

Abstract
Molecular beacon DNA probes, containing one to four pyrene monomers on the 5′ end and the quencher DABCYL on the 3′ end, were engineered and employed for real-time probing of DNA sequences. In the absence of a target sequence, the multiple-pyrene labeled molecular beacons (MBs) assumed a stem-closed conformation resulting in quenching of the pyrene excimer fluorescence. In the presence of target, the beacons switched to a stem-open conformation which separated the pyrene label from the quencher molecule and generated an excimer emission signal proportional to the target concentration. Steady-state fluorescence assays resulted in a sub-nanomolar limit of detection in buffer, while time-resolved signaling enabled low-nanomolar target detection in cell growth media. It was found that the excimer emission intensity could be scaled by increasing the number of pyrene monomers conjugated to the 5′ terminal. Each additional pyrene monomer resulted in substantial increases in the excimer emission intensities, quantum yields, and excited-state lifetimes of the hybridized MBs. The long fluorescence lifetime (~40 ns), large Stokes shift (130 nm), and tunable intensity of the excimer make this multiple-pyrene moiety a useful alternative to traditional fluorophore labeling in nucleic acid probes. In addition, this excimer complex serves as an efficient FRET donor for red-emitting fluorophores, such as TMR, for further extending the Stokes shift of the fluorescent complex.
Introduction
Hairpin oligonucleotide probes, or molecular beacons (MBs), are utilized in many types of DNA/RNA targeting applications including intracellular mRNA monitoring, real-time PCR, gene microarrays, and biosensors.1–8 Presently, there is a strong interest in enhancing the fluorescence signaling sensitivity of MBs for real-time probing of nucleic acids in biological samples. MBs typically consist of a 25–35-mer nucleotide sequence with a complementary 5–6 base region at each end to which a fluorophore and non-fluorescent quencher molecule are covalently attached. In its natural conformation, the complimentary ends of the oligonucleotide hybridize and form a short stem bringing the fluorophore and quencher moieties into close proximity. This close spatial proximity induces fluorescence resonance energy transfer (FRET) from the excited-state fluorophore to the quencher molecule, thus quenching the fluorescence signal from the donor fluorophore.1,3,9 In the presence of a DNA/RNA target, the loop strand of the MB hybridizes with the complimentary target sequence, causing the stem to open and the termini to separate. The spatial separation of the fluorophore and quencher restores emission from the excited-state fluorophore and generates a fluorescence signal proportional to the amount of target present. In traditional MB signaling, the detection sensitivity is directly limited by the fluorescence intensity generated upon the opening of a single MB stem when target binding occurs. Fluorophores such as FAM and TMR, are often employed in MBs due to having intrinsically high quantum yields, small molecular weights, and high chemical stability and solubility. Despite their appealing properties, these fluorophore labeled MBs are limited to the fluorescence output of a single dye molecule in the stem-open state, which in turn limits the signaling range and prevents lower target detection limits from being attained. Higher signaling sensitivities are often achieved through FRET and other excited-state interactions involving two or more fluorophores and monitoring their emission channels.9–14 Okamura et al.15 showed that double-labeling organic fluorophores as fluorescence donors could improve the FRET efficiency in linear probes by increasing the acceptor fluorescence range. However, the emission intensity of multiple-fluorophore complexes in similar FRET-based probes is limited by the significant self-quenching of the donor fluorescence as observed in the background signal. Recently, novel fluorescent materials including quantum dots16 and amplifying fluorescent polymers17 have been explored as signaling elements in molecular beacons for signal amplification.
We have modified a fluorophore-quencher labeling scheme in MBs incorporating an assembly of pyrene monomers, which both enhances fluorescence output and enables a time-resolved signaling approach for improved background discrimination. In close enough proximity, an excited-state pyrene monomer and ground-state monomer can form an excimer state which fluoresces at a substantially longer wavelength than the monomer emission.18–20 It has also been shown that the intensity of the excimer emission can be enhanced by both inter and intramolecular aggregation of pyrene monomers due to a greater probability of dimerization. Previous bis-pyrene labeled oligonucleotides have been used to probe RNA folding and DNA duplex formation by monitoring the monomer and excimer emission fluctuations that arise from local base stacking and quenching effects.21–24 Both the dimer forming and base intercalating phenomena of these bis-pyrene labels have made them versatile in probing oligonucleotide interactions. In stem forming oligonucleotide probes, such as molecular beacons and molecular beacon aptamers, pyrene monomers have been labeled at opposite terminals to facilitate an excimer-monomer switching mechanism that is controlled by the bound and unbound conformations of the oligonucleotide.25,26 The sensitivity of these singly-labeled pyrene probes is typically more dependent on the excimer emission due to the inherently high autofluorescence and light scattering in the pyrene monomer emission region.
We report the spectroscopic application of an excimer signaling multiple-pyrene label in molecular beacon DNA probes. Pyrene monomers linked symmetrically to the 5′ end of an MB sequence favors the formation of intramolecular excimers when excited. Unlike previous pyrene labeled DNA probes which rely on base-stacking interactions and excited-state switching mechanisms, the multiple-pyrene MBs provide a sensitive excimer-FRET signaling mechanism making use of the long fluorescence lifetime and large Stokes shift of the pyrene excimer. The conjugation of additional monomers at the 5′ terminal allows the fluorescence intensity of the excimer to be magnified, thus yielding a higher fluorescence signal for each molecular recognition event. In close range to longer wavelength fluorophores, the multiple-pyrene label is also shown to be a FRET donor to further extend the Stokes shift of the fluorescent complex.
Materials and Methods
Chemicals and Reagents
Deoxyribonucleotides, dendrimers, 5′ amino-modifiers, and 3′ DABCYL-CPGs were purchased from Glen Research (Sterling, Va). Four MBs were synthesized with the following 32-mer sequence: 5′-CCTAGCTCTAAATCACTATGGTCGCGCTAGG-3′ (bold indicates stem region). Each was labeled with a single DABCYL molecule at the 3′ terminal and between one and four pyrene molecules at the 5′ terminal (MB-1P, MB-2P, MB-3P, and MB-4P). The target cDNA was synthesized with the sequence complimentary to the loop sequence of the MB (non-bold region). Pyrenebutyric acid was purchased from Aldrich. Dimethylformamide, dicyclocarbodiimide, and dimethylaminopyridine were purchased from Fisher. A solution of 0.1 M triethylamine acetate (TEAA) was used as HPLC buffer A and HPLC-grade acetonitrile (Fisher) was used as buffer B. Purified oligonucleotides were dissolved in DNase-free water (Fisher). Buffer-based detection assays were conducted at room temperature in a Tris-HCl buffer (20mM Tris-HCl, 25mM NaCl, 5mM MgCl2, pH: 7.4). DMEM (Mediatech, Herndon, VA) was used as cell media for time-resolved DNA detection assays. 9,10-diphenylanthracene in cyclohexane (ΦR = 0.95, η = 1.423) was used as a reference standard in quantum yield measurements for the pyrene labeled MBs.
Instruments
DNA synthesis was performed on an ABI3400 DNA/RNA synthesis. A ProStar HPLC (Varian) with an Econsil C-18 column (5U, 250 × 4.6 mm) was used for probe purification. Absorbances were measured on a Carry-Bio300 UV/Vis spectrometer (Varian) and used to calculate probe concentration. A Fluorolog-Tau-3 spectrofluorometer was used for all steady-state hybridization assays with an excitation of 350 nm. Time-resolved emission was measured on an OB920 (Edinburgh Analytical Instruments) by time correlated single photon counting using a nitrogen flash lamp at 337 nm for excitation.
Synthesis and Purification of Pyrene Labeled MBs
Oligonucleotides were synthesized using solid-state phosphoramidite chemistry on a 1 μmol scale. Each MB sequence began on column with a 3′ DABCYL molecule covalently attached to a control-pore glass (CPG) substrate. After synthesizing each sequence, the 5′ nucleotides of MB-2P, MB-3P, and MB-4P were coupled to dendrimeric linker phosphoramidites bearing DMT protected hydroxyl groups. Doubler and trebler phosphoramidites were used for MB-2P and MB-3P, respectively. For MB-4P, two levels of branching were constructed by coupling a single doubler to the 5′ base and an additional doubler to each of the two symmetric branches. An amine group was added to each branch by coupling a 5′ amino-modifier C6 linker phosphoramidite to the deprotected hydroxyl groups. The 5′ terminal of MB-1P contained only a single amino-modifier C6 linker. The columns were then flushed slowly with 15 ml of dimethylformamide (DMF). The CPG containing the MB-1P sequence was released from the column into a 1 ml solution of DMF containing 14.4 mg (50 μmol) pyrenebutyric acid, 10.3 mg (50 μmol) dicyclocarbodiimide, and 6.1 mg (50 μmol) dimethylaminopyridine. This step was repeated for each MB sequence with the quantity of coupling reagents scaled by the number of pyrene molecules to be added to the 5′ terminal (i.e. 200 μmol for MB-4P). The coupling reactions proceeded at room temperature for approximately 6 hours and the resulting solutions were washed three times with DMF to remove uncoupled pyrene. The CPG pellets were then added to 3 ml solutions of 50% v/v ammonium hydroxide/methylamine and incubated at 60° C for 15 minutes to cleave heterocyclic amine groups, cyanoethyl groups, and CPG from the oligonucleotide sequences. The mixtures were centrifuged and the colorless supernatants were retained. The impure oligonucleotides were precipitated in ethanol and isolated in solid form. Each MB was twice purified using gradient elution RP-HPLC on a C-18 column with a flow rate of 1 ml/min. Buffer B was increased linearly from 20% to 70% over a 30 minute period, while absorbance and fluorescence intensities were monitored. Samples which had absorbance peaks at 260, 350, and 490 nm and emission at 398 nm with 350 nm excitation were collected and dried in a SpeedVac. For each MB, the peak with the longest retention time, indicating the maximum pyrene coupling, was retained and used for experiments. The purified MBs were identified by MALDI-TOF mass spectrometry using a Voyager DE mass spectrometer (applied Biosystems): MB-1P 10389 (found), 10386 (calculated); MB-2P 11189 (found), 11189 (calculated); MB-3P 11663 (found), 11661 (calculated); MB-4P 12797 (found), 12795 (calculated).
Quantum Yield Determination
The quantum yields of pyrene labeled MBs were determined using 9,10-diphenylanthracene in cyclohexane as a reference (ΦR = 0.95). The pyrene labeled oligonucleotides were dissolved in aqueous solution with 5 units of DNase I to fully separate the quenching effect of DABCYL on the pyrene complexes. Concentrations of the reference and probes were adjusted to yield an approximate absorbance intensity of less than 0.05 to avoid inner filter effects. An excitation of 346 nm was used and fluorescence emission was integrated from 350–700 nm. The quantum yields were calculated from the following formula
where the subscripts F and R denote sample and reference respectively; Φ, ∫ I, A, and n are fluorescence quantum yield, integrated fluorescence intensity, absorbance, and refractive index of the solvent, respectively. In this case, n=1.333, and nR=1.423.
Results and Discussion
Design and Synthesis of Multiple-Pyrene Labeled MBs
Previous DNA probes have incorporated pyrene derivates into various positions around a nucleotide in order to monitor pyrene-base and pyrene-pyrene interactions resulting from target binding.21–35 A method of assembling multiple quenchers using dendrimer linkers was applied here to assemble multiple pyrene molecules together at the 5′ end of a DNA sequence.36 With a single DABCYL (4-(4-(dimethylamino)phenylazo)benzoic acid) quencher molecule on the 3′ end, two to four pyrene molecules were linked to the 5′ end (MB-2P, MB-3P, and MB-4P) by varying the arrangement of carbon linkers (see Figure 8, supporting information). The pyrene labeled MB reported here (MB-2P) contains a dual-pyrene label on the 5′ end and a single DABCYL molecule on the 3′ end (Figure 1a).
Figure 1.

(a) Structural representation of MB-2P with pyrene monomers labeled to dual 5′ linker chains and DABCYL molecule labeled to 3′ chain. (b) Schematic of MB-2P hybridization with complimentary target. (c) Emission spectra of 1 μM MB-2P with increasing concentrations of cDNA in buffer solution. The excimer signal increases upon addition of cDNA and reaches maximum intensity at 1 eq. of target (1 μM cDNA). (buffer: 20mM Tris-HCl, 25mM NaCl, 5mM MgCl2, pH: 7.4)
The dual-pyrene moiety positions the pyrene monomers in close proximity through two symmetric 18 atom linkers covalently attached at the 5′ terminal. Figure 1b schematically shows the hybridization of MB-2P with target DNA. In the beacon’s stem-open conformation, intense excimer fluorescence is generated as a result of the dimerization of the two adjacent pyrene monomers. Increasing the concentration of MB-2P in the hybridized state did not change the excimer/monomer emission ratio, confirming that dimer formation was due to intramolecular interactions. The long length of the alkane linkers is favorable, because it helps (i) minimize pyrene intercalation within the base stacking region of the DNA duplex and (ii) prevent monomer interactions with bases on the 5′ stem, thus avoiding fluorescence quenching effects. Unlike excimer-monomer switching mechanisms, the dual-pyrene functionality makes use of only the excimer emission which offers significant advantages over traditional fluorophores in spectroscopic signaling applications. Despite having more synthesis steps than traditional MBs, the strong hydrophobicity of the dual-pyrene label is remarkably effective in enhancing the purity of the MB, which in turn minimizes the residual fluorescence and target binding competition from non-labeled DNA sequences.
The DABCYL chromophore (λmax = 490 nm) was selected as a quencher, since it exhibits sufficient spectral overlap with the excimer emission of pyrene (480 nm) and provides an efficient non-radiative sink for the pyrene excimer. In addition, DABCYL has minor absorption in the pyrene monomer emission region, which helps further diminish the excimer emission, since the excited-state monomer is the precursor for the excimer state. DABCYL thus serves as an efficient excimer quencher in these multiple-pyrene MBs.
Steady-State Fluorescence Measurements to Detect Target DNA
To test the target signaling performance of MB-2P, steady-state fluorescence experiments were performed. Figure 1c shows the emission spectra of MB-2P at 350 nm excitation with varying concentrations of target DNA in buffer solution. The emission of MB-2P without target was indistinguishable from the background light scattering in the excimer emission range, indicating a high quenching efficiency by the DABCYL label. Two small peaks at 375 and 398 nm represented the fraction of unquenched monomer fluorescence in the dual-pyrene label. When titrated with target DNA, the excimer emission of MB-2P increased, reaching a maximum intensity with one equivalent of target. The spectra also show a slight, but steady increase in the emission peaks at 375 and 398 nm, which was due to the fluorescence of the pyrene monomer fraction being simultaneously restored when the MB hybridized to the target. As high as a 29-fold excimer signal enhancement was attained when equimolar target sequence was introduced into the MB solution. The sensitivity of this steady-state signaling mechanism is comparable with most FRET-based MBs with a limit of detection in the sub-nanomolar range. For example, a FAM labeled MB with the same sequence and quencher label exhibited a signal enhancement of approximately 15-fold under the same experimental conditions. The signal-to-background ratio of MB-2P was further enhanced by adding optimal amounts of divalent cations and DMF to the buffer. Hybridization results of MB-2P indicated that covalently bringing two pyrene molecules together allows efficient formation of an excimer complex. Furthermore, the excimer emission can be quenched in the same way as other organic fluorophores by common quencher molecules such as DABCYL. Thus, this pyrene assembly may be utilized in a variety of molecular probing platforms.
Time-Resolved Excimer Measurements
Differentiating the fluorescence signals between the probe and background species is often difficult with steady-state measurements. Alternatively, time resolved fluorescence monitoring can provide better signaling sensitivity in complex samples, but is dependent on the fluorescent lifetime and Stokes shift of the signaling agent. Lanthanide chelates (luminophores) are currently being explored as labeling agents in LRET-based probes due to their extraordinarily long fluorescence lifetimes and large Stokes shifts (up to 300 nm).37 Since most organic fluorophores possess lifetimes less than 10 ns, their use for such time-gated analysis is precluded. However, the pyrene excimer is unique among organic fluorophores, possessing a long fluorescence lifetime (40 ns) and large Stokes shift (130 nm). Retaining this lifetime and Stokes shift property in the multiple-pyrene label is critical for maximizing signaling sensitivity in complex biological samples.
Time-resolved fluorescence signaling of MB-2P was first investigated in buffer solution (Figure 2). Without target, the fluorescence signal of the quenched dual-pyrene label was retained longer than the buffer signal at both 398 and 480 nm. The background emission increase of MB-2P was noticeably more pronounced in the monomer channel due to both a weaker monomer quenching efficiency (less absorption overlap from DABCYL molecule at 398 nm) and substantially longer emission lifetime of the pyrene monomer (100 ns) compared to the light scattering from the buffer (< 7 ns). The lifetime of MB-2P at 480 nm was 33 ns (70% fluorescence contribution) with most of the signal originating from the unquenched excimer fraction in the dual-pyrene label. In the presence of excess cDNA, the fluorescence decay of MB-2P changed very little in the monomer channel (Figure 2a), whereas the excimer emission intensity increased significantly over the full time scale (Figure 2b). The unquenched excimer emission of the dual-pyrene label was easily differentiated from the buffer and background emissions over a sustained time period after excitation. A maximum signal-to-background enhancement of 25 was achieved in the 90–100 ns time interval. The lifetime of target-hybridized MB-2P increased to 39 ns (87.5%), indicative of the unquenched excited-state of the pyrene excimer in solution. This value is approximately one order magnitude longer than the fluorescence lifetimes of most biological species, thus making time-resolved fluorescence signaling feasible in complex biological samples.
Figure 2.

Fluorescence decay traces after 337 nm excitation of 1 μM MB-2P (blue) and MB-2P with target DNA (red) in buffer solution. Fluorescence emissions were monitored at a) the pyrene monomer wavelength (398 nm) and at b) the pyrene excimer wavelength (480 nm).
Nucleic Acid Detection in Cell Media
To evaluate its potential effectiveness in bioassays, MB-2P was tested in Dulbecco’s cell growth media. Figure 3 shows the steady-state emission spectra of MB-2P in the presence and absence of target in both buffer solution and cell growth media.
Figure 3.
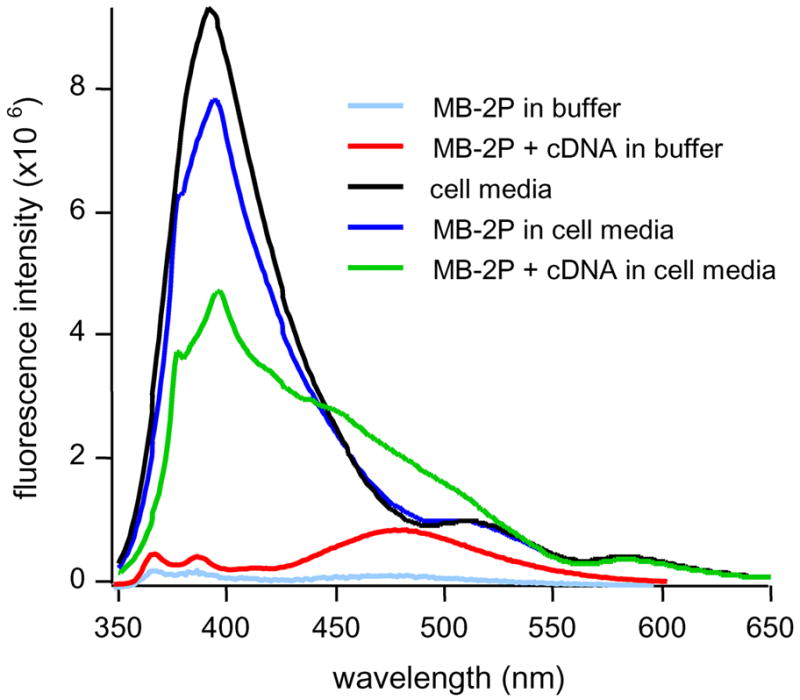
Steady-state emission spectra of 500 nM MB-2P comparing background fluorescence in cell media and buffer. Excimer signal enhancement in cell media is poor compared to buffer.
In the cell growth media, the high autofluorescence dominated the luminescence spectra from 400 to 500 nm. To discriminate the dual-pyrene excimer emission from the high background fluorescence, time-gated photon counting was employed. Figure 4a compares the emission intensities of MB-2P in the presence of target at different times after excitation. At 0 ns, the spectrum is dominated by the autofluorescence of the medium and resembles the steady-state luminescence spectrum shown in Figure 3. At 40 ns, the spectrum closely resembled the steady-state spectrum of MB-2P in buffer with a dominant pyrene excimer emission peak at approximately 480 nm. At 100 ns after the excitation pulse, the luminescence spectrum is dominated by the pyrene monomer fluorescence, because of the longer fluorescence lifetime of the pyrene monomer compared to the excimer.
Figure 4.

(a) Time-resolved emission spectra of 500 nM MB-2P with 5 μM cDNA in cell growth media. The fluorescence emission was recorded at 0 ns (black), 40 ns (red), and 270 ns (blue) time gates, (b) Emission decay at 480 nm of 500 nM MB-2P (blue) and MB-2P with increasing concentrations of cDNA (red) in cell growth media. Autofluorescence decay trace is black. Signal-to-background is maximized in 60–110 ns time interval.
The sensitivity of the time-resolved excimer signaling approach was investigated for quantifying DNA in cell growth media. It followed from the time-resolved emission spectra that once the autofluorescence had decayed away (less than 40 ns after excitation), a local emission maximum for MB-2P was observed in the pyrene excimer emission channel. The fluorescence decays of MB-2P were thus appropriately monitored at this channel (480 nm) with varying concentrations of target cDNA in the cell media (Figure 4b). The total photon counts increased proportionally with cDNA concentration and a clear signal separation was obtained for the fully hybridized MB-2P. A maximum signal separation was observed in a time window of 60 – 110 ns. In this time window, much of the excimer emission still occurred, while most of the background autofluorescence had decayed. These results show time-resolved analysis of the excimer fluorescence to be a sensitive and robust method for detecting low nanomolar DNA concentrations in complex biological media without the need for sample pretreatment, extractions, or target amplification.
Tunable Intensity through Multiple-Pyrene Labeling
Another feature of the multiple-pyrene label is that the excimer intensity can be scaled,34 to a large extent, by the local pyrene density at the terminal of the MB. Unlike pyrene, other fluorophores will self-quench if intermolecular distances are too short. It has been shown that double-labeling organic fluorophores to the terminals of free oligonucleotides significantly decreases the fluorescence intensity relative to single-fluorophore labeled oligonucleotides.15 While most commercial organic fluorophores have Stokes shifts ranging from 10 and 40 nm, the pyrene excimer exhibits an exceedingly long Stokes shift (approx. 130 nm) with no overlap between the monomer absorption (350 nm) and excimer emission (480 nm) bands. This peak separation allows the pyrene excimer to avoid the effects of self-quenching even at high concentrations and labeling densities. More importantly, pyrene excimer formation requires the complexation of an excited-state molecule with a ground state molecule. Increasing the density of pyrene molecules assembled at the MB terminal enhances the probability of forming excited-state dimers. In polypyrenylalkanes, it has been shown that intramolecular excimer formation is favored both by the thermodynamic stabilization of the dimer and the long excited-state lifetime of the pyrene monomer during which excimer formation proceeds.20 In the dendrimeric pyrene assemblies reported here, the long length and symmetric branching arrangement of the alkane linkers provide more flexibility for the pyrene molecules to interact equivalently and thus not be limited to nearest-neighbor interactions, such as with rigid, linear assemblies. This may further enhance excimer formation since any one of the pyrene monomers, when excited, can dimerize with any of the ground-state monomers with equal probability. The expanded excimer intensity range of these multiple-pyrene MBs may offer improved signaling sensitivity in fluorescence-based detection methods.
In addition to MB-2P, we synthesized MBs with 1,3, and 4 pyrene monomers conjugated at the 5′ end to investigate the effect of pyrene assemblies on the excimer signaling performance in MBs.
The emission spectra of the pyrene labeled beacons in the presence of excess cDNA showed a substantial increase in excimer intensity with an increased number of pyrene monomers in the label (Figure 5a). The monomer emission intensity at 376 and 398 nm also increased, but to a much lesser extent than the excimer emission for the MBs bearing 2, 3, and 4 pyrenes. The increased excimer/monomer emission ratio for each additional pyrene (Table 1) indicates excimer formation was more favorable with higher densities of pyrene labeled at the terminal. The fluorescence quantum yields of the different MBs were determined (Table 1). The quantum yield increased with an increasing number of pyrenes in the label, reaching a value of 0.20 for MB-4P. Such a high quantum yield, together with a remarkable extinction coefficient (56,000×4=224,000 cm−1M−1), make the 4-pyrene label an exceptionally bright UV excitable fluorophore. The fluorescence decays were also monitored to investigate if the increased excimer signals from the MBs were sustained after pulse excitation. Figure 5b shows the fluorescence decays at 480 nm for the multiple-pyrene MBs in the presence of excess cDNA. Each additional pyrene monomer resulted in slower excimer decay, with an increase in fluorescence lifetime of 8 ns for MB-4P relative to MB-2P (Table 1). This increased excimer lifetime has the potential to provide higher background discrimination, and thus higher signaling sensitivity, in both steady-state and time-resolved bioassays.
Figure 5.

a) Steady-state emission spectra of MB-1P (black), MB-2P (blue), MB-3P (red), and MB-4P (green) with excess cDNA in buffer, b) Fluorescence decay of MB-2P (black), MB-3P (red), and MB-4P (green) at 480 nm. [MB]=200 nM, [cDNA]=2μM
Table 1.
Photophysical and performance properties of MBs with different numbers of pyrene molecules in the label.
| Probe | Φa | E/Mb | τ(%)c | Tmd | S/Be |
|---|---|---|---|---|---|
| MB-1P | 0.009 | 0.13 | - | ~60* | 1* |
| MB-2P | 0.037 | 1.54 | 39.0 (87.5) | ~61.5 | 29 |
| MB-3P | 0.091 | 3.04 | 43.7 (94.9) | ~61.5 | 14 |
| MB-4P | 0.198 | 6.23 | 47.4 (96.4) | ~61.5 | 12 |
fluorescence quantum yield
ratio of excimer and monomer fluorescence
fluorescence lifetime; the % intensity contributions are given in brackets
melting temperatue in °C
ratio of signal and background fluorescence.
Hybridization experiments indicate that although overall excimer intensity increased with the number of pyrene molecules, the background signal from the free MB also increased, as the steady-state signal-to-background ratio of MB-4P was nearly three fold lower than that of MB-2P (Table 1). It was apparent that the excimer quenching efficiency of the single DABCYL molecule decreased as the number of pyrene monomers increased. This was likely due in part to a larger overall separation between the pyrene and quenching moieties, which reduced the energy transfer efficiency. Another potential reason may be that the emission intensity exceeded the quenching capacity of the single DABCYL molecule. Two possibilities for improving the quenching efficiency in the stem-closed MBs include (i) matching the lengths of the linkers connecting the quencher and pyrene molecules and (ii) replacing the single quencher with a superquencher assembly36 to increase the number of available energy acceptors. These modifications will be evaluated in future work.
These results suggest a way to build a bright fluorophore by assembling multiple pyrene molecules together in a way that both preserves probe functionality and enhances excimer formation. While previous pyrene assemblies have been made use of linear polypeptide35 and polynucleotide34 backbones, these dendrimeric linkages bring multiple pyrene monomers together in close proximity to further enhance the probability of excimer formation. Of course, solubility of the product may be a problem for some biological applications because of the hydrophobicity of the pyrene assembly. Therefore, water soluble pyrene derivatives, such as Cascade Blue, may be better suited for these pursuits. Our initial testing indicates that this fluorophore exhibits similar excimer forming properties as pyrene with the excimer emission peak around 510nm (Figure 9, Supporting Information). Also, to alleviate the quenching of pyrene by oxygen common to biological environments, alkynylpyrene derivatives38 have been shown to retain high fluorescence intensity in aerated environments and thus have potential to be employed in similar excimer signaling probes as presented here.
Stem Stability Analysis
Considering the long length of the dendrimeric linkers and hydrophobicity of the pyrene molecules, the ability of the MB stem to open freely in the presence of cDNA was likely to be impacted by a combination of stabilizing and destabilizing interactions between the multiple-pyrene moiety and either the quencher moiety or bases within the stem helix.
We evaluated the rate of stem opening by comparing the hybridization kinetics of MB-1P and MB-4P in buffer solution (Figure 6a). The excimer fluorescence intensity was monitored vs. time for each MB (monomer fluorescence for MB-1P and excimer fluorescence for MB-4P) in the presence of complimentary target. As shown in Figure 6a, under the same conditions, the half-time to open MB-1P was approximately 76 seconds while MB-4P required 71 seconds. This result suggests the multiple-pyrene labels have little effect on the hybridization kinetics of the MB to its target DNA. A thermal denaturation analysis (Figure 6b) was also conducted to compare the relative stem stabilities in each MB. The excimer fluorescence intensity was monitored with increasing temperature as the equilibrium shifted from the stem-closed to stem-open conformation (monomer emission used for MB-1P). The melting point for MB-1P was 60.1 °C and approximately 1.5 °C lower than any of the multiple-pyrene MBs. There was no clear difference in stability, however, between the MBs containing multiple pyrenes conjugated to the terminal. This indicates that the multiple-pyrene assemblies may be self-aggregating and thus causing the stem stability to be independent of additional pyrene monomers in the label (beyond 2). In addition, the multiple pyrene label may exhibit more affinity toward the quencher moiety than a single pyrene linker, which may account for the small difference in melting temperatures between the single and multiple pyrene MBs.
Figure 6.

a) Hybridization kinetics of MB-1P and MB-4P. Three hybridization trials were conducted for each beacon (a,b,c) [MB]=100nM, [cDNA]=1μM. b) Thermal denaturation profiles of multiple-pyrene labeled beacons. Monomer intensity (376 nm) was monitored for MB-1P (blue) and excimer intensity (480 nm) was monitored for MB-2P (black), MB-3P (red), and MB-4P (green) [MB]=100nM
Excimer-FRET Signaling
For bioanalysis applications, fluorophores with long Stokes shifts are ideal for resolving the fluorescence signal against the shorter-wavelength autoluminescence.
To extend the emission wavelength beyond the pyrene excimer emission, multiple-pyrene-fluorophore assemblies were explored as alternative FRET-based signaling agents that employ the pyrene excimer as a fluorescence donor. Tetramethylrhodamine (TMR) was chosen as a fluorescence acceptor due to its absorption in the 500–550 nm region coinciding with the excimer emission range. Two compounds were synthesized each containing 1 TMR molecule and 2 or 3 pyrene monomers respectively attached to linkers spaced 17 atoms from a central carbon atom. A compound containing 1 pyrene monomer and 1 TMR molecule was also synthesized as a control. The close distance of each dye within the complex is to ensure a high probability of both pyrene excimer formation and FRET between the excimer and acceptor fluorophore. Figure 7 shows the steady-state emission spectrum of the pyrene-TMR complexes excited at 344 nm. Major emission peaks at 590 nm were observed for both the 2P-1TMR and 3P-1TMR complexes. The pyrene monomer and excimer emission peaks were also visible, although the excimer emission intensity was low relative to the monomer and TMR intensities. This suggests that FRET may be occurring between the pyrene excimer state and TMR thus resulting in a combined Stokes shift of more than 240 nm. For the control complex, 1P-1TMR, there was no excimer emission and only minor TMR emission was observed due to direct excitation. This indicates that FRET was not feasible when only a single pyrene monomer was in range of the acceptor fluorophore. Multiple-pyrene conjugates incorporating other red-emitting fluorophores, such as Texas Red, may also have a similar FRET capability and provide an even larger Stokes shift between excitation and acceptor emission. These results suggest the potential application of multiple-pyrene assemblies as fluorescent donors in FRET-based molecular probes. The increased separation between the pyrene excitation and fluorescence channels provided by suitable acceptor fluorophores will make it easier to discriminate between true and false-positive signals in complex biological media.
Figure 7.

(a) Chemical structure of 3-pyrene-TMR conjugate, (b) Emission spectra of pyrene-TMR conjugates excited at 345 nm in buffer and normalized to the pyrene monomer emission to approximate equal concentrations. The spectra of 2 and 3-pyrene conjugates reveal a high TMR emission relative to excimer emission, indicating FRET between the pyrene excimer and TMR. TMR emission from 1-pyrene conjugate originates from direct excitation of TMR.
Conclusions
We have designed and applied multiple-pyrene molecular beacons for the detection of target nucleic acids in real-time, homogeneous assays. The dual-pyrene labeled beacon (MB-2P) produces an excimer emission in the presence of target DNA and offers high detection sensitivity and selectivity comparable to other fluorophore labeled DNA probes. The long lifetime (~40 ns) and large Stokes shift (130 nm) of the pyrene excimer favors a time-resolved analysis approach which easily discriminates between the stem-open and stem-closed conformations of the MB in cellular media. Monitoring the excimer emission decay of MB-2P resulted in a sensitive method for detecting low concentrations of target DNA under conditions of high autofluorescence (cell growth media) without the need for sample pretreatment or washing steps. Whereas previous multiple-fluorophore labeling in molecular probes resulted in decreased fluorescence intensities, multiple-pyrene labeling enables a unique tunable intensity feature in which the excimer intensity is scaled by the number of pyrene monomers in the label. Both the excimer intensity and quantum yield of MB-4P (hybridized) were 6 fold higher than for MB-2P with little change in the pyrene monomer intensity. The high emission intensities achieved by these multiple-pyrene assemblies may be utilized in similar probes for expanding the dynamic signaling range in both steady-state and time-resolved fluorescence assays. Multiple-pyrene-fluorophore complexes were also prepared to illustrate the use of the pyrene excimer as a FRET donor to further extend the Stokes shift and thus potentially enable higher signaling resolution in autofluorescent environments. The integration of multiple-pyrene assemblies in molecular beacons presents a useful application of the pyrene excimer for targeting biomolecules with increased sensitivity and using straightforward spectroscopic methods.
Acknowledgments
This work was supported by NIH grants, 2 R01 GM066137 and U54 NS 058185, ONR grant N00014-07-1-0982 and Florida State Center of Excellence. The authors at Columbia thank NSF and NIH for generous support of this research through grants CHE 04-15516 and HG002806-03.
Reference List
- 1.Tyagi S, Kramer FR. Nat Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]; Tyagi S, Bratu DP, Kramer FR. Nat Biotechnol. 1998;16:49–53. doi: 10.1038/nbt0198-49. [DOI] [PubMed] [Google Scholar]
- 2.Fang XH, Mi YM, Li JWJ, Tan W. Cell Biochemistry and Biophysics. 2002;37:71–81. doi: 10.1385/CBB:37:2:071. [DOI] [PubMed] [Google Scholar]
- 3.Tan W, Wang K, Drake TJ. Curr Opin Chem Biol. 2004;8:547–553. doi: 10.1016/j.cbpa.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 4.Yang CJ, Medley CD, Tan W. Curr Pharm Biotechnol. 2005;6:445–452. doi: 10.2174/138920105775159322. [DOI] [PubMed] [Google Scholar]
- 5.Liu X, Farmerie W, Schuster S, Tan W. Anal Biochem. 2000;283:56–63. doi: 10.1006/abio.2000.4656. [DOI] [PubMed] [Google Scholar]
- 6.Peng XH, Cao ZH, Xia JT, Carlson GW, Lewis MM, Wood WC, Yang L. Cancer Res. 2005;65:1909–1917. doi: 10.1158/0008-5472.CAN-04-3196. [DOI] [PubMed] [Google Scholar]
- 7.Chen W, Martinez G, Mulchandani A. Anal Biochem. 2000;280:166–172. doi: 10.1006/abio.2000.4518. [DOI] [PubMed] [Google Scholar]
- 8.Perlette J, Tan W. Anal Chem. 2001;73:5544–5550. doi: 10.1021/ac010633b. [DOI] [PubMed] [Google Scholar]
- 9.Tyagi S, Marras SA, Kramer FR. Nat Biotechnol. 2000;18:1191–1196. doi: 10.1038/81192. [DOI] [PubMed] [Google Scholar]
- 10.Zhang P, Beck T, Tan W. Angew Chem Int Ed Engl. 2001;40:402–405. doi: 10.1002/1521-3773(20010119)40:2<402::AID-ANIE402>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 11.Bonnet G, Tyagi S, Libchaber A, Kramer FR. Proc Natl Acad Sci U S A. 1999;96:6171–6176. doi: 10.1073/pnas.96.11.6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jockusch S, Marti AA, Turro NJ, Li Z, Li X, Ju J, Stevens N, Akins DL. Photochem Photobiol Sci. 2006;5:493–498. doi: 10.1039/b600213g. [DOI] [PubMed] [Google Scholar]
- 13.Marti AA, Jockusch S, Li Z, Ju J, Turro NJ. Nucleic Acids Res. 2006;34:e50. doi: 10.1093/nar/gkl134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsourkas A, Behlke MA, Xu Y, Bao G. Anal Chem. 2003;75:3697–3703. doi: 10.1021/ac034295l. [DOI] [PubMed] [Google Scholar]
- 15.Okamura Y, Kondo S, Sase L, Suga T, Mise K, Furusawa I, Kawakami S, Watanabe Y. Nucleic Acids Res. 2000;28:e107. doi: 10.1093/nar/28.24.e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim JH, Morikis D, Ozkan M. Sensors and Actuators B: Chemical. 2004;102:315–319. [Google Scholar]
- 17.Yang CYJ, Pinto M, Schanze K, Tan WH. Angew Chem Int Ed. 2005;44:2572–2576. doi: 10.1002/anie.200462431. [DOI] [PubMed] [Google Scholar]
- 18.Birks JB. Photophysics of Aromatic Molecules (Wiley Monographs in Chemical Physics) 1979. p. 704. [Google Scholar]
- 19.Nohta H, Satozono H, Koiso K, Yoshida H, Ishida J, Yamaguchi M. Anal Chem. 2000;72:4199–4204. doi: 10.1021/ac0002588. [DOI] [PubMed] [Google Scholar]
- 20.Snare MJ, Thistlethwaite PJ, Ghiggino KP. J Am Chem Soc. 1983;105:3328–3332. [Google Scholar]
- 21.Kostenko E, Dobrikov M, Pyshnyi D, Petyuk V, Komarova N, Vlassov V, Zenkova M. Nucleic Acids Res. 2001;29:3611–3620. doi: 10.1093/nar/29.17.3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewis FD, Zhang Y, Letsinger RL. J Am Chem Soc. 1997;119:5451–5452. [Google Scholar]
- 23.Yamana K, Iwai T, Ohtani Y, Sato S, Nakamura M, Nakano H. Bioconjug Chem. 2002;13:1266–1273. doi: 10.1021/bc025530u. [DOI] [PubMed] [Google Scholar]
- 24.Yamana K, Fukunaga Y, Ohtani Y, Sato S, Nakamura M, Kim WJ, Akaike T, Maruyama A. Chem Commun (Camb) 2005:2509–2511. doi: 10.1039/b502033f. [DOI] [PubMed] [Google Scholar]
- 25.Fujimoto K, Shimizu H, Inouye M. J Org Chem. 2004;69:3271–3275. doi: 10.1021/jo049824f. [DOI] [PubMed] [Google Scholar]
- 26.Yang CJ, Jockusch S, Vicens M, Turro NJ, Tan W. Proc Natl Acad Sci U S A. 2005;102:17278–17283. doi: 10.1073/pnas.0508821102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ebata K, Masuko M, Ohtani H, Jibu M. Photochem Photobiol Sci. 2006;62:836–839. doi: 10.1111/j.1751-1097.1995.tb09144.x. [DOI] [PubMed] [Google Scholar]
- 28.Kierzek R, Li Y, Turner D, Bevilacqua PC. J Am Chem Soc. 1993;115:4985–4992. [Google Scholar]
- 29.Masuko M, Ohtani H, Ebata K, Shimadzu A. Nucleic Acids Res. 1998;26:5409–5416. doi: 10.1093/nar/26.23.5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okamoto A, Tainaka K, Nishiza K, Saito I. J Am Chem Soc. 2005;127:13128–13129. doi: 10.1021/ja053609e. [DOI] [PubMed] [Google Scholar]
- 31.Seo JY, Hwang GT, Kim BH. Nucleic Acids Symp. 2005;49:135–136. doi: 10.1093/nass/49.1.135. [DOI] [PubMed] [Google Scholar]
- 32.Yamana K, Iwase R, Furutani S, Tsuchida H, Zako H, Yamaoka T, Murakami A. Nucleic Acids Res. 1999;27:2387–2392. doi: 10.1093/nar/27.11.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paris PL, Langenhan JM, Kool ET. Nucleic Acids Res. 1998;26:3789–3793. doi: 10.1093/nar/26.16.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cuppoletti A, Cho Y, Park JS, Strassler C, Kool ET. Bioconjug Chem. 2005;16:528–534. doi: 10.1021/bc0497766. [DOI] [PubMed] [Google Scholar]
- 35.Tong G, Lawlor JM, Tregear GW, Haralambidis J. J Am Chem Soc. 1995;117:12151–12158. [Google Scholar]
- 36.Yang CJ, Lin H, Tan W. J Am Chem Soc. 2005;127:12772–12773. doi: 10.1021/ja053482t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lakowicz JR. Principles of Fluorescent Spectroscopy. 1999. p. 87. [Google Scholar]
- 38.Maeda H, Maeda T, Mizuno K, Fujimoto K, Shimizu H, Inouye M. Chemistry. 2006;12:824–831. doi: 10.1002/chem.200500638. [DOI] [PubMed] [Google Scholar]