

One of the most fascinating subjects in chemistry and biology is structural and functional comparison between heme-copper centers in heme copper oxidases (HCOs) and heme-non-heme iron centers in bacterial NO reductases (NORs). Both contain a heterobinuclear center with a heme in close proximity to either a copper (in HCOs) or a non-heme iron (in NOR).1,2 While the heme-copper center catalyzes four-electron reduction of O2 to H2O,3–5 the heme-non-heme iron center promotes two-electron reduction of NO to N2O (2NO + 2H+ + 2e− = N2O + H2O).6,7 The structural similarities between HCOs and NORs suggest that they might have evolved from a common phylogeny.8 Therefore, while copper was selected by nature for O2 reduction, iron is preferred for NO reduction, indicating that the presence of a CuB or an FeB site is a prerequisite for enzyme catalysis. Therefore, an interesting issue is why a protein is efficient at O-O bond cleavage when using a copper ion and is proficient at N-N bond formation when using an iron ion.
An entry point to addressing the above issue is cross reactivity between the two enzymes. Previous studies have shown that a bacterial NOR from Paracoccus denitrificans has HCO activity.9 Several families of HCOs, such as the ba3 and caa3 oxidases from Thermus thermophilus and cytochrome cbb3 oxidase from Pseudomonas stutzeri, have displayed NOR reactivity, while other families, such as cytochrome oxidase from bovine heart, show no NOR activity.10–13 The reasons for the different reactivities between HCO and NOR and among HCOs from different species still remain to be clarified. A contributing factor is difficulty in replacing one metal ion with another in native HCOs or NORs. Synthetic models that mimic the native enzymes both structurally and functionally, are rare in the literature.14–16
To provide insights into the reduction of O2 by HCO, a CuB center has been designed into wild-type Mb (called CuBMb, Figure 1A) to create a binuclear heme-copper center as a small model protein.17 Studies of the CuB Mb have shown that the CuB center plays a critical role in O2 binding and reduction,17 and modulation of redox potential of the heme when the heme and copper is coupled.20 In addition, proton delivery, perhaps through a hydrogen bonding network, is important in heterolytic O–O bond cleavage.18,19 Finally both the heme type19 and presence of chloride20,21 also play a role in its O2 reduction. Here, we report the effects of metal ions in the CuB center on the reaction of NO with CuBMb. This study shows that the redox property and the oxidation state of metal ions in the CuB center can exert significant structural and reactivity changes for NO reduction and the implications of such interactions in HCOs are discussed.
Figure 1.
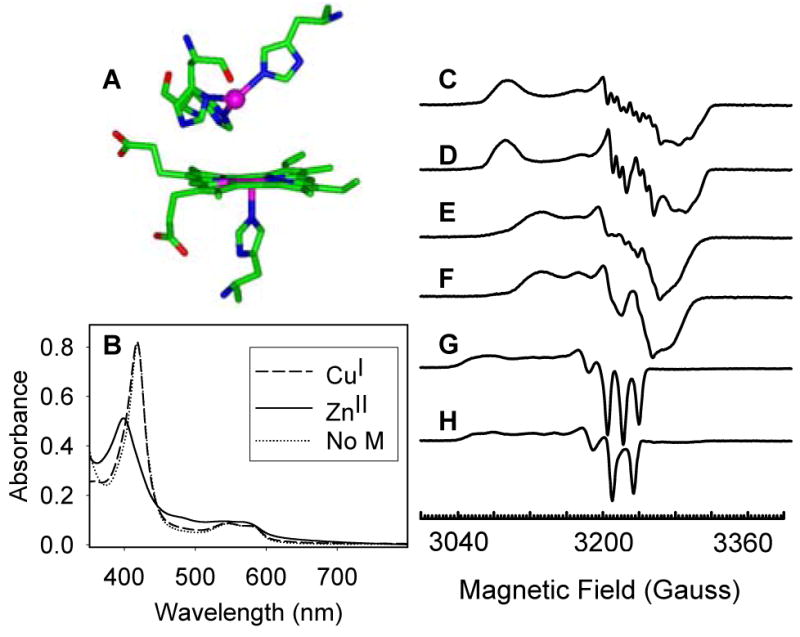
(A) The active site of a computer model of CuBMb; (B) UV-Vis spectra of ferrous CuBMb-NO in the absence of metal ions (dotted line); in the presence of Cu(I) (dashed line) and in the presence of Zn(II) (solid line); EPR spectra of (C) ferrous-CuBMb-NO and (D) ferrous-CuBMb-15NO in the absence of metal ions; (E) ferrous-CuBMb-NO and (F) ferrous-CuBMb- 15NO in the presence of copper; (G) ferrous-CuBMb-NO and (H) ferrous-CuBMb- 15NO in the presence of Zn(II). Samples were recorded in 20 mM Tris pH 8 at 45 K and 0.2 mW power; microwave frequency, 9.050 GHz.
The absorption spectrum of ferrous-CuBMb-NO, prepared from the reaction of met-CuBMb with excess dithionite and NaNO2 under Ar, displays a Soret band at 420 nm and visible absorption bands at 548 and 583 nm (Figure 1B), similar to that of ferrous-WTswMb-NO.22,23 The EPR spectra of ferrous CuBMb-NO and CuBMb-15NO, prepared using NaNO2 and Na15NO2 respectively, displayed g values at 2.090, 2.003 and 1.972 (Figure 1C and 1D). The hyperfine splitting from both bound NO and the proximal histidine nitrogen can be clearly observed, indicating the formation of a six-coordinate ferrous heme-nitrosyl species.24
The effect of copper on the binding of NO to ferrous CuBMb was studied under the same conditions. As shown in Figures 1E and 1F, the EPR spectra of ferrous-CuBMb-NO in the presence of copper showed shifted g values at 2.067, 2.006 and 1.97. Although the hyperfine splitting from NO is still clearly observed, the hyperfine splitting from the proximal histidine becomes less resolved in comparison to the Cu-free species, probably due to a weakening of the proximal heme Fe-His bond after the binding of Cu. In the presence of Zn(II), the UV-Vis spectrum of ferrous CuBMb-NO showed a Soret band at 399 nm, a charge-transfer peak at 484 nm (as a shoulder), and visible bands at 543 nm and 568 nm, which are characteristic of the formation of a five-coordinate ferrous heme-NO species (Figure 1B).25,26 This conclusion is further supported by the characteristic g values (2.107, 2.032 and 2.009) and the hyperfine splitting pattern of the EPR spectra of the ferrous CuBMb-NO and CuBMb-15NO species in the presence of Zn(II) (Figures 1G and 1H), which is similar to the EPR spectrum of five-coordinate ferrous Hemoglobin-NO.24 Thus, although the binding of Cu(I) in the CuB center can only weaken the proximal heme Fe-His bond, the bound Zn(II) caused the complete cleavage of the Fe-His bond and formation of a five-coordinate heme-NO complex. As a control experiment, the EPR spectra of WTswMb-NO showed little change in the presence of Cu(I) or Zn(II) (see Supporting Information). These results demonstrated that the bound Cu(I) and Zn(II) in CuBMb play different roles on the binding of NO to heme.
To examine the reactivity of CuBMb towards NO reduction, purified NO was added to a solution containing CuBMb under He in the presence of metal ions and ascorbate/tetramethyl-p-phenylenediamine (TMPD). In the presence of Cu(II), CuBMb catalyzes the reduction of NO to N2O, as evidenced by the appearance of a second peak at a longer retention time in the GC, which corresponds to a 44 MW peak (N2O) in the MS (Figure 2). In addition, the relative GC peak intensity of N2O:NO increases as a function of time, indicating further reduction of NO to N2O as the reaction proceeds. The turnover number of NO reduction was calculated to be ~ 2 mol NO·mol CuBMb −1 ·min−1, close to the 3 mol NO·mol enzyme−1 ·min−1 reported for the ba3 oxidases from Thermus thermophilus.10–13 Under identical conditions, the catalytic reduction of NO was not observed with CuBMb alone, with Cu(II) alone, or with WTswMb in the presence of Cu(II) (Figure 2). These results demonstrated that Cu(I) plays a critical role in the reduction of NO to N2O in CuBMb.
Figure 2.
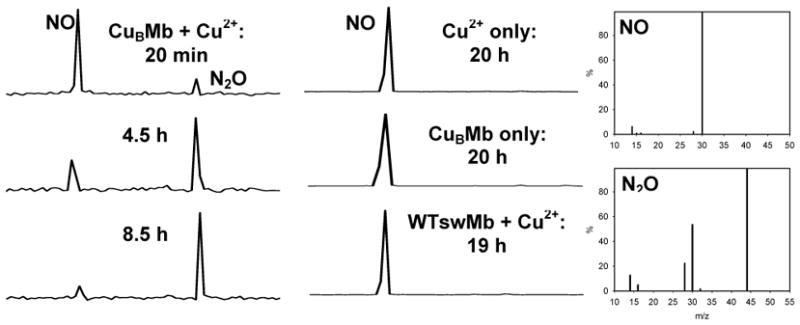
GC/MS chromatogram of NO reduction by CuBMb and Cu2+The GC peaks have been normalized.
It has been shown that HCOs from different species show either substantial NO reduction activity or no activity. The results presented here strongly suggest that the designed heme-copper center in CuBMb is a close structural and functional model of HCOs with NO reduction activity. Our small and well-characterized protein model which possesses no other chromophores, and an easily substitutable CuB metal binding site, allowed us to gain new insights. First, no NO reduction was observed in the absence of metal ions, or in the presence of redox-inactive Zn(II) in the designed CuB center, even with a large excess of reductant (see Supporting Information). Catalytic reduction of NO by CuBMb occurred only in the presence of Cu(I). These results demonstrate that electron transfer from the CuB center is essential for NO reduction. Second, it has been shown that NO can labilize the heme Fe-His bond in both heme proteins and model compounds.27,28 Binding of NO to the reduced binuclear center in NOR results in formation of a five-coordinate ferrous heme-NO complex,29,30 and formation of a five-coordinate heme-NO species has been detected and proposed as a key intermediate in the reduction of NO by cytochrome cbb3 oxidase.13 In contrast to these observations, it has been suggested that 5-coordinate ferrous heme-NO complexes could represent dead-end products incompetent in N2O production.6 Our UV-Vis and EPR studies indicated that Cu(I) binding to CuBMb further weakens the heme Fe-His bond, in addition to the NO trans effect, suggesting that bond weakening, not necessarily bond cleavage into a 5-coordinate species, is a contributing factor in NO reduction. Finally, the binding of Zn(II) to the CuB center of the same CuBMb protein produced a five-coordinate heme-NO species, resulting from the cleavage of the proximal heme Fe-His bond (Figure 1). Although the binding of metal ions in the designed CuB center can cause conformational changes, the different effects of Cu(I) and Zn(II) on the structure of ferrous-CuBMb-NO probably result from their different oxidation states, with higher metal ion oxidation states facilitating greater weakening of the proximal heme Fe-His bond via increasing interactions with the Fe bound NO. Therefore, reduced Fe(II) in NOR has the desirable features of both Zn(II), which is capable of further weakening of the heme Fe-His bond necessary for NO reduction and Cu(I), which possesses redox activity. These reasons may be why iron has been chosen by nature for the reduction of NO to N2O.
Supplementary Material
Experimental details for EPR and GC/MS measurements, as well as the EPR spectra of WTswMb-NO in the presence of Cu(I) and Zn(II). This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This material is based on work supported by National Institutes of Health Grant GM62211. We thank Dr. Mark Nilges for help with EPR experiments and Furong Sun for help on GC/MS data collection.
References
- 1.Cheesman MR, Zumft WG, Thomson AJ. Biochemistry. 1998;37:3994–4000. doi: 10.1021/bi972437y. [DOI] [PubMed] [Google Scholar]
- 2.Hendriks J, Warne A, Gohlke U, Haltia T, Ludovici C, Luebben M, Saraste M. Biochemistry. 1998;37:13102–13109. doi: 10.1021/bi980943x. [DOI] [PubMed] [Google Scholar]
- 3.Babcock GT, Wikström M. Nature. 1992;356:301–309. doi: 10.1038/356301a0. [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Horsman JA, Barquera B, Rumbley J, Ma J, Gennis RB. J Bacteriol. 1994;176:5587–5600. doi: 10.1128/jb.176.18.5587-5600.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferguson-Miller S, Babcock GT. Chem Rev. 1996;96:2889–2907. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 6.Averill BA. Chem Rev. 1996;96:2951–2964. doi: 10.1021/cr950056p. [DOI] [PubMed] [Google Scholar]
- 7.Richardson DJ, Watmough NJ. Curr Opin Chem Biol. 1999;3:207–219. doi: 10.1016/S1367-5931(99)80034-9. [DOI] [PubMed] [Google Scholar]
- 8.Zumft WG. Microbiol Mol Biol Rev. 1997;61:533–616. doi: 10.1128/mmbr.61.4.533-616.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fujiwara T, Fukumori Y. J Bacteriol. 1996;178:1866–1871. doi: 10.1128/jb.178.7.1866-1871.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giuffre A, Stubauer G, Sarti P, Brunori M, Zumft WG, Buse G, Soulimane T. Proc Natl Acad Sci U S A. 1999;96:14718–14723. doi: 10.1073/pnas.96.26.14718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pinakoulaki E, Ohta T, Soulimane T, Kitagawa T, Varotsis C. J Am Chem Soc. 2005;127:15161–15167. doi: 10.1021/ja0539490. [DOI] [PubMed] [Google Scholar]
- 12.Forte E, Urbani A, Saraste M, Sarti P, Brunori M, Giuffre A. Eur J Biochem. 2001;268:6486–6490. doi: 10.1046/j.0014-2956.2001.02597.x. [DOI] [PubMed] [Google Scholar]
- 13.Pinakoulaki E, Stavrakis S, Urbani A, Varotsis C. J Am Chem Soc. 2002;124:9378–9379. doi: 10.1021/ja0271633. [DOI] [PubMed] [Google Scholar]
- 14.Kim E, Chufan EE, Kamaraj K, Karlin KD. Chem Rev. 2004;104:1077–1133. doi: 10.1021/cr0206162. [DOI] [PubMed] [Google Scholar]
- 15.Collman JP, Boulatov R, Sunderland CJ, Fu L. Chem Rev. 2004;104:561–588. doi: 10.1021/cr0206059. [DOI] [PubMed] [Google Scholar]
- 16.Wasser IM, Huang H-w, Moeenne-Loccoz P, Karlin KD. J Am Chem Soc. 2005;127:3310–3320. doi: 10.1021/ja0458773. [DOI] [PubMed] [Google Scholar]
- 17.Sigman JA, Kwok BC, Lu Y. J Am Chem Soc. 2000;122:8192–8196. [Google Scholar]
- 18.Sigman JA, Kim HK, Zhao X, Carey JR, Lu Y. Proc Natl Acad Sci U S A. 2003;100:3629–3634. doi: 10.1073/pnas.0737308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang N, Zhao X, Lu Y. J Am Chem Soc. 2005;127:16541–16547. doi: 10.1021/ja052659g. [DOI] [PubMed] [Google Scholar]
- 20.Zhao X, Yeung N, Wang Z, Guo Z, Lu Y. Biochemistry. 2005;44:1210–1214. doi: 10.1021/bi0479151. [DOI] [PubMed] [Google Scholar]
- 21.Zhao X, Nilges MJ, Lu Y. Biochemistry. 2005;44:6559–6564. doi: 10.1021/bi047465c. [DOI] [PubMed] [Google Scholar]
- 22.Bolard J, Garnier A. Biochim Biophys Acta. 1972;263:535–549. doi: 10.1016/0005-2795(72)90034-7. [DOI] [PubMed] [Google Scholar]
- 23.Farmer PJ, Sulc F. J Inorg Biochem. 2005;99:166–184. doi: 10.1016/j.jinorgbio.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 24.Morse RH, Chan SI. J Biol Chem. 1980;255:7876–7882. [PubMed] [Google Scholar]
- 25.Reynolds MF, Parks RB, Burstyn JN, Shelver D, Thorsteinsson MV, Kerby RL, Roberts GP, Vogel KM, Spiro TG. Biochemistry. 2000;39:388–396. doi: 10.1021/bi991378g. [DOI] [PubMed] [Google Scholar]
- 26.Couture M, Adak S, Stuehr DJ, Rousseau DL. J Biol Chem. 2001;276:38280–38288. doi: 10.1074/jbc.M105341200. [DOI] [PubMed] [Google Scholar]
- 27.Wayland BB, Olson LW. J Am Chem Soc. 1974;96:6037–6041. doi: 10.1021/ja00826a013. [DOI] [PubMed] [Google Scholar]
- 28.Burstyn JN, Yu AE, Dierks EA, Hawkins BK, Dawson JH. Biochemistry (N Y) 1995;34:5896–5903. doi: 10.1021/bi00017a019. [DOI] [PubMed] [Google Scholar]
- 29.Moeenne-Loccoz P, de Vries S. J Am Chem Soc. 1998;120:5147–5152. [Google Scholar]
- 30.Kumita H, Matsuura K, Hino T, Takahashi S, Hori H, Fukumori Y, Morishima I, Shiro Y. J Biol Chem. 2004;279:55247–55254. doi: 10.1074/jbc.M409996200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details for EPR and GC/MS measurements, as well as the EPR spectra of WTswMb-NO in the presence of Cu(I) and Zn(II). This material is available free of charge via the Internet at http://pubs.acs.org.