In conclusion, this study has demonstrated that the 2-methoxymethylenecyclopent-4-ene-1,3-dione structural moiety is responsible for the antifungal activity of coruscanone A analogs, with the styryl-like moiety being an ideal supplemental group, presumably contributing to enzyme binding. The putative pharmacophore may act like maleimides as a Michael acceptor12 reacting with the target to form an irreversible covalent adduct, while additional experimental evidence is needed to support this hypothesis. Although compounds with improved antifungal activity have not yet been found, a meaningful SAR for coruscanone A analogs has been established based on the limited number of compounds synthesized by a rationally designed classical medicinal chemistry approach. This has laid a solid foundation for further lead optimization of this class of compounds by a systematic chemical modification including the synthesis of water-soluble compounds to improve their overall pharmaceutical properties. An effort aimed at elucidating the antifungal mechanism of action of coruscanone A analogs using genomic profiling and biochemical approaches is also underway in our group.
Experimental Section
Melting points were measured with a Thomas-Hoover capillary melting point apparatus and are uncorrected. NMR spectra were recorded on Bruker DPX-300, DRX-400 or Varian Mercury-400BB instruments. Chemical shifts are expressed in ppm relative to the solvent residue signals. ESI-FTMS were measured on a Bruker-Magnex BioAPEX 30es ion cyclotron high-resolution HPLC-FT spectrometer by direct injection into an electroscopy interface. High resolution TOFMS were measured on an Agilent series 1100 SL equipped with an ESI source. Flash column chromatography was done on silica gel (40 μm, J. T. Baker) and reversed-phase silica gel (RP-18, 40 μm, J. T. Baker). TLC was performed on silica gel sheets (Silica Gel 60 F254, Merck, Germany) and reversed-phase plates (RP-18 F254S, Merck, Germany). Analytical HPLC used to determine the purity of target compounds was conducted on an ODS (Discovery®) column (250 × 4.6 mm, 10 μm) using two solvents systems, (a) 70% CH3CN in H2O and (b) 80% MeOH in H2O, and UV detection at 254 nm for a 20-minutes run. The results are expressed by a retention time in minutes and a relative content in percentage. All the reagents were obtained from the commercial vendors in appropriate grades and were used without further purification.
General Procedure for Preparation of Bromomethyl Ketones 5–8
To a solution of the α-methyl ketone (2 mmol) in dry THF (20 mL) at room temperature under nitrogen was slowly added a solution of the pyrrolidone hydrotribromide (2.4 mmol) in dry THF (40mL) in 1 hr. The mixture was continued to stir at room temperature for 24 hr. After completion of the reaction, Excess pyrrolidone hydrotribromide was removed by filtration. The filtrate was concentrated to dryness. The resulting residue was dissolved in Et2O, washed with brine and dried (Na2SO4). Removal of the solvents afforded crude product, which was chromatographed on silica gel eluting with Et2O-hexanes (1:6) to give the bromomethyl ketone.
Bromomethyl Styryl Ketone (5)
Yield, 77% from styryl methyl ketone, see ref. 3 for spectroscopic data.
(E)-1-Bromo-4-(4-chlorophenyl)but-3-en-2-one (6)
Yield, 70% from 4-(4-chlorophenyl)but-3-en-2-one, pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.65 (1H, d, J = 16 Hz, β-CH=), 7.51 (2H, d, J = 8 Hz, H-3, 5), 7.38 (2H, d, J = 8 Hz, H-2, 6), 6.92 (1H, d, J = 16 Hz, α-CH=), 4.06 (2H, s, CH2-Br).
(E)-1-Bromo-4-(furan-2-yl)but-3-en-2-one (7)
Yield, 59% from 4-(furan-2-yl)but-3-en-2-one, colorless oil; 1H NMR (400 MHz, CDCl3) δ 7.55 (1H, br s, H-5), 7.48 (1H, d, J = 16 Hz, β-CH=), 6.87 (1H, d, J = 16 Hz, α-CH=), 6.75 (1H, d, J = 3.2 Hz, H-3), 6.53 (1H, m, H-4), 4.05 (2H, s, CH2-Br).
(E)-1-Bromo-4-(thiophen-2-yl)but-3-en-2-one (8)
Yield, 59% from 4-(thiophen-2-yl)but-3-en-2-one, colorless oil; 1H NMR (400 MHz, CDCl3) δ 7.78 (1H, dd, J = 16, 3.2 Hz, β-CH=), 7.43 (1H, br s, H-5), 7.32 (1H, br s, H-3), 7.06 (1H, br d, H-4), 6.71 (1H, dd, J = 16, 3.2 Hz, α-CH=), 4.02 (2H, s, CH2-Br).
(E)-1-Bromo-4-[4-O-(t-butyldimethylsilyl) phenyl] but-3-en-2-one (9)
Yield, 65% from TBS- protected (E)-4-hydoxyphenylbut-3-en-2-one that was prepared by a standard procedure,20 colorless oil; 1H NMR (400 MHz, CDCl3): δ 7.64 (1H, d, J = 16 Hz, β-CH=), 7.47 (2H, d, J = 8 Hz, H-3, 5), 6.84 (2H, d, J = 8 Hz, H-2, 6), 6.80 (1H, d, J = 16 Hz, α-CH=), 4.05 (2H, s, CH2Br), 0.97 (9H, s, 3× Me), 0.21 (6H, s, 2× Me).
General Procedure for Preparation of Phosphonium Salts 10–15
To a refluxed solution of the triphenylphosphine (1 mmol) in dry THF (25 ml) under nitrogen was slowly added bromomethyl ketone derivative (1 mmol). The reaction mixture was refluxed for 2–4 hr. After completion, reaction was cooled to room temperature. The white precipitate was collected by the filtration and washed with Et2O.
[2-Oxo-4-phenyl-(3E)-butenyl]triphenyl Phosphonium Bromide (10)
Yield, 86% from 5, see ref. 3 for spectroscopic data.
[2-Oxo-4-(4-chlorophenyl)-(3E)-butenyl]triphenyl Phosphonium Bromide (11)
Yield, 90% from 6, white powder; 1H NMR (400 MHz, CDCl3) δ 8.46 (1H, d, J = 16 Hz, β-CH=),7.90-7.64 (15H, m, Ph), 7.71(2H, d, J = 8 Hz, H-3, 5), 7.31 (2H, d, J = 8 Hz, H-2, 6), 7.04 (1H, d, J = 16 Hz, α-CH=), 5.91 ( 2H, d, J = 12 Hz, CH2-P).
[2-Oxo-4-(furan-2-yl)-(3E)-butenyl]triphenyl Phosphonium Bromide (12)
Yield, 56% from 7, white powder; 1H NMR (400 MHz, CDCl3) δ 8.31 (1H, d, J = 16 Hz, β-CH=), 7.89-7.58 (15H, m, Ph), 7.48 (1H, br s, H-5), 7.03 (1H, d, J = 3.6 Hz, H-3), 6.70 (1H, dd, J = 16, 2.4 Hz, α-CH=), 6.45 (1H, m, H-4), 5.82 (2H, d, J = 12 Hz, CH2-P).
[2-Oxo-4-(thiophen-2-yl)-(3E)-butenyl]triphenyl Phosphonium Bromide (13)
Yield, 56% from 8, white powder; 1H NMR (400 MHz, CDCl3) δ 8.75 (1H, d, J = 16 Hz, β-CH=), 7.89-7.63 (17H, m), 7.42 (1H, br d, J = 4 Hz, H-3), 7.03 (1H, br s, H-4), 6.64 (1H, d, J = 16 Hz, α-CH=), 5.82 (2H, d, J = 12 Hz, CH2-P).
(2-Oxo-2-phenylethyl)triphenyl Phosphonium Bromide (14)
Yield, 62% from commercially available 2-bromo-1-phenylethanone, white powder; 1H NMR (400 MHz, CDCl3) δ 8.35 (2H, d, J = 7 Hz, H-2, 6), 7.89–7.95, 7.72, 7.56, 7.48 (18H, m), 6.34 (2H, d, J = 12 Hz, CH2-P).
[2-Oxo-4-(4-t-butyldimethylsilyloxyphenyl)-(3E)-butenyl]triphenyl Phosphonium Bromide (15)
Yield, 75% from 9, pale yellow powder; 1H NMR (400 MHz, CDCl3) δ 8.28 (1H, d, J = 16 Hz, β-CH=), 7.90-7.63, (17H, m), 6.96 (1H, d, J = 16 Hz, α-CH=), 6.81 (2H, d, J = 8 Hz, H-2, 6), 5.83 (2H, d, J = 12 Hz, CH2-P), 0.95 (9H, s, Me × 3), 0.19 (6H, s, Me × 2).
General Procedure for Preparation of Phosphoranes 16–21
To a suspension of the above phosphonium salt 6 (1 mmol) in H2O and MeOH (1:1) was added a solution of 2N NaOH (10 mL). The mixture was stirred at room temperature overnight. After evaporation of the MeOH, the suspension was extracted with CHCl3. The organic layer was washed with the brine, dried (Na2SO4), and concentrated to dryness to give corresponding phosphorane.
4-Phenyl-1-(triphenylphosphoranylidene)-(3E)-buten-2-one (16)
Yield, 91% from 10, yellow crystal, see ref. 3 for spectroscopic data.
4-(4-Chlorophenyl)-1-(triphenylphosphoranylidene)-(3E)-buten-2-one (17)
Yield, 80% from 11, colorless oil; 1H NMR (400 MHz, CDCl3) δ 7.71-7.40 (17H, m), 7.34 (1H, d, J = 16 Hz, β-CH=), 7.26 (2H, d, J = 8 Hz, H-2, 6), 6.85 (1H, d, J = 16 Hz, α-CH=), 4.06 (1H, br d, J = 23 Hz, CH=P).
4-(Furan-2-yl)-1-(triphenylphosphoranylidene)-(3E)-buten-2-one (18)
Yield, 93% from 12, pale yellow powder; 1H NMR (400 MHz, CDCl3): δ 7.70-7.31 (17H, m), 7.18 (1H, d, J = 16 Hz, β-CH=), 6.81 (1H, d, J = 16 Hz, α-CH=), 6.36 (1H, m, H-4), 3.98 (1H, br d, J = 24 Hz, CH=P).
4-(Thiophen-2-yl)-1-(triphenylphosphoranylidene)-(3E)-buten-2-one (19)
Yield, 93% from 13, pale yellow powder; 1H NMR (400 MHz, CDCl3) δ 7.71-7.47 (16H, m), 7.16 (1H, br d, H-5), 7.06 (1H, br d, H-3), 6.95 (1H, m, H-4), 6.71 (1H, d, J = 16 Hz, α-CH=), 3.97 (1H, br d, J = 24 Hz, CH=P).
2-Phenyl-(triphenylphosphoranylidene)ethan-2-one (20)
Yield, 90% from 14, pale yellow powder; 1H NMR (400 MHz, CDCl3) δ 7.97 (2H, br d, H-2, 6), 7.75-7.35 (18H, m, Ph, H-3, 4, 5), 4.43 (1H, d, J = 24 Hz, CH=P).
4-(4-t-Butyldimethylsilyloxyphenyl)-1-(triphenylphosphoranylidene)-(3E)-buten-2-one (21)
Yield, 80% from 15, yellow oil; 1H NMR (400 MHz, CDCl3): δ 7.68-7.37 (19H, m, Ph, H-3, 5, α-CH=, β-CH=), 6.77(2H, br d, H-2, 6), 3.97 (1H, br d, CH=P), 0.97 (9H, s, Me × 3), 0.18 (6H, s, Me × 2).
General Procedure for Preparation of Butenolides 22–29
A solution of maleic anhydride derivative (1.2 mmol) in benzene (10 mL) was added to a refluxed solution of a phosphorane (1 mmol) in benzene (50 mL) under nitrogen. After completion of the reaction (monitored by TLC), the reaction mixture was evaporated to dryness and the residue was chromatographed on silica gel eluting with hexane-acetone (4:1) to yield the desired product.
(5E)-3-Methyl-5-[(E)-2-oxo-4-phenylbut-3-enylidene]furan-2(5H)-one (22)
Yield, 73% from 2-methylmalei anhydride (2) and 16, see ref. 3 for spectroscopic data.
(5Z)-3,4-Dimethyl-5-[(E)-2-oxo-4-phenylbut-3-enylidene]furan-2(5H)-one (23)
Yield, 96% from 2,3-dimethylmaleic anhydride (3) and 16, yellow powder; 1H NMR (400 MHz, CDCl3) δ 7.72 (1H, d, J = 16 Hz, H-4′), 7.65 (2H, m, H-6′, 10′), 7.56 (1H, d, J = 16 Hz, H-3′), 7.42 (3H, m, H-7′, 8′, 9′), 5.78 (1H, s, H-1′), 2.13 (3H, s, Me), 2.03 (3H, s, Me); 13C NMR (100 MHz, CDCl3) δ 187.5, 168.9, 155.8, 148.4, 144.0, 134.7, 130.7, 129.0 (2C), 128.4, 128.7 (2C), 125.2, 106.2.
(5E)-5-[(E)-2-Oxo-4-phenylbut-3-enylidene]-3-phenylfuran-2(5H)-one (24)
Yield, 56% from 2-phenylmaleic anhydride (4) and 16, yellow powder; 1H NMR (400 MHz, CDCl3) δ 8.57 (1H, s, H-3), 8.01 (2H, br d), 7.68 (1H, d, J = 16 Hz, H-4′), 7.61 (2H), 7.47-7.43 (6H), 6.92 (1H, d, J = 16 Hz, H-3′), 6.56 (1H, s, H-1′).
(5E)-5-[(E)-4-(4-Chlorophenyl)-2-oxobut-3-enylidene]-3-methylfuran-2(5H)-one (25)
Yield, 56% from 2 and 17, pale yellow powder; 1H NMR (400 MHz, CDCl3) δ 8.09 (1H, s, H-3), 7.58 (1H, d, J = 16 Hz, H-4′), 7.51(2H, d, J = 8 Hz, H-7′,9′), 7.38 (2H, d, J = 8 Hz, H-6′,10′), 6.83 (1H, d, J = 16 Hz, H-3′), 6.43 (1H, s, H-1′), 2.10 (3H, s, Me).
(5E)-5-[(E)-4-(Furan-2-yl)-2-oxobut-3-enylidene]-3-methylfuran-2(5H)-one (26)
Yield, 50% from 2 and 18, pale yellow powder; 1H NMR (400 MHz, CDCl3) δ 8.10 (1H, s, H-3), 7.53 (1H, br d, H-8′), 7.41 (1H, d, J = 16 Hz, H-4′), 6.76 (1H, d, J = 16 Hz, H-3′), 6.74 (1H, br d, H-7′), 6.51 (1H, br d, H-6′), 6.37 (1H, s, H-1′), 2.09 (3H, s, Me); HRESIMS m/z [M+H]+ 231.0652 (calcd for C13H11O4, 231.0652).
(5E)-3-Methyl-5-[(E)-2-oxo-4-(thiophen-2-yl)but-3-enylidene]furan-2(5H)-one (27)
Yield, 50% from 2 and 19, pale yellow powder; 1H NMR (400 MHz, CDCl3) δ 8.10 (1H, s, H-3), 7.76 (1H, d, J = 15.6 Hz, H-4′), 7.44 ( 1H, br d, H-8′), 7.35 (1H, br d, H-7′), 7.08 (1H, brd, H-6′), 6.67 (1H, d, J = 15.6 Hz, H-3′), 6.39 (1H, s, H-1′), 2.10 (3H, s, Me); HRESIMS m/z [M+H]+ 247.0424 (calcd for C13H11O3S, 247.0423).
(5E)-3-Methyl-5-(2-oxo-2-phenylethylidene)furan-2(5H)-one (28)
Yield, 56% from 2 and 20, pale yellow powder; 1H NMR (400 MHz, CDCl3) δ 8.10 (1H, s, H-3), 7.95 (2H, d, J = 7 Hz), 7.59 (1H, m), 7.51 (2H, d, J = 7 Hz), 6.90 (1H, s, H-1′), 2.11 (3H, s, Me); HRESIMS m/z [M+H]+ 215.0703 (calcd for C13H11O3, 215.0703).
(5E)-5-[(E)-4-(t-Butyldimethylsilyloxyphenyl)-2-oxobut-3-enylidene]-3-methylfuran-2(5H)-one (29)
Yield, 40% from 2 and 21, pale yellow powder; 1H NMR (400 MHz, CDCl3) δ 8.10 (1H, s, H-3), 7.59 (1H, d, J = 15 Hz, H-4′), 7.48 (2H, br d), 6.86 (2H, d, J = 8 Hz), 6.75 (1H, d, J = 15 Hz, H-3′), 6.45 (1H, s, H-1′), 2.10 (3H, s, Me), 0.98 (9H, s, Me × 3), 0.22 (6H, s, Me × 2).
General Procedure for Preparation of Cyclopentenediones 30–37
A solution of the butenolide (1 mmol) in dry MeOH (20 mL) was added to a solution of NaOMe (10 mmol) in MeOH (5 mL) and the resulting orange solution was stirred at room temperature for 3–6 hr and then poured into ice water (100 mL) and acidified to pH 1.0 with 2 M HCl. The methanol was removed by evaporation, and the suspension was extracted with ether. Evaporation of the dried extracts gave the crude products, which was generally crystallized from MeOH to afford the product.
2-[(E)-1-Hydroxy-3-phenylallylidene]-4-methylcyclopent-4-ene-1,3-dione (30)
Yield, 62% from 22, see ref. 3 for physical and spectroscopic data including NMR signal assignments.
2-[(E)-1-Hydroxy-3-phenylallylidene]-4,5-dimethylcyclopent-4-ene-1,3-dione (31)
Yield, 82% from 23, pale yellow powder; 1H NMR (400 MHz, CDCl3) δ 12.05 (1H, br s, OH), 7.73 (2H, br s), 7.58 (2H, br d), 7.37 (3H, br s), 1.96 (6H, br s, 2 × Me); 13C NMR (100 MHz, CDCl3) δ 200.9, 192.1, 166.2, 151.4, 147.4, 142.3, 134.9, 130.5, 128.9 (2C), 128.5 (2C), 117.7 and 102.8 [for C-1, 3, 6, 4, 5, 8, 9, 12, 11 (13), 10 (14), 7, 2, respectively], 9.0 (Me), 8.4 (Me); HRESIMS m/z [M+H]+ 255.1014 (calcd for C16H15O3, 255.1016).
2-[(E)-1-Hydroxy-3-phenylallylidene]-4-phenylcyclopent-4-ene-1,3-dione (32)
Yield, 47% from 24, yellow powder; 1H NMR (400 MHz, benzene-d6, Z/E-isomers) δ 12.65 (br s), 8.13/8.06 (1H each, d, J = 16 Hz, H-7), 7.82 (1H × 2, d, J = 16 Hz, H-8), 7.76 (2H × 2, m, H-10, 14), 7.34 (2H × 2, m), 7.07 (3H × 2, m), 6.98 (3H × 2, m), 6.54 (1H × 2, s, H-5); 13C NMR (100 MHz, CDCl3, Z/E-isomers) δ 200.6, 199.8, 191.2, 191.1, 169.0, 168.9, 153.7, 150.8, 143.5, 143.4, 137.7, 134.9, 134.8, 134.5, 131.1 (2C), 131.0 (2C), 130.4, 130.0 (2C), 129.4, 129.2 (2C), 129.0 (2C), 128.9 (2C), 128.8 (4C), 128.7 (4C), 117.7, 117.5, 104.4, 104.2; HRESIMS m/z [M+H]+ 303.1001 (calcd for C20H15O3, 303.1016).
2-[(E)-3-(4-Chlorophenyl)-1-hydroxyallylidene]-4-methylcyclopent-4-ene-1, 3-dione (33)
Yield, 55% from 25, yellow powder; 1H NMR (400 MHz, benzene-d6, Z/E-isomers) δ 12.70/12.40 (1H each, br s, OH), 7.91/7.84 (1H each, d, J = 16 Hz, H-7), 7.58 (1H × 2, d, J = 16 Hz, H-8), 6.94 (2H × 2, br d), 6.90 (2H × 2, br d), δ 5.98 (1H × 2, s, H-5), 1.53 and 1.47 (3H each, s, Me-4); 13C NMR (100 MHz, CDCl3, Z/E-isomers) δ 201.1, 200.5, 192.1, 191.6, 167.4, 167.1, 158.2, 154.3, 141.5, 141.4 (2C), 140.9, 137.1 (2C), 136.6, 133.3, 129.7 (4C), 129.2 (4C), 118.1, 118.0, 103.5, 103.3, 10.5, 10.3; HRESIMS m/z [M+H]+ 275.0470 (calcd for C15H12O3Cl, 275.0470).
2-[(E)-3-(Furan-2-yl)-1-hydroxyallylidene]-4-methylcyclopent-4-ene-1,3-dione (34)
Yield, 50% from 26, yellow powder; 1H NMR (400 MHz, benzene- 12.64/12.35 (1H each, br s, OH), d6, Z/E-isomers) δ 8.03/7.98 (1H each, d, J = 16 Hz, H-7), 7.48 (1H × 2, d, J = 16 Hz, H-8), 6.82 (1H × 2, br d, H-12), 6.05 (1H × 2, br d, H-10), 5.95 (1H × 2, br d, H-11), 5.85 (1H × 2, s, H-5), 1.52 and 1.45 (3H each, s, Me); 13C NMR (100 MHz, CDCl3, Z/E-isomers) δ 201.1, 200.6, 192. 0, 191.5, 167.5, 167.3, 157.9, 153.9, 151.7 (2C), 145.7, 145.6, 140.6, 136.7, 129.0, 128.9, 116.03, 115.99, 115.4, 115.3, 112.8 (2C), 103.2, 103.0, 11.3, 10.5; HRESIMS m/z [M+H]+ 231.0641(calcd for C13H11O4, 231.0652).
2-[(E)-1-Hydroxy-3-(thiophen-2-yl)allylidene]-4-methylcyclopent-4-ene-1,3-dione (35)
Yield, 50% from 27, yellow powder; 1H NMR (400 MHz, CDCl3, Z/E-isomers) δ 12.08/11.98 (1H each, br s, OH), 7.90 (1H × 2, d, J = 16 Hz, H-7), 7.51/7.49 (1H each, brd, J = 16 Hz, H-8), 7.50 (1H × 2, br s, H-12), 7.37 (1H × 2, br d, H-10), 7.11 (1H × 2, m, H-11), 6.68/6.60 (1H each, s, H-5), 2.11/2.12 (3H each, s, Me); 13C NMR (100 MHz, CDCl3, Z/E-isomers) δ 201.1, 200.5, 192.1, 191.6, 167.5, 167.3, 157.9, 154.0, 140.6 (2C), 136.8 (2C), 135.6, 135.5, 131.6, 131.5, 130.0, 129.9, 128.4 (2C), 116.4, 116.3, 103.0, 102.8, 11.3, 10.5; HRESIMS m/z [M+H]+ 247.0411 (calcd for C13H11O3S, 247.0423).
2-[Hydroxy(phenyl)methylene]-4-methylcyclopent-4-ene-1,3-dione (36)
Yield, 52% from 28, pale yellow powder; 1H NMR (400 MHz, CDCl3, Z/E-isomers) δ 8.06 (2H × 2, d, J = 8 Hz, H-8, 12), 7.59 (1H × 2, t, J = 8 Hz, H-10), 7.50 (2H × 2, t, J = 8 Hz, H-9, 11), 11), 6.74/6.59 (1H each, q, J = 1.2 Hz, H-5), 2.12/2.09 (3H each, s, J = 1.2 Hz, Me); 13C NMR (100 MHz, CDCl3, Z/E-isomers) δ 202.7, 202.1, 190.3, 189.8, 173.8, 173.5, 157.5, 153.0, 140.4, 136.2, 133.0, 131.2, 129.9 (2C), 129.7 (4C),128.0 (4C), 103.2, 102.9, 11.4, 10.4; HRESIMS m/z [M+H]+ 215.0681 (calcd for C13H11O3, 215.0703).
2-[(E)-3-[4-O-(t-Butyldimethylsilyl)]phenyl-1-methoxyallylidene]-4-methylcyclopent-4-ene-1,3-dione (37)
Yield, 50% from 29, pale yellow oil; 1H NMR (400 MHz, CDCl3, Z/E-isomers) δ 12.1/12.0 (1H each, br s, OH), 7.76 (1H × 2, d, J = 16 Hz, H-7), 7.61/7.58 (1H each, d, J = 16 Hz, H-8), 7.56 (2H × 2, dd, J = 8, 2 Hz, H-10, 14), 6.89 (2H × 2, d, J = 8 Hz, H-11, 13), 6.67/6.68 (1H each, q, J = 1.6 Hz, H-5), 2.12/2.11(3H each, d, J =1.6 Hz, Me), 1.02 (6H × 2, s, Me), 0.25 (6H × 2, s, 2 × Me).
General Procedure for Alkylation of Cyclopentenediones 30–37
To a solution of the cyclopentenedione (1 mmol) in dry acetone (20 mL) under argon was added K2CO3 (1.5 mmol). After refluxing for 5 minutes, Me2SO4 or Et2SO4 (1.2 mmol) was added to the deep yellow-colored solution, and reflux was continued for 1–3 hr. After completion of the reaction, K2CO3 was removed by filtration. The filtrate was concentrated to dryness and the residue was subjected to reversed phase RP-18 column chromatography using aqueous MeOH (60–80%) to give the desired product.
2-[(E)-1-Methoxy-3-phenylallylidene]-4-methylcyclopent-4-ene-1,3-dione (1, Coruscanone A)
Yield, 93% from methylation of 30, see ref. 3 for physical and spectroscopic data including NMR signal assignments. Anal. HPLC [tR (min) (content)]: 5.65 (97.7%) from solvent a, or 5.72 (99.0%) from solvent b.
2-[(E)-1-Methoxy-3-phenylallylidene]-4,5-dimethylcyclopent-4-ene-1,3-dione (38)
Yield, 60% from methylation of 31, yellow powder; 1H NMR (400 MHz, CDCl3) δ 8.04 (1H, d, J = 16 Hz, H-7), 7.61 (2H, br d), 7.55 (1H), 7.33 (3H, m), 4.18 (3H, s, OMe), 1.99 (6H, br s, Me); 13C NMR (100 MHz, CDCl3) δ 194. 6, 192.2, 167.3, 150.8, 150.3, 141.9, 135.6, 130.1, 128.9 (2C), 128.4 (2C), 121.3, 108.6, 64.5, 9.02 (2C); HRESIMS m/z [M+H]+ 269.1171 (calcd for C17H17O3, 269.1172); Anal. HPLC [tR (min) (content)]: 6.56 (99.5%) from solvent a, or 5.72 (99.6%) from solvent b.
2-[(E)-1-Methoxy-3-phenylallylidene]-4-phenylcyclopent-4-ene-1, 3-dione (39)
Yield, 92% from methylation of 32, yellow powder; 1H NMR (400 MHz, benzene-d6, Z/E-isomers) δ 8.48/8.35( 1H each, d, J = 16 Hz, H-7), 7.84 (2H × 2, m, H-2′, 6′), 7.62 (1H × 2, d, J = 16 Hz, H-8), 7.45 (3H × 2, m), 7.11 (3H × 2, m), 7.02 (3H × 2, m), 6.78/6.76 (1H each, s, H-5), 4.0/3.92 (3H each, s, OMe); 13C NMR (100 MHz, CDCl3, Z/E-isomers) δ 194.0, 193.4, 191.3, 191.1, 170.0, 169.9, 153.0, 152.8, 143.2, 143.0, 138.4, 138.1, 135.5 (2C), 130.8, 130.7, 130.4 (2C), 130.0, 129.1 (4C), 129.0 (3C), 128.9 (4C), 128.8 (2C), 128.6 (4C), 121.4, 121.2, 110.0, 109.8, 64.8, 64.7; HRESIMS m/z [M+H]+ 317.1160 (calcd for C21H17O3, 317.1172); Anal. HPLC [tR (min) (content)]: 9.56 (99.5%) from solvent a, or 11.70 (99.7%) from solvent b.
2-[(E)-3-(4-Chlorophenyl)-1-methoxyallylidene]-4-methylcyclopent-4-ene-1,3-dione (40)
Yield, 95% from methylation of 33, yellow powder; 1H NMR (400 MHz, benzene-d6, Z/E-isomers) δ 8.30/8.23 (1H each, d, J = 16 Hz, H-7), 7.39 (1H × 2, d, J = 16 Hz, H-8), 7.08 (2H × 2, d, J = 7.6 Hz, H-11, 13), 6.94 (2H × 2, d, J = 7.2 Hz, H-10, 14), 6.22/6.19 (1H each, s, H-5), 3.98/3.93 (3H each, s, OMe), 1.60 (6H, br s, H-6); 13C NMR (100 MHz, CDCl3, Z/E-isomers) δ195.2, 194.0, 192.4, 191.6, 168.4, 168.2, 157.0, 156.5, 141.0 (2C), 140.7, 140.4, 136.1 (2C), 134.0 (2C),129.6 (2C), 129.5 (2C), 129.2 (4C), 121.9, 121.7, 108.9, 108.8, 64.8, 64.7, 11.2, 11.1; HRESIMS m/z [M+H]+ 289.0611(calcd for C16H14O3Cl, 289.0626); Anal. HPLC [tR (min) (content)]: 7.68 (96.7%) from solvent a, or 7.46 (99.4%) from solvent b.
2-[(E)-3-(Furan-2-yl)-1-methoxyallylidene]-4-methylcyclopent-4-ene-1,3-dione (41)
Yield, 90% from methylation of 34, yellow powder; 1H NMR (400 MHz, benzene-d6, Z/E-isomers) δ 8.42/8.35 (1H each, d, J = 16 Hz, H-7), 7.33 (1H × 2, d, J = 16 Hz, H-8), 6.89 (1H × 2, brd, H-12), 6.20 (1H × 2, br d), 6.15 (1H × 2, br d), 5.91 (1H × 2, br s, H-5), 3.94/3. 89 (3H each, s, OMe), 1.59/1.58 (3H each, s, Me); 13C NMR (100 MHz, CDCl3, Z/E-isomers) δ 195.0, 193.9, 192.5, 191.7, 168.6, 168.4, 156.6, 156.4, 152.2 (2C), 145.3 (2C), 140.4, 140.2, 128.8 (2C), 119.2, 119.0, 115.2 (2C), 112.7 (2C), 108.7, 108.6, 64.6, 64.5, 11.1, 11.0; HRESIMS m/z [M+H]+ 245.0793 (calcd for C14H13O4, 245.0809); Anal. HPLC [tR (min) (content)]: 4.22 (98.7%) from solvent a, or 3.88 (98.0%) from solvent b.
2-[(E)-1-Methoxy-3-(thiophen-2-yl)allylidene]-4-methylcyclopent-4-ene-1,3-dione (42)
Yield, 90% from methylation of 35, yellow powder; 1H NMR (400 MHz, benzene-d6, Z/E-isomers) δ 8.29/8.23 (1H each, d, J = 16 Hz, H-7), 7.66 (1H, d, J = 16 Hz, H-8), 6.85 (1H × 2, br d), 6.71(1H × 2, br d), 6.54 (1H × 2, br d), 6.19/6.16 (1H each, s, H-5), 3.94/3.89 (3H each, s, OMe), 1.60/1.59 (3H each, s, Me); 13C NMR (100 MHz, CDCl3, Z/E-isomers) δ 195.2, 194.0, 192.5, 191.7, 168.6, 168.4, 156.7, 156.3, 141.3 (2C), 140.5, 140.2, 135.3, 135.2, 130.9, 130.8, 129.3 (2C), 128.3 (2C), 120.4, 120.2, 108.4 (2C), 64.7, 64.6, 11.15, 11.1; HRESIMS m/z [M+H]+ 231.0564 (calcd for C14H13O3S, 261.0580); Anal. HPLC [tR (min) (content)]: 4.77 (95.8%) from solvent a, or 4.64 (96.0%) from solvent b.
2-[Methoxy(phenyl)methylene]-4-methylcyclopent-4-ene-1, 3-dione (43)
Yield, 88% from methylation of 36, pale yellow powder; 1H NMR (400 MHz, CDCl3, Z/E-isomers) δ 7.50 (3H × 2, m), 7.29 (2H × 2, m), 6.70/6.57 (1H each × 2, s, H-5), 3.77/3.76 (3H each × 2, s, OMe), 2.06/1.95 (3H each × 2, s, Me); 13C NMR (100 MHz, CDCl3, Z/E-isomers) δ 193.7, 193.5, 193.0, 192.3, 171.7, 171.5, 156.6, 156.4, 140.4, 140.3, 131.1, 131.0, 130.8,130.7, 128.9 (4C), 128.7 (4C), 107.8(2C), 59.4, 59.2, 11.22, 11.17; HRESIMS m/z [M+H]+ 229.0839 (calcd for C14H13O3, 229.0859); Anal. HPLC [tR (min) (content)]: 2.98 (97.7%) from solvent a, or 2.95 (98.1%) from solvent b.
2-[(E)-3-[4-O-(t-Butyldimethylsilyl)phenyl]-1-methoxyallylidene]-4-methylcyclopent-4-ene-1,3-dione (44)
Yield, 85% from methylation of 37, yellow oil; 1H NMR (400 MHz, CDCl3, Z/E-isomers) δ 7.83/7.81 (1H each, d, J = 16 Hz, H-7), 7.58 (1H × 2, d, J = 16 Hz, H-8), 7.52 (2H × 2, d, J = 8 Hz, H-11, 13), 6.84 (2H × 2, d, J = 8 Hz, H-10, 14), 6.68/6.66 (1H each, s, H-5), 4.18 (3H × 2, s, OMe), 2.08 (3H × 2, s, Me), 0.98 (9H × 2, s, 3 × Me), 0.22 (6H × 2, s, 2 × Me).
2-[(E)-3-(4-Hydroxyphenyl)-1-methoxyallylidene]-4-methylcyclopent-4-ene-1,3-dione (45)
To a solution of 44 (20 mg) in dry THF (10 mL) tetra-t-butyl ammonium fluoride (TBAF) was added under N2. After stirring for 3 hr at room temperature, the reaction mixture was extracted with Et2O. The organic layers were separated, dried (Na2SO4) and evaporated to give the crude product, which was purified by column chromatography on silica gel using 30% EtOAc in hexanes as pale orange powder (11.2 mg in a yield of 80%); 1H NMR (400 MHz, CDCl3, Z/E-isomers) δ 7.84/7.83 (1H each, d, J = 16 Hz, H-7), 7.61 (1H × 2, d, J = 16 Hz, H-8), 7.56 (2H × 2, d, J = 8 Hz, H-10, 14), 6.96 (2H × 2, m, H-11, 13), 6.70/6.68 (1H each, s, H-5), 6.63/6.47 (1H each, br s, OH), 4.21 (3H × 2, s, OMe), 2.10/2.09 (3H each, s, Me); HRESIMS m/z [M+H]+ 271.0935 (calcd for C16H15O4, 271.0965); Anal. HPLC [tR (min) (content)]: 3.21 (96.4%) from solvent a, or 3.84 (98.8%) from solvent b.
2-[(E)-1-Methoxy-3-(4-methoxyphenyl)allylidene]-4-methylcyclopent-4-ene-1,3-dione (46)
To a solution of 45 (1 mmol) in dry acetone (20 mL) under argon was added K2CO3 (1.5 mmol). After refluxing for 5 min, MeI (1.2 mmol) was added to the deep yellow-colored solution, and reflux continued for 3 hr. After filtration, the filtrate was concentrated to dryness to give a residue, which was purified by flash chromatography using 30% EtOAc in hexanes to give 46 as yellow powder in a yield of 90%; 1H NMR (400 MHz, CDCl3, Z/E-isomers) δ 7.87/7.85 (1H each, d, J = 16 Hz, H-7), 7.62 (1H × 2, d, J = 16 Hz, H-8), 7.59 (2H × 2, d, J = 8 Hz, H-10, 14), 6.93 (2H × 2, d, J = 8 Hz, H-11, 13), 6.70/6.68 (1H each, q, J = 1.6 Hz, H-5), 4.21 (3H × 2, s, OMe), 3.86 (3H × 2, s, Ar-OMe), 2.10/2.09 (3H each, s, Me); 13C NMR (100 MHz, CDCl3, Z/E-isomers) δ 195.4, 194.3, 192.6, 191.9, 169.6, 169.4, 161.6 (2C), 156.5, 156.1, 142.78, 142.76, 140.2, 140.0, 130.3 (4C), 128.3 (2C), 118.9, 118.8, 114.4 (4C), 108.2, 108.1, 64.6, 64.5, 55.4 (2C), 11.1. 11.0; HRESIMS m/z [M+H]+ 285.1088 (calcd for C17H17O4, 285. 1121; Anal. HPLC [tR (min) (content)]: 5.31 (97.7%) from solvent a, or 5.67 (99.3%) from solvent b.
2-[(E)-1-Ethoxy-3-phenylallylidene]-4-methylcyclopent-4-ene-1,3-dione (47)
Yield, 92% from ethylation of 30, yellow needles (MeOH), mp 75–76 °C; 1H NMR (400 MHz, CDCl3, Z/E-isomers) δ 8.02/8.00 (1H each, d, J = 16 Hz, H-7), 7.61 (3H × 2, m, H-8, 10, 14), 7.36 (3H × 2, m, H-11, 12, 13), 6.67/6.66 (1H each, s, H-5), 4.45 (2H × 2, m, OCH2CH3), 2.06 (3H × 2, s, Me), 1.45 (3H × 2, m, OCH2CH3); 13C NMR (100 MHz, CDCl3, Z/E-isomers) δ 195.7, 194.5, 192.9, 192.1, 168.1, 167.9, 156.9, 156.5, 143.0 (2C), 140.7, 140.4, 135.7 (2C), 130.5 (2C), 129.1 (4C), 128.7 (4C), 122.0, 121.9, 109.4, 109.3, 74.0, 73.9, 16.0 (2C), 11.41, 11.36; HRESIMS m/z [M+H]+ 269.1155 (calcd for C17H17O3, 269.1172); Anal. HPLC [tR (min) (content)]: 5.82 (97.4%) from solvent b.
2-[(E)-1-Ethoxy-3-phenylallylidene]-4, 5-dimethylcyclopent-4-ene-1,3-dione (48)
Yield, 83% from ethylation of 31, yellow powder; 1H NMR (400 MHz, CDCl3) δ 8.08 (1H, d, J = 16 Hz, H-7), 7.68 (1H, d, J = 16 Hz, H-8), 7.58 (2H, m, H-10, 14), 7.36 (3H, m, H-11, 12, 13), 4.42 (2H, m, OCH2CH3), 1.99 (3H, s, Me), 1.45 (3H, t, OCH2CH3); 13C NMR (100 MHz, CDCl3, Z/E-isomers) δ 194.7, 192.3, 166.2, 150.6, 150.2, 141.9, 135.7, 130.0, 128.8 (2C), 128.4 (2C), 121.9, 109.2, 73.4, 15.7, 8.9; Anal. HPLC [tR (min) (content)]: 9.11 (96.7%) from solvent a, or 9.50 (99.0%) from solvent b.
General Procedure for Acylation of Cyclopentenediones 30 and 31
A solution of the cyclopentenedione (50 mg) in pyridine (2 mL) and Ac2O (2 mL) was kept at room temperature for 24 hr. In the case of using the acylating reagents benzoyl chloride and cinnamoyl chloride, the solution of cyclopentenedione and a catalytic amount of DMAP in pyridine was first cooled to 0°C. After addition of the acylating reagent, the reaction mixture was brought to room temperature and kept for 24 hr. The reaction mixture was poured into ice water and extracted with Et2O. The Et2O layer was washed with brine, dried (Na2SO4), and evaporated to dryness. The residue was chromatographed on silica gel eluting with hexane-acetone (6:1) to afford the product.
2-[(E)-1-Acetoxy-3-phenylallylidene]-4-methylcyclopent-4-ene-1, 3-dione (49)
Yield, 76% from 30, yellow powder; 1H NMR (400 MHz, CDCl3, Z/E-isomers) δ 8.14/8.13 (1H each, d, J = 16 Hz, H-7), 7.61 (2H × 2, m, H-10, 14), 7.45-7.39 (4H × 2, m, H-11, 12, 13, H-8), 6.88/6.82 (1H each, q, J = 1.2 Hz, H-5), 2.49/2.48 (3H each, s, OAc), 2.12/2.08 (3H each, d, J = 1.2 Hz, Me); 13C NMR (100 MHz, CDCl3, Z/E-isomers) δ 193.3, 192.0 (2C), 190.6, 167.52, 167.47, 159.2, 158.9, 155.7, 155.5, 143.2, 143.1, 142.7, 142.3, 134.9 (2C), 130.8 (2C), 129.0 (4C), 128.72 (2C), 128.68 (2C), 119.4, 119.3, 113.4 (2C), 20.91, 20.88, 11.4, 11.3; HRESIMS m/z [M+H]+ 283.0964 (calcd for C17H15O4, 283.0965); Anal. HPLC [tR (min) (content)]: 5.06 (97.8%) from solvent a, or 4.67 (100%) from solvent b.
2-[(E)-1-Acetoxy-3-phenylallylidene]-4, 5-dimethylcyclopent-4-ene-1, 3-dione (50)
Yield, 90% from 31, yellow powder, mp 214–216 °C; 1H NMR (400 MHz, CDCl3) δ 8.18 (1H, d, J = 16 Hz, H-7), 7.62 (2H, m, H-10, 14), 7.42-7.39 (4H, m, H-8, 11, 12, 13), 2.50 (3H, s, OAc), 2.06/2.02 (3H each, s, Me); 13C NMR (100 MHz, CDCl3): δ 192.7, 191.5, 167.7, 154.3, 153.4, 153.0, 142.3, 135.1, 130.6, 129.0 (2C), 128.6 (2C), 119.5, 113.0, 20.9, 9.24, 9.15. HRESIMS m/z [M+H]+ 297.1125 (calcd for C18H17O4, 297.1121); Anal. HPLC [tR (min) (content)]: 5.94 (99.6%) from solvent a, or 5.72 (99.6%) from solvent b.
2-[(E)-1-Benzoxy-3-phenylallylidene]-4, 5-dimethylcyclopent-4-ene-1, 3-dione (51)
Yield, 77% from 31, yellow powder; 1H NMR (400 MHz, CDCl3) δ 8.30 (1H, m, d, J = 16 Hz, H-7), 8.27 (2H, d, J = 7.2 Hz, H-2′, 6′), 7.71 (1H, t, J = 7.2 Hz), 7.63-7.56 (4H, m), 7.45 (1H, d, J = 16 Hz, H-8), 7.39 (4H, m), 2.09/2.00 ( 3H each, s, Me); 13C NMR (100 MHz, CDCl3) δ 192. 7, 191.2, 163.5, 154.6, 153.5, 153.2, 142.4, 135.1, 133.9, 130.6, 130.5 (2C), 129.0 (2C), 128.8 (3C), 128.6 (2C), 119.6, 113.1, 9.3, 9.2; HRESIMS m/z [M+H]+ 359.1263 (calcd for C23H19O4, 359.1278); Anal. HPLC [tR (min) (content)]: 10.11 (99.5%) from solvent a, or 10.79 (99.5%) from solvent b.
2-[(E)-1-Cinnamoyloxy-3-phenylallylidene]-4, 5-dimethylcyclopent-4-ene-1, 3-dione (52)
Yield, 78% from 31, yellow powder; 1H NMR (400 MHz, CDCl3) δ 8.23 ( 1H, d, J = 16 Hz, H-7), 7.94 (1H, d, J = 16 Hz, β-H of cinnamoyl), 7.63 (3H, m, H-10, 14), 7.44-7.38 (8H, m), 6.74 (1H, d, J = 16 Hz, α-H of cinnamoyl), 2.07/2.00 (3H each, s, Me); 13C NMR (100 MHz, CDCl3) δ 193.0, 191.5, 163.7, 154.5, 153.5, 153.3, 148.0, 142.5, 135.4, 134.2, 131.1, 130.8, 129.2 (4C), 128.8 (4C), 119.8, 116.5, 113.4, 9.5, 9.4; Anal. HPLC [tR (min) (content)]: 12.33 (97.6%) from solvent a, or 10.49 (97.9%) from solvent b.
2-(1-Methoxyethylidene)-4-methyl-cyclopent-4-ene-1,3-dione (53)
To a solution of 2-methylmaleic anhydride (2) (8.0 g, 71.4 mmol) in dry 1,2-dichloroethane (200 mL) at room temperature under N2 is added anhydrous AlCl3 (32.3 g, 133.4 mmol). The suspension was cooled to 0 °C and α-methylvinyl acetate (10.7 g, 107.1 mmol) was added dropwise over 15 minutes. The mixture was subsequently refluxed for 1.5 hr. After cooling, the mixture was poured to ice water containing 10% HCl (300 mL). The aqueous layer was extracted with CHCl3 (150 mL × 3). The combined organic layers were washed with brine (150 mL) and H2O (200 mL × 4) to neutral, dried (Na2SO4), and concentrated to dryness to yield a residue (8.5 g), which was chromatographed on silica gel eluting with hexane-EtOAc (1:2) to afford 2-(1-hydroxyethylidene)-4-methylcyclopent-4-ene-1,3-dione (5.97 g, 55%) as pale yellow powder, 1H NMR (400 MHz, CDCl3, E/Z-isomers) δ 11.8 (br s, OH), 6.50/6.42 (br s, H-5), 2.22 (s, Me), 1.91 (s, Me-4); 13C NMR (100 MHz, CDCl3, E/Z-isomers) δ 200.4/199.6/192.1/191.7 (CO), 176.9/176.7 (C-6), 157.8/153.4 (C-4), 140.5/136.2 (C-5), 104.2 (2C, C-2), 17.9 (vinylic Me), 11.2/10.2 (Me-4); HRTOFMS m/z [M + H]+ 153.0585 (calcd for C8H9O3, 153.0546). Following the general procedure for methylation of cyclopentenediones, the title compound was obtained from the above compound in a yield of 46% as yellow powder; 1H NMR (400 MHz, CDCl3, E/Z-isomers) δ 6.61/6.59 (br s, H-5), 4.01 (s, OMe), 2.62 (br s, vinylic Me), 2.03/2.05 (s, Me-4); 13C NMR (100 MHz, CDCl3, E/Z-isomers) δ 195.4/194.5/193.0/192.5 (C=O), 173.8/173.5 (C-6), 155.8/155.2 (C-4), 139.7/139.1 (C-5), 107.2 (2C, C-2), 56.0/55.9 (OMe), 14.05/13.99 (vinylic Me), 11.0/10.8 (Me-4); HRTOFMS m/z [M + H]+ 167.0682 (calcd for C9H11O3, 167.0703). Anal. HPLC [tR (min) (content)]: 2.51 (97.6%) from solvent a, or 2.66 (98.5%) from solvent b.
2-Methoxy-1,4-bis-methoxymethoxybenzene (55)
A solution of methoxyhydroquinone (54) (1 g, 7.14 mmol) in dry THF (30 mL) was added to an ice-cooled, stirred suspension of NaH (0.68 g of 60% dispersion in mineral oil, 17.00 mmol) in dry THF (100 mL). After stirring for 10 min at 0 °C, chloromethoxymethyl ether (2.29 g, 28.44 mmol) was added and stirring continued for 24 hr. Crushed ice and water (200 mL) were added to the reaction mixture. The aqueous phase was extracted with Et2O (200 mL × 3). The organic layers were combined, washed with H2O (150 mL), dried (Na2SO4), and concentrated to dryness. The residue was chromatographed on silica gel using CHCl3 as solvent to give 55 (1.23 g, yield, 75%) as colorless oil, 1H NMR (CDCl3, 400 MHz) δ 7.03 (1H, d, J = 8.7 Hz), 6.63 (1H, d, J = 2.6 Hz), 6.56 (1H, dd, J = 8.7, 2.6 Hz), 5.13 (2H, s), 5.11 (2H, s), 3.84 (3H, s), 3.50 (3H, s), 3.47 (3H, s); 13C NMR (CDCl3, 100 MHz) δ 153.5 (s), 151.2 (s), 141.8 (s), 118.2 (d), 107.5 (d), 102.4 (d), 96.6 (t), 95.5 (t), 56.5 (q), 56.3 (q), 56.2 (q).
1-Bromo-4-methoxy-2,5-bis-methoxymethoxybenzene (56)
To a stirred solution of 55 (1 g, 4.386 mmol) and NaHCO3 (368 mg, 4.386 mmol) in MeOH (100 mL) at room temperature was added slowly a solution of pyridium hydrobromide perbromide (C5H5N+HBr3−) (1.47 g, 4.596 mmol) in MeOH (50 mL) in 25 min. The reaction was complete immediately after the addition. The reaction mixture was diluted with ice water (150 mL) and extracted with Et2O (125 mL × 4). The combined organic layers were washed with brine and H2O, dried (MgSO4), and concentrated to dryness to give 56 (1.2 g, yield, 89%) as a purple powder, 1H NMR (CDCl3, 400 MHz) δ 7.30 (1H, s), 6.78 (1H, s), 5.16 (2H, s), 5.12 (2H, s), 3.84 (3H, s), 3.52 (3H, s), 3.49 (3H, s); 13C NMR (CDCl3, 100 MHz) δ 150.2 (s), 149.6 (s), 142.2 (s), 121.5 (d), 102.7 (d), 102.5 (s), 96.4 (t), 96.3 (t), 56.6 (q), 56.4 (q), 56.3 (q).
1-Methoxy-2,5-bis-methoxymethoxy-4-methylbenzene (57)
To a solution of 56 (0.88 g, 2.857 mmol) in dry THF (30 mL) at −78 °C was added MeLi (2.45 mL of 1.4 M in Et2O, 3.43 mmol). After stirring at −78 °C for 1 hr, MeI (2.03 g, 14.296 mmol) was added to the resultant white cloudy suspension. The solution was stirred at −78 °C for 1 hr, and then warmed to room temperature. The reaction mixture was diluted with saturated aqueous NH4Cl (20 mL) and water (20 mL). The organic layer was separated and the aqueous layer was extracted with Et2O (50 mL × 3). The combined organic layers were washed with brine and H2O, dried (MgSO4), and concentrated to dryness to yield 57 (0.62 g, yield, 90%) as a colorless oil, 1H NMR (CDCl3, 300 MHz) δ 6.88 (1H, s), 6.68 (1H, s), 5.08 (4H, s), 3.78 (3H, s), 3.46 (3H, s), 3.44 (3H, s), 2.12 (3H, s); 13C NMR (CDCl3, 75 MHz) δ 151.1 (s), 148.9 (s), 141.3 (s), 120.3 (d), 119.5 (s), 101.5 (s), 96.6 (t), 95.8 (t), 56.5 (q), 56.4 (q), 56.2 (q), 15.8 (q).
1-(2-Methoxy-3,6-bis-methoxymethoxy-5-methylphenyl)-2-phenylethanol (58)
To a solution of 57 (0.59 g, 2.438 mmol) in dry THF (5 mL) at −78 °C was added BuLi (1.46 mL of 2.5 M in hexane, 3.657 mmol). After stirring at −78 °C for 1 hr, the solution was warmed to 0 °C and stirring continued at 0 °C for 5 hr. The solution was again warmed to 25 °C and stirred for 3 hr. The resultant white cloudy suspension was cooled to 0 °C. Phenylacetaldehyde (0.37 g, 3.657 mmol) was added slowly over 10 min and stirring continued for 30 min. The reaction was quenched with saturated aqueous NH4Cl (20 mL). The organic layer was separated and the aqueous layer was extracted with Et2O (30 mL × 3). The combined organic layers were washed with brine and H2O, dried (MgSO4), and concentrated to dryness. The residue was chromatographed on silica gel using CH2Cl2 and then CH2Cl2–MeOH (50:1) to yield 58 (0.55 g, yield, 66%) as an off-white oil, 1H NMR (CDCl3, 300 MHz) δ 7.25 (5H, m, H-2″-6″), 6.92 (1H, s, H-4′), 5.28 (1H, dd, J = 8.4, 5.5 Hz, H-1), 5.16 (2H, s, MeOCH2O-3′), 4.96/4.89 (1H each, ABq, J = 5.6 Hz, MeOCH2O-6′), 3.90 (3H, s, MeO-2′), 3.60 (3H, s, MeOCH2O-6′), 3.52 (3H, s, MeOCH2O-4′), 3.29 (1H, dd, J = 13.6, 8.4 H-2a), 3.18 (1H, dd, J = 13.7, 5.5 Hz, H-2b); 13C NMR (CDCl3, 75 MHz) δ 149.2 (s, C-6′), 147.1 (s, C-2′), 146.9 (s, C-3′), 139.8 (s, C-1″), 130.8 (s, C-1′), 129.9 (2C, d, C-3″,5″), 128.6 (2C, d, C-2″,6″), 127.0 (s, C-5′), 126.6 (s, C-4″), 118.6 (d, C-4′), 100.4 (d, MeOCH2O-6′), 95.9 (d, MeOCH2O-3′), 70.3 (d, C-1), 61.6 (q, MeO-2′), 57.9 (q, MeOCH2O-6′), 56.7 (q, MeOCH2O-3′), 44.6 (t, C-2), 17.4 (q, Me-5′); the above NMR signal assignments were facilitated by a 2D HMBC experiment.
2-Methoxy-5-methyl-3-styrylbenzene-1,4-diol (59)
A solution of 58 (100 mg, 0.291 mmol) and 3N HCl (3 mL) in dioxane (10 mL) was stirred under argon at 50 °C for 8 hr. The reaction mixture was diluted with H2O (20 mL) and then extracted with Et2O (30 mL × 3). The combined organic layers were washed with brine and H2O, dried (MgSO4), and concentrated to dryness to give 59 (71 mg, yield, 95%) as an off-white powder, 1H NMR (CDCl3, 300 MHz) δ 7.55 (2H, br d, J = 7.2 Hz H-2″,6″), 7.39 (2H, br t, J = 7.1 Hz, H-3″,5″), 7.32 (1H, d, J = 7.5 Hz, H-4″), 7.29 (1H, d, J = 16.3 Hz, H-1′), 7.20 (1H, d, J = 16.9 Hz, H-1′), 6.71 (1H, s, H-5), 3.76 (3H, s, MeO-2), 2.22 (3H, s, Me-5).
2-Methoxy-5-methyl-3-styryl-1,4-benzoquinone (60)
A solution of 59 (70 mg, 0.273 mmol) and DDQ (160 mg, 0.705 mmol) in CH2Cl2 (15 mL) and H2O (2 mL) was stirred under argon at room temperature for 12 hr. The reaction mixture was diluted with H2O (20 mL) and then extracted with CH2Cl2 (30 mL × 3). The combined organic layers were washed with brine and H2O, dried (MgSO4), and concentrated to dryness. The residue was chromatographed on silica gel using CH2Cl2 as solvent to yield 60 (45 mg, yield, 65%) as red powder; UV (CH3CN), λmax (log ε) 278 (4.35), 292 (4.35) nm; IR (KBr) νmax, 1652, 1611, 1559, 1448, 1336, 1305, 1230, 1135, 965, 885, 750, 692 cm−1; 1H NMR (CDCl3, 300 MHz) δ 7.87 (1H, d, J = 16.6 Hz, H-8), 7.60 (2H, d, J = 7.2 Hz, H-11,15), 7.43 (2H, t, J = 7.2 Hz, H-12,14), 7.35 (1H, t, J = 7.2 Hz, H-14), 7.25 (1H, d, J = 16.6 Hz, H-9), 6.54 (1H, q, J = 1.5 Hz, H-6), 4.19 (3H, s, OMe), 2.13 (3H, d, J = 1.4 Hz, H-7); 13C NMR (CDCl3, 75 MHz) δ 188.0 (s, C-1), 183.6 (s, C-4), 154.3 (s, C-2), 146.0 (s, C-5), 138.9 (d, C-9), 138.0 (s, C-10), 132.1 (s, C-6), 128.9 (d, C-12,14), 128.8 (d, C-13), 127.3 (d, C-11,15), 126.2 (C-4), 117.6 (C-8), 61.3 (OMe), 16.2 (C-7); ESIMS m/z [M (C15H11O3) + H]+ 255.
In Vitro Antifungal Susceptibility Testing
A modified version of the CLSI (formerly NCCLS) methods was used for susceptibility testing.21 Organisms were obtained from either the American Type Culture Collection (Manassas, VA) [Candida albicans ATCC 90028, Cryptococcus neoformans ATCC 90113, and Aspergillus fumigatus ATCC 90906] or provided by Spencer Redding/Ted White – C. albicans isolates #1 (fluconazole-susceptible) and #17 (fluconazole-resistant).11 Samples were serially-diluted in 20% DMSO/saline and transferred in duplicate to 96 well flat bottom microplates. Inocula were prepared by correcting the OD630 of microbe suspensions in incubation broth [RPMI 1640/2% dextrose/0.03% glutamine/MOPS at pH 6.0 (Cellgro) for C. albicans, Sabouraud Dextrose for C. neoformans, and 5% Alamar Blue™/RPMI 1640 broth (2% dextrose, 0.03% glutamine, buffered with 0.165M MOPS at pH 7.0) for A. fumigatus] to afford final target inocula of: C. albicans: 1.0 × 104, C. neoformans: 1.0 × 105, and A. fumigatus: 3.0 × 104 CFU/ml. Drug controls [amphotericin B (ICN Biomedicals, Ohio) and fluconazole (Sequoia Research Products Ltd., UK) were included in each assay. All organisms were read at 630 nm using the EL-340 Biokinetics Reader (Bio-Tek Instruments, Vermont) prior to and after incubation (C. albicans at 37 °C for 18–24 h; C. neoformans and A. fumigatus at 30 °C for 72 h) except A. fumigatus, which was read at 544ex/590em using the Polarstar Galaxy Plate Reader (BMG LabTechnologies, Germany). Percent growth was calculated and plotted versus test concentration to afford the IC50 (sample concentration that affords 50% growth of the organism). The minimum inhibitory concentration (MIC) is defined as the lowest test concentration that allows no detectable growth. Minimum fungicidal concentrations (MFCs) were determined by removing 5 μl from each clear (or blue) well, transferring to agar and incubating as previously mentioned. The MFC was defined as the lowest test concentration that allows no growth of the organism on agar.
In Vitro Cytotoxicity Assay
The mammalian cells Vero (African green monkey kidney fibroblast) and LLC-PK-1 (Pig kidney epithelial) used in this study were obtained from ATCC (Manassas, VA). Cytotoxicity was determined by Neutral Red method22 up to a highest concentration of 10 μg/mL. The detailed procedure was described in a previous paper.23
Supplementary Material
1H and/or 13C NMR spectra of compounds 33, 38–43, 45–47, 49, 52, 53, and 60. This material is available free of charge via the Internet at http://pubs.acs.org.
Figure 2.
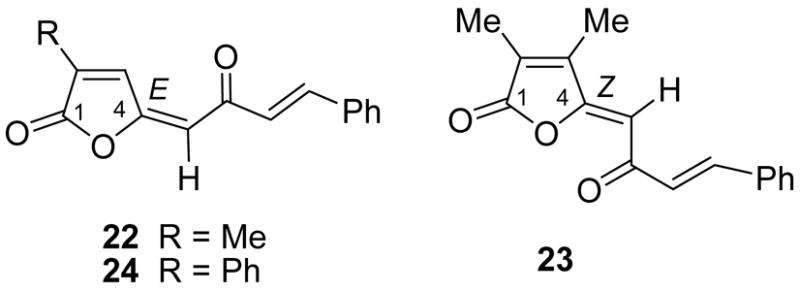
Key butenolide intermediates 22–24.
Figure 3.

Synthetic coruscanone A analogs.
Scheme 1.

General Reaction Scheme for Preparation of Coruscanone A (1) Analogs
(R1,R2: H, CH3, alkyl, or phenyl; R3: aromatic, styryl, heteroaromatic, etc.; R4:CH3, alkyl, acyl, etc)
Scheme 2.

Synthesis of Benzoquinone 60a
a Reagents and conditions: (a) MeOCH2Cl/NaH, THF, 0 °C, 24 hr, 75%. (b) C5H5NHBr3/MeOH/NaHCO3, r.t., 25 min, 89%. (c) (i) MeLi/THF, −78 °C, 1 hr; (ii) MeI, −78 °C, 1 hr, 90%. (d) (i) BuLi/THF, −78 °C, 1 hr, then warm to 0 °C, 5 hr, r.t., 3 hr; (ii) PhCH2CHO, 0 °C, 0.5 hr; 66%. (e) 3N HCl/dioxane, 50 °C, 3 hr, 95%. (f) DDQ/CH2Cl2-H2O, r.t., 12 hr, 65%.
Scheme 3.

Proposed Mechanism for Inhibition of Human Chymase by 61
Scheme 4.

Possible Interaction Mechanism of 1 With the Target
Acknowledgments
The authors wish to thank Dr. Ted White and Dr. Spencer Redding for kindly providing the fluconazole-resistant Candida albicans strains, Ms. Marsha Wright for biological testing, Dr. D. Chuck Dunbar and Dr. Bharthi Avula for running MS spectra, and Dr. Ameeta K. Agarwal, Dr. Mohammad K. Ashfaq and Dr. Susan P. Manly for helpful discussion. This work was supported by the NIH, NIAID, Division of AIDS, Grant No. AI 27094 and the USDA Agricultural Research Service Specific Cooperative Agreement No. 58-6408-2-0009.
References
- 1.(a) Georgopapadakou NH, Walsh TJ. Antifungal agents: chemotherapeutic targets and immunologic strategies. Antimicrob Agents Chemother. 1996;40:279–291. doi: 10.1128/aac.40.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kontoyiannis DP, Mantadakis E, Samonis G. Systemic mycoses in the immunocompromised host: an update in antifungal therapy. J Hosp Infect. 2003;53:243–258. doi: 10.1053/jhin.2002.1278. [DOI] [PubMed] [Google Scholar]; (c) White TC, Bowden RA, Marr KA. Clinical, cellular, and molecular factors that contribute to antifungal drug resistance. Clin Microbiol Rev. 1998;11:382–402. doi: 10.1128/cmr.11.2.382. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wong-Beringer A, Kriengkauykiat J. Systemic antifungal therapy: new options, new challenges. Pharmacotherapy. 2003;23:1441–1462. doi: 10.1592/phco.23.14.1441.31938. [DOI] [PubMed] [Google Scholar]
- 2.Newman DJ, Cragg GM, Snader KM. Natural products as sources of new drugs over the period 1981–2002. J Nat Prod. 2003;66:1022–1037. doi: 10.1021/np030096l. [DOI] [PubMed] [Google Scholar]
- 3.Li XC, Ferreira D, Jacob MR, Zang Q, Khan SI, Elsohly HN, Nagle DG, Smille TJ, Khan IA, Walker LA, Clark AM. Antifungal cyclopentenediones from Piper coruscans. J Am Chem Soc. 2004;126:6872–6873. doi: 10.1021/ja048081c. [DOI] [PubMed] [Google Scholar]
- 4.(a) Lee HH. The structure of lucidone and methyllucidone. Tetrahedron Lett. 1968:4243–4246. [Google Scholar]; (b) Ng S, Lee HH, Bennett G. 13C NMR study on linderones and lucidones. Magn Resn Chem. 1990;28:337–342. [Google Scholar]
- 5.(a) Iwataki I, Shibuya M, Nakada A, Mizuno M. 2-(Aminomethylene)-4-cyclopentene-1,3-diones as agricultural fungicides. JP 53101336. [Google Scholar]; (b) Kataoka M, Naruse N, Kawabe N, Hosoi K, Ohno M, Okuda S, Eguchi J, Tosaya M, Umezawa H, Takeuchi T. β-Trione compounds as fungicides. JP51001633. [Google Scholar]; (c) Kawada H, Hayashi S, Kasugai A, Shigematsu T. Benzylidenecyclopentene microbicide. JP 52079022. [Google Scholar]; (b) Lee DL, Michaely WJ. Herbicidal 2-(2-substittued benzoyl)-1,3-cyclopentanedione. US 4681621. [Google Scholar]; (e) Simonov VV, Anishchenkom AF, Popova EN, Dunaeva TP, Gazizov RT, Simonov VD. Halogenated spirans. DE 2804271. [Google Scholar]
- 6.Clemo NG, Geodge DR, Pattenden G. Synthesis of calythrone and related cyclopentene-1,3-diones via rearrangement of 4-ylidenebutenolides. J Chem Soc, Perkin Trans 1. 1981:1448–1453. [Google Scholar]
- 7.Nilsson M. Preparation of 2-acetylcyclopent-4-ene-1,3-diones from maleic anhydrides and isopropenyl acetate. Acta Chem Scand. 1964;18:441–446. [Google Scholar]
- 8.(a) Suzuki Y, Kono Y, Inoue T, Sakurai A. A potent antifungal benzoquinone in etiolated sorghum seedlings and its metabolites. Phytochemistry. 1998;47:997–1001. [Google Scholar]; (b) Haraguchi H, Taniguchi M, Tanaka T, Oi S, Hashimoto K. Citrinin, an electron acceptor having antifungal activity. Agric Biol Chem. 1989;53:1741–1742. [Google Scholar]; (c) Wang HJ, Gloer KB, Gloer JB, Scott JA, Malloch D. Anserinones A and B: new antifungal and antibacterial benzoquinones from the coprophilous fungus Podospora Anserina. J Nat Prod. 1997;60:629–631. doi: 10.1021/np970071k. [DOI] [PubMed] [Google Scholar]; (d) Meazza G, Dayan FE, Wedge DE. Activity of Quinones on Colletotrichum Species. J Agric Food Chem. 2003;51:3824–3828. doi: 10.1021/jf0343229. [DOI] [PubMed] [Google Scholar]
- 9.Kalinin AV, Da Silva AJM, Lopes CC, Lopes RSC, Snieckus V. Directed ortho metalation and cross coupling links. Carbamoyl rendition of the Baker-Venkataraman rearrangement. Regiospecific route to substituted 4-hydroxycoumarins. Tetrahedron Lett. 1998;39:4995–4998. [Google Scholar]
- 10.(a) Bruce JM, Creed D, Dawes K. Light-induced and related reactions of quinines. Part VIII. Some (2-hydroxyalkyl)-, phenethyl, (2-ethoxycarbonylethyl)-, and styryl-1,4-benzoquinones. J Chem Soc C. 1971:3749–3756. [Google Scholar]; (b) Irngartinger H, Stadler B. Synthesis, structures and topochemistry of 2-monovinyl-substituted 1,4-benzoquinones. Eur J Org Chem. 1998:605–626. [Google Scholar]; (c) Brehm I, Meier H. Formation and reactivity of 2-styryl-1,4-benzoquinones. Eur J Org Chem. 2001:3307–3311. [Google Scholar]; (d) Brehm I, Hinneschiedt S, Meier H. 1,4-Benzoquinones with styryl substituents. Eur J Org Chem. 2002:3162–3170. [Google Scholar]
- 11.(a) Pfaller MA, Rhine-Chalberg J, Redding SW, Smith J, Farinacci G, Fothergill AW, Rinaldi MG. Variations in fluconazole susceptibility and electrophoretic karyotype among oral isolates of Candida albicans from patients with AIDS and oral candidiasis. J Clin Microbiol. 1994;32:59–64. doi: 10.1128/jcm.32.1.59-64.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) White TC. Increased mRNA levels of ERG16, CDR, and MDR1 correlate with increases in azole resistance in Candida albicans isolates from a patient infected with human immunodeficiency virus. Antimicrob Agents Chemother. 1997;41:1482–1487. doi: 10.1128/aac.41.7.1482. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) White TC. The presence of an R467K amino acid substitution and loss of allelic variation correlate with an azole-resistant lanosterol 14α-demethylase in Candida albicans. Antimicrob Agents Chemother. 1997;41:1488–1494. doi: 10.1128/aac.41.7.1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Griffiths DG, Partis MD, Sharp RN, Beechey RB. N-Polymethylenecarboxy-maleimides – A new class of probes for membrane sulphydryl groups. FEBS Lett. 1981;134:261–263. doi: 10.1016/0014-5793(81)80615-1. [DOI] [PubMed] [Google Scholar]; (b) Partis MD, Griffiths DG, Roberts GC, Beechey RB. Cross-linking of protein by ω-maleimido alkanoyl N-hydroxysuccinimido esters. J Protein Chem. 1983;2m:263–277. [Google Scholar]
- 13.Aoyama Y, Konoike T, Kanda A, Naya N, Nakajima M. Total synthesis of human chymase inhibitor methyllinderone and structure-activity relationships of its derivatives. Bioorg Med Chem Lett. 2001;11:1695–1697. doi: 10.1016/s0960-894x(01)00265-7. [DOI] [PubMed] [Google Scholar]
- 14.McGoverns SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 15.Ahn BZ, Sok SE. Michael acceptors as a tool for anticancer drug design. Curr Pharm Design. 1996;2:247–262. [Google Scholar]
- 16.Zeller KP, Mewes K, Ehninger G, Blanz J. Formation of reactive intermediates by cytochrome P-450 mediated oxidation of the anti-cancer drug mitoxantrone. Pure Appl Chem. 1994;66:2415–2418. [Google Scholar]
- 17.(a) Powers JC, Asgian JL, Ekici OD, James KE. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem Rev. 2002;102:4639–4750. doi: 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]; (b) Ekici OD, Goetz MG, James KE, Li ZZ, Rukamp BJ, Asgian JL, Caffrey CR, Hansell E, Dvorak J, McKerrow JH, Potempa J, Travis J, Mikolajczyk J, Salvesen GS, Powers JC. Aza-peptide Michael acceptors: A new class of inhibitors specific for caspases and other Clan CD cysteine proteases. J Med Chem. 2004;47:1889–1892. doi: 10.1021/jm049938j. [DOI] [PubMed] [Google Scholar]; (c) Dragovich PS, Prins TJ, Zhou R, Johnson TO, Hua Y, Luu HT, Sakata SK, Brown EL, Maldonado FC, Tuntland T, Lee CA, Fuhrman SA, Zalman LS, Patick AK, Matthews DA, Wu EY, Bennett MG, Borer C, Nayyar NK, Moran T, Chen L, Rejto PA, Rose PW, Guzman MC, Dovalsantos EZ, Lee S, McGee K, Mohajeri M, Liese A, Tao J, Kosa MB, Liu B, Batugo MR, Gleeson J-PR, Wu ZP, Liu J, Meador JW, Ferre RN. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 8. Pharmacological optimization of orally bioavailable 2-pyridone-containing peptidomimetics. J Med Chem. 2003;46:4572–4585. doi: 10.1021/jm030166l. [DOI] [PubMed] [Google Scholar]; (d) Ekici OD, Li ZZ, Campbell AJ, James KE, Asgian JL, Mikolajczyk J, Salvesen GS, Ganesan R, Jelakovic S, Gruetter MG, Powers JC. Design, synthesis, and evaluation of aza-peptide Michael acceptors as selective and potent inhibitors of Caspases-2, -3, -6, -7, -8, -9, and -10. J Med Chem. 2006;49:5728–5749. doi: 10.1021/jm0601405. [DOI] [PubMed] [Google Scholar]
- 18.(a) Prestera T, Holtzclaw WD, Zhang Y, Talalay P. Chemical and molecular regulation of enzymes that detoxify carcinogens. Proc Natl Acad Sci U S A. 1993;90:2965–2969. doi: 10.1073/pnas.90.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dinkova-Kostova AT. Protection against cancer by plant phenylpropenoids: induction of mammalian anticarcinogenic enzymes. Mini-Rev Med Chem. 2002;2:595–610. doi: 10.2174/1389557023405558. [DOI] [PubMed] [Google Scholar]; (c) Gao X, Talalay P. Induction of phase 2 genes by sulforaphane protects retinal pigment epithelial cells against photooxidative damage. Proc Natl Acad Sci U S A. 2004;101:10446–10451. doi: 10.1073/pnas.0403886101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rinaldi R, Eliasson E, Swedmark S, Morgenstern R. Reactive intermediates and the dynamics of glutathione transferases. Drug Metab Dispos. 2002;30:1053–1058. doi: 10.1124/dmd.30.10.1053. [DOI] [PubMed] [Google Scholar]
- 20.Greene TW, Wuts PGM. Protective groups in organic synthesis. 2. John Wiley & Sons; New York: 1999. pp. 77–80. [Google Scholar]
- 21.(a) NCCLS. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts; Approved Standard M27-A2. National Committee on Clinical Laboratory Standards. 2002;22(15) [Google Scholar]; (b) NCCLS. Reference method for broth dilution antifungal susceptibility testing of filamentous fungi; approved standard, M38-A. National Committee on Clinical Laboratory Standards. 2002;22(16) [Google Scholar]
- 22.Borenfreund E, Puerner J. Toxicity determined in vitro morphological alterations and neutral red absorption. Toxicol Lett. 1985;24:119–124. doi: 10.1016/0378-4274(85)90046-3. [DOI] [PubMed] [Google Scholar]
- 23.Yang CR, Zhang Y, Jacob MR, Khan SI, Zhang YJ, Li XC. Antifungal activity of C-27 steroidal saponins. Antimicrob Agents Chemother. 2006;50:1710–1714. doi: 10.1128/AAC.50.5.1710-1714.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1H and/or 13C NMR spectra of compounds 33, 38–43, 45–47, 49, 52, 53, and 60. This material is available free of charge via the Internet at http://pubs.acs.org.