

Abstract
Non-substrate interaction of thrombin with fibrinogen promotes sequential cleavage of fibrinopeptides A and B (fpA and fpB) from the latter resulting in its conversion into fibrin. The recently established crystal structure of human thrombin in complex with the central part of human fibrin clarified the mechanism of this interaction. Here, we reveal new details of the structure and present the results of molecular modeling of the fpA- and fpB-containing portions of the Aα and Bβ chains, not identified in the complex, in both fibrinogen and protofibrils. The analysis of the results reveals that in fibrinogen the fpA-containing portions are in a more favorable position to bind in the active site cleft of bound thrombin. Surface plasmon resonance experiments establish that the fpB-containing portions interact with the fibrin-derived dimeric D-D fragment, suggesting that in protofibrils they bind to the newly formed DD regions bringing fpB into the vicinity of bound thrombin. These findings provide a coherent rationale for the preferential removal of fpA from fibrinogen at the first stage of fibrin assembly and the accelerated cleavage of fpB from protofibrils/fibrils at the second stage.
Thrombin-mediated conversion of the plasma protein fibrinogen to the insoluble fibrin matrix is the major event in blood clotting. Fibrinogen consists of two identical disulfide-linked subunits, each of which is formed by three non-identical polypeptide chains, Aα, Bβ and γ (1,2). The NH2-terminal portions of all six chains form the central E region of the molecule while their COOH-terminal portions form two identical distal D regions and two αC-domains (1–4). The Aα and Bβ chains start with the 16-residue fibrinopeptide A and the 14-residue fibrinopeptide B sequence (fpA and fpB)1, respectively. Proteolytic removal of fpA and fpB with thrombin exposes in each E region two pairs of polymerization sites (knobs) “A” and “B” starting with the Gly-Pro-Arg and Gly-His-Arg sequences, respectively. The interaction between these knobs and complementary polymerization sites (holes) “a” and “b” located in the D regions of neighboring molecules (D:E:D interaction) results in spontaneous polymerization of monomeric fibrin to the insoluble fibrin polymer (clot) (2,3,5).
Fibrin assembly is a highly ordered process that occurs in two stages. At the first stage, monomeric molecules assemble in a half-staggered manner through the D:E:D interactions to produce two-stranded protofibrils. At the second stage, protofibrils aggregate laterally to make thicker fibers that coalesce to form a three-dimensional network of fibrin clot. Numerous studies indicate that fpA is cleaved from fibrinogen much faster that fpB and that removal of fpA triggers formation of protofibrils, while removal of fpB coincides with their lateral aggregation (2,6–10). It has also been shown that fpB release, which is very slow at the start of the reaction, is accelerated upon polymer formation (7–9,11,12). Such a delay in fpB cleavage is necessary for normal fibrin assembly (13). It is also connected with the formation of different type of clots, consisting of fibrin type I and type II (7). Fibrin I, in which only the fpAs are removed, is less compact and is more readily digested by plasmin while fibrin II, lacking both fpA and fpB, is more compact and more resistant to fibrinolysis (7). Despite numerous studies, the mechanism underlying sequential cleavage of the fibrinopeptides is not completely understood.
To remove the fibrinopeptides thrombin binds to the central region of fibrinogen, not only through the active site cleft, but also through its fibrinogen-binding exosite I (14,15). The interaction via this exosite plays a major role in substrate recognition and removal of fpA and fpB, and it may be involved in their sequential cleavage. Recently, a complex between thrombin and a fragment Eht derived from the central region of fibrinogen was crystallized and its X-ray structure was solved (16). Although the locations of the NH2-terminal portions of the Aα and Bβ chains in the complex were not identified, the structure established the mode of the specific non-substrate interaction between these molecules via the thrombin exosite I (16). In the present work the structure interpretation has been extended and molecular modeling of a possible arrangement of these portions in fibrinogen and a protofibril, both with bound thrombin, has been performed. The resulting three-dimensional models establish the structural basis for the preferential removal of fpA at the first stage of fibrin assembly and provide a coherent explanation for the accelerated cleavage of fpB at the second stage.
EXPERIMENTAL PROCEDURES
Analysis of Electron Density and Model Building
The electron density was calculated based on the diffraction data obtained previously from a crystal of the Eht-thrombin complex (16). Building of the additional NH2-terminal residues of the Aα and Bβ chains in the Eht-thrombin complex was performed with the program XtalView (17) using 2Fo−Fc and Fo−Fc electron density maps contoured at 1.0σ and 2.0σ levels, respectively. The residues were added one by one extending the chains for no more then one residue prior to each cycle of refinement. The result of each addition was subjected to conventional least-square minimization of atomic coordinates with the program CNS (18) and validated by calculating an omit map. The crystallographic parameters and the refinement statistics are presented in Table 1. The statistics for the new coordinates are similar overall to that reported earlier (16). However, the electron density map in the new areas that have been interpreted is markedly improved.
Table 1.
Crystallographic parameters and refinement statistics
| Data collection | |
| Space group | P31 |
| Unit cell dimensions: | |
| a, b (Å) | 76.2 |
| c (Å) | 192.4 |
| Resolution (Å) | 20.0–3.65 (3.78-3.65) |
| I/σ (I) | 3.4 (1.9) |
| Completeness (%) | 93.4 (87.6) |
| Rsym (%) | 0.191 (0.389) |
| Redundancy | 2.2 (2.1) |
| Refinement | |
| Resolution (Å) | 20.0–3.65 (3.78-3.65) |
| No. reflections | 25,847 (2,432) |
| Rwork/Rfree | 22.1 (26.2)/29.0 (31.6) |
| No. atoms | |
| protein | 6,834 |
| PPACK | 60 |
| B-factor(Å2) | 27.5 |
| rms deviations from ideal values: | |
| bond length (Å) | 0.013 |
| bond angles (deg) | 1.88 |
Values in parentheses are for the highest-resolution shell.
Molecular Modeling
The Aα20–28 segments connecting the newly built NH2-terminal residues of the Aα chains with Arg19 of the fpA variant bound to thrombin molecules of the Eht-thrombin complex were built manually using computer graphics. Individual residues were added starting at AαCys28 and extending the chains towards AαArg19. The initial model was refined with short-term molecular dynamics at a constant temperature using the program CNS.
The entire NH2-terminal portions of the Aα and Bβ chains in fibrinogen were initially generated as extended polypeptides. To randomize their conformations several manual perturbations of the main chain dihedral angles were applied, followed by a 5 ps molecular dynamics calculation with simulated annealing.
The docking of the fibrin(ogen) E region into the dimeric DD region was carried out manually guided by the topology of the molecular surfaces. The structure of the cross-linked D dimer in complex with the synthetic peptides mimicking polymerization knobs (19) (PDB entry 1FZB) was used as a template. The docking space was restricted to the area allowing the binding of knobs A into their complementary holes on the surface of D-D. The relative positions of the individual components in the manually built model were then subjected to final adjustment by a rigid body dynamics procedure with the program X-PLOR (20), followed by energy minimization of the individual atomic positions.
The model illustrating possible localization of the NH2-terminal portions of the Bβ chains in a protofibril was prepared by manually arranging these portions on the surface of the dimeric D region. The initial arrangement was then adjusted with short term molecular dynamics with CNS to relieve bad contacts and steric clashes.
Figures - All figures were prepared with PyMol (21).
Fibrinogen Fragments and Peptides
The D1 fragment was prepared from plasmin digest of fibrinogen, while the D-D:E1 complex and D-D dimer were prepared from a plasmin digest of factor XIIIa-cross-linked fibrin as described earlier (22,23). The recombinant dimeric (Bβ1-66)2 fragment mimicking the dimeric arrangement of the Bβ chain in fibrinogen and its H16P, P18V mutant, Mut-(Bβ1-66)2, were produced in E. coli and purified as described (24). The truncated variant of the mutant, Mut-(β18-66)2, was prepared by treatment of Mut-(Bβ1-66)2 with thrombin (24). Although thrombin was expected to cleave only fpB (Bβ1-14), when the digestion mixture was left for more then 1h, the major degradation product was Mut-(β18-66)2. This was determined by NH2-terminal sequence analysis performed with a Hewlett-Packard model G 1000S sequencer. Synthetic peptides Gly-Pro-Arg-Pro and Gly-His-Arg-Pro mimicking polymerization knobs “A” and “B”, respectively, were purchased from Sigma.
Fluorescence Study
Fluorescence measurements of thermal unfolding of the D-D:E1 complex and the D-D dimer loaded with the synthetic peptides were performed by monitoring the ratio of the fluorescence intensity at 370 nm to that at 330 nm with excitation at 280 nm in an SLM 8000-C fluorometer. The temperature was controlled with a circulating water bath programmed to raise the temperature at ~1 °C/min. The concentrations of the D-D:E1 complex and the D-D dimer were 0.12 μM and 0.16 μM, respectively.
Surface Plasmon Resonance
The interaction of the Bβ chain fragment variants with the immobilized monomeric D1 fragment and dimeric D-D was studied by surface plasmon resonance using the BIAcore 3000 biosensor (Biacore AB, Uppsala, Sweden), which measures the association/dissociation of proteins in real time. Immobilization of D1 and D-D to the CM5 sensor chip was performed using the amine coupling kit (BIAcore AB, Uppsala, Sweden) according to the recommended procedure. Briefly, D1 at 25 μg/ml or D-D at 50 μg/ml, both in 10 mM sodium acetate, pH 5.0, were injected onto the chip surface to achieve the immobilization level of ~ 1000 or ~ 2000 response units (RU), respectively. Binding experiments were performed in binding buffer, HBS-P (BIAcore AB, Uppsala, Sweden), containing 1 mM CaCl2, at 10 μl/min flow rate. The Bβ chain fragment variants were injected at different concentrations and the association between them and immobilized D1 or D-D was monitored by the change in the SPR response; the dissociation was measured upon replacement of the ligand solution for the binding buffer without ligand. To regenerate the chip surface, complete dissociation of the complex was achieved by adding a solution containing 0.1 M sodium acetate, pH 4.0, and 2 M urea for 2 min followed re-equilibration with the binding buffer. Experimental data were analyzed using BIAevaluation 3.2 software supplied with the instrument. Kinetic constants, kass and kdiss, were estimated by global analysis of the association/dissociation curves using the 1:1 Langmurian interaction model, and the dissociation equilibrium constant (Kd) was calculated as Kd = kdiss/kass. The values were examined for self-consistency of the data as described in (25).
RESULTS
Analysis of Electron Density
Although our previous study (16) established the crystal structure of a complex between thrombin and the fibrinogen-derived thrombin-treated Eht fragment, locations of the NH2-terminal portions of the latter including Aα and Bβ chain residues 17–31 and 15–55, respectively, were not identified. In that study, while refining the X-ray structure of the complex, electron density in the vicinity of the first clearly observed NH2-terminal residues of the Aα and Bβ chains, AαAsp32 and BβGlu56, was not interpreted because of its partial disorder. In the present study the interpretation of this electron density has been extended. By reanalyzing the electron density three more residues, Lys29, Asp30 and Ser31, in each Aα chain (Fig. 1A) have been added. Even though we did not interpret the density between two AαLys29 residues, it most likely corresponds to the disulfide bridge formed by two AαCys28 residues. Therefore, these residues were added to the model (Fig. 1B). The locations of the AαCys28 residues imply antiparallel directions for the Aα chain backbones. This in turn suggests that the electron density in the vicinity of each Aα Asp30 most likely corresponds to two AαSer26 of the antiparallel Aα chains. These residues were also added to the model, as well as the neighboring AαAla27 whose electron density was not observed. It should be noted that in the crystal structure of chicken fibrinogen (26) (PDB entry 1M1J, which replaced the original entry 1JFE), in which positions of homologous AαCys28 and the neighboring AαSer27 residues were identified, the Aα chains also have a similar configuration (Fig. 1B).
Fig. 1. Stereoview of the annealed composite omit electron density map of the regions surrounding the NH2-terminal portions of the Aα (panels A and B) and Bβ (panel C) chains in the complex of thrombin with the Eht fragment.

The map is contoured at 0.8 σ. Panels A and C show the newly built residues; panel B highlight those added through the modeling efforts. Carbon atoms of the newly built and modeled residues are colored in white while those built earlier (16) are in blue for the Aα chains and green for the Bβ chains; nitrogen and oxygen atoms are in blue and red. Panel B shows also the Aα chains of chicken fibrinogen superimposed with the homologous chains of the Eht fragment. Chicken structure (26) is represented by Cα tracing (yellow); the first two identified residues, AαSer27 and AαCys28, are also shown by wireframe model with their side chains.
Similarly, by reanalyzing the electron density in the vicinity of BβGlu56, two more residues in the NH2-terminal portion of each Bβ chain, BβLys54 and BβVal55, were added to the model (Fig. 1C). Although the final composite omit map displayed additional density adjacent to these residues, we did not attempt to interpret it due to its substantial disorder. The locations of all newly identified NH2-terminal residues in the Eht-thrombin complex are shown in Fig. 2A. We next modeled possible conformations of the missing NH2-terminal portions of the Aα and Bβ chains, Aα1–25 and Bβ1–53, respectively.
Fig. 2. Arrangement of the NH2-terminal portions of the Aα chains in the structure of the complex of thrombin with the Eht fragment.

Panel A shows a ribbon diagram of the thrombin-Eht complex with the newly built Aα26-31 and Bβ54-55 residues shown by sticks colored by atom types: blue for nitrogens, red for oxygens, orange for sulfurs, and white for carbons; locations of Bβ54-55 residues are also indicated by the dotted circles. The thrombin-bound fpA variant (28) (PDB entry 1UCY) is shown by magenta (Aα7-16) and yellow (Aα17-19) sticks. Dotted lines indicate the distance between AαArg19 and AαSer26 (see text). Panel B illustrates the solvent accessible surface of the complex with the newly modeled Aα20-25 connecting segments shown by white sticks. In both panels the Aα, Bβ and γ chains of Eht are colored in blue, green and red, respectively; thrombin molecules are in beige.
Modeling of the NH2-terminal Portions of the Aα and Bβ chains
Since several structures of thrombin in complex with fpA variants are available (15,27–30) (PDB entries 1FPH, 1BBR, 1UCY, 1YCP, and 1DM4, respectively), the conformation of the NH2-terminal portions of the Aα chains in the thrombin-Eht complex was modeled assuming that their fpAs interact with thrombin in a manner similar to that observed in these structures. The atomic coordinates of one of the variants (28) (PDB entry 1UCY), which includes the fpA residues Aα7-16 and the flanking Gly17-Pro18-Arg19 composing polymerization knob “A”, were selected as a template. This variant was docked to the thrombin molecules of the Eht-thrombin complex (Fig. 2A) and then the remaining 6-residue segment, Aα20-25, was added to connect Arg19, the last identified COOH-terminal residue of the fpA variant, with AαSer26, the newly built NH2-terminal residues of the Aα chains of the Eht fragment. The resulting model is shown in Fig. 2B.
The modeling of the NH2-terminal portions of the Aα chains was tightly restricted by the length of the connecting Aα20–25 segment (21.8 Å) and by the distance between the main chain carbonyl and nitrogen atoms of AαArg19 and AαSer26, respectively (~20 Å). An additional conformational restriction for the Aα20–25 segments comes from the topology of the molecular surface around the disulfide bridge. There are two pairs of wall-like structures, each formed by the Aα and Bβ chain residues (α- and β-walls in Fig. 3A), which sterically constrain the modeled segments forcing their accommodation in the canyon-like groove between them. In this configuration, several side chains of each segment can potentially interact through contacts (Fig. 3B), which may further assist their localization in the groove. Most of the contacts could be formed with the Aα and Bβ chain residues composing the canyon walls. Particularly, NZ of AαLys29 is surrounded by a number of polar oxygen atoms, namely, by Oγ of AαSer26 and by the main chain carbonyl oxygen atoms of AαGln25 and AαHis24. The side chain of AαGln25 may form hydrogen bonds with the main chain oxygen and nitrogen atoms of BβArg57 and BβAla59, respectively, and AαHis24 may form a salt bridge with AαAsp30. This salt bridge seems to cause a bend pointing the chain towards the active site cleft of bound thrombin. In addition, the side chains of fibrinogen AαGlu22 and thrombin Arg73 may form a salt bridge.
Fig. 3. Topology of the molecular surface around the modeled segments of the Aα chains in the thrombin-Eht complex (A) and potential contacts between these segments and the complex (B).

Panel a shows the solvent accessible surface of the thrombin-Eht complex with the Aα20-28 segments (shown by sticks) located in the groove between the wall-like structures denoted as α- and β-walls. The color scheme is the same as in Fig. 2, namely, the Aα, Bβ and γ chains of Eht are in blue, green and red, respectively, thrombin molecules are in beige; the Aα20-25 segments are in white, AαArg19 of the thrombin-bound fpA variant is in yellow, and the Aα26-28 segment are colored by atom types. Panel B shows a ribbon diagram of the thrombin-Eht complex in the same projection as that in panel A with a potential set of polar contacts between individual residues of the Aα22-30 segments and the bulk of the complex. The residues involved in contact formation are represented by ball-and-sticks and colored according to their atom types; interatomic contacts are shown by dashed lines.
The contacts described above imply that in fibrinogen these segments should be bound to the body of the molecule through interactions with His24, Gln25 and Ser26 even when thrombin is not present, while the remaining Aα1-23 residues, whose structure in solution is not known, could be unbound or “free-swimming”, i.e. in a random conformation. This was taken into account in the modeling of a possible arrangement of the entire 1-25 portions of the Aα chains in fibrinogen. To model such an arrangement, we used the structure of the Eht fragment as a template for the newly built Aα26-31 residues, to which we added the Aα24-25 segment in the conformation presented in Fig. 3. The remaining Aα1-17 (fpA) and Aα17-23 segments were generated in a random conformation. A possible arrangement of the entire Aα1-25 portions in fibrinogen is depicted in Fig. 4A.
Fig. 4. Putative location of the NH2-terminal portions of the Aα and Bβ chains in fibrinogen.

Panels A and B represent the ribbon diagram of fibrinogen and solvent accessible surface of a complex of fibrinogen with thrombin, respectively. The newly modeled portions of the Aα and Bβ chains are presented in both panels by sticks. The model in panel A was generated by superimposing the chicken fibrinogen structure (26), which was used as a template, with that of the Eht fragment (16), followed by replacement of the overlapping regions with those from the latter. In the model, the Aα, Bβ and γ chains derived from Eht are shown in blue, green and red, respectively, those derived from chicken fibrinogen are in gray. The complete NH2-terminal portions of the Aα and Bβ chains including fpA and fpB (both shown in magenta) were modeled as described in the text. Panel B represents the same fibrinogen molecule in complex with two thrombin molecules that were docked to its central region in the way that they appear in the structure of the thrombin-Eht complex (Fig. 2). The complete Aα, Bβ and γ chains are in blue, green and red, respectively. Thrombin molecules are in beige, their catalytic triad is highlighted in red. The vertical lines denote approximate boundaries between the fibrinogen D and E regions.
Modeling of the missing Bβ1-53 portions was more problematic since the structure of thrombin-bound fpB was not available, and the polypeptides to be modeled were much larger. Since the cleavage of both fpA and fpB requires that they fit into the active site cleft of thrombin, they should adopt similar conformations when bound to the cleft, at least in the area adjacent to the cleavage site. With such conformations, the distance between the last residue of fpB (Arg14) and the first newly built Bβ chain residue (Lys54) was found to be about 36 Å, which is much shorter than the length of the extended Bβ15-53 segment to be modeled (143.8 Å). Since there were no apparent conformational restrictions for positioning these segments on the surface of Eht, no attempt was made to locate them in the Eht-thrombin complex. As to the folding status of these portions, it has been previously proposed, based mainly on the secondary structure prediction, that in fibrinogen they adopt a specific conformation (31). However, residues corresponding to these portions were not identified in the crystal structures of chicken fibrinogen (26) or in the Eht-thrombin complex (16). Furthermore, the well-established fact that the NH2-terminal portions of the Bβ chains are easily cleaved by proteases (1) is in agreement with their lacking a compact structure. Thus, since there was no experimental evidence for a folded structure in these portions, we generated them in a random conformation (Fig. 4A).
To clarify a possible mechanism for cleavage of fibrinopeptides by thrombin, two thrombin molecules were added to the model of fibrinogen presented in Fig. 4A, as they appear in the structure of the thrombin-Eht complex (16). The resulting complex of fibrinogen with thrombin (Fig. 4B) clearly shows that the NH2-terminal portions of the fibrinogen Aα chains are in more favorable positions to occupy the active site cleft of thrombin than those of the Bβ chains. This provides a plausible explanation for the preferential removal of fibrinopeptides A at the first stage of fibrin assembly, during which protofibrils are formed. Since numerous studies have established that polymer formation accelerates removal of fibrinpopeptides B (7–9,11,12), we next modeled a protofibril, which represents the simplest form of a fibrin polymer, to clarify the underlying mechanism.
Modeling of a Protofibril
A number of previous attempts to model protofibrils have been carried out after crystal structures of fibrinogen and fibrin(ogen)-derived fragments were established (19,32–34). The most detailed model of a protofibril was suggested by Yang et al (5). It incorporated the low-resolution crystal structures of chicken fibrinogen and the fibrin-derived dimeric D fragment (D dimer or D-D) complexed with synthetic peptides mimicking the polymerization knobs. In this model, the D and E regions were placed distant from one another and all contacts between them (D:E:D interactions) were limited to those between knobs “A” and “B” and the complementary holes “a” and “b”. However, such a mode of interaction is in poor agreement with certain experimental observations. First, it was found that in the fibrin-derived D-D:E1 complex, which mimics the conformation and interactions of the D and E regions in (proto)fibrils (35), knobs “B” are exposed (35) suggesting that in this complex they are not involved in the D:E:D interactions. Second, it was shown that the D:E:D interactions in fibrin and in the D-D:E1 complex causes conformational changes in the D regions resulting in the exposure of their tPA- and plasminogen-binding sites (22). At the same time, binding of the synthetic peptides Gly-Pro-Arg-Pro and Gly-His-Arg-Pro, which mimic knobs “A” and “B”, respectively, to the complementary holes of the isolated D dimer was not sufficient for exposing the binding sites (22). Further, denaturation studies revealed that the D regions in fibrin polymer and in the D-D:E1 complex had increased thermal stability due to the D:E:D interactions (22,36–38). To test whether the synthetic knobs could cause such stabilization, the thermal stability of the D-D:E1 complex was compared with that of the D dimer loaded with the Gly-His-Arg-Pro and/or Gly-Pro-Arg-Pro peptides. When these species were heated in the fluorometer while monitoring the ratio of fluorescence intensity at 370 nm to that at 330 nm as a measure of the spectral shift that accompanies unfolding, no thermal stabilization of D-D was observed in the presence of 16 μM (a 100-fold molar excess over D-D) of either one or both peptides (Fig. 5). This peptide concentration is equal to the Kd for the interaction of Gly-Pro-Arg-Pro with the D1 fragment reported earlier (39). Since the similarity in the conformations of D1 and D-D (19) suggests similarity in their affinities to Gly-Pro-Arg-Pro, at this peptide concentration 50% of the binding sites in D-D should be saturated. Further increases in the Gly-Pro-Arg-Pro concentration to 160 μM (a 1000-fold molar excess over D-D), at which more than 90% of the binding sites should be saturated, did not change the thermal stability of D-D (Fig. 5, open triangles). Altogether, these observations suggest that the interaction between the D and E regions in protofibrils is not limited to the knob-to-hole binding, i.e. these regions may form additional contacts and therefore should be in a closer proximity to each other. This was taken into account in subsequent modeling of the D:E:D contacts.
Fig. 5. Influence of the E1 fragment and the synthetic peptides mimicking knobs “A” and “B” on the stability of the fibrin-derived D dimer.

Solid and dashed curves represent fluorescence-detected melting of the dimeric D-D fragment at 0.16 μM and the D-D:E1 complex at 0.12 μM, respectively. Melting of 0.16 μM D-D in the presence of a 100-fold molar excess of Gly-Pro-Arg-Pro or Gly-Pro-Arg-Pro and Gly-His-Arg-Pro are shown by open and filled circles, respectively, while that in the presence of a 1000-fold excess of Gly-Pro-Arg-Pro is shown by open triangles. All experiments were performed in 50 mM glycine buffer, pH 8.6, with 0.5 mM Ca2+.
In modeling the D:E:D contacts, fpA was first removed from the structure of the E region of fibrinogen presented in Fig. 4 to expose knobs “A”. Then the dimeric D fragment (19) was positioned in a way that permitted docking of these knobs into the complementary holes “a” (Fig. 6A). With such an arrangement, the central portion of the E region and the D dimer exhibit apparent surface complementarities. Namely, there is a ridge in the middle of the E region which seems to be complementary to the crevice formed by the γ chains of the D dimer. Assuming that the D:E:D interaction uses these complementarities, the structure of the E region was then docked to that of the D dimer. In the resulting structure (Fig. 6B), which actually corresponds to that of the D-D:E1 complex and represents an essential portion of a protofibril (Fig. 6B, bottom diagram), the ridge interacts with the crevice burying about 600 Å2 of its solvent accessible area. Such a contact area could be sufficient to account for the increased thermal stability and function-related conformational changes discussed above.
Fig. 6. Putative arrangement of the D and E regions and location of the NH2-terminal portions of the Bβ chains in a protofibril.
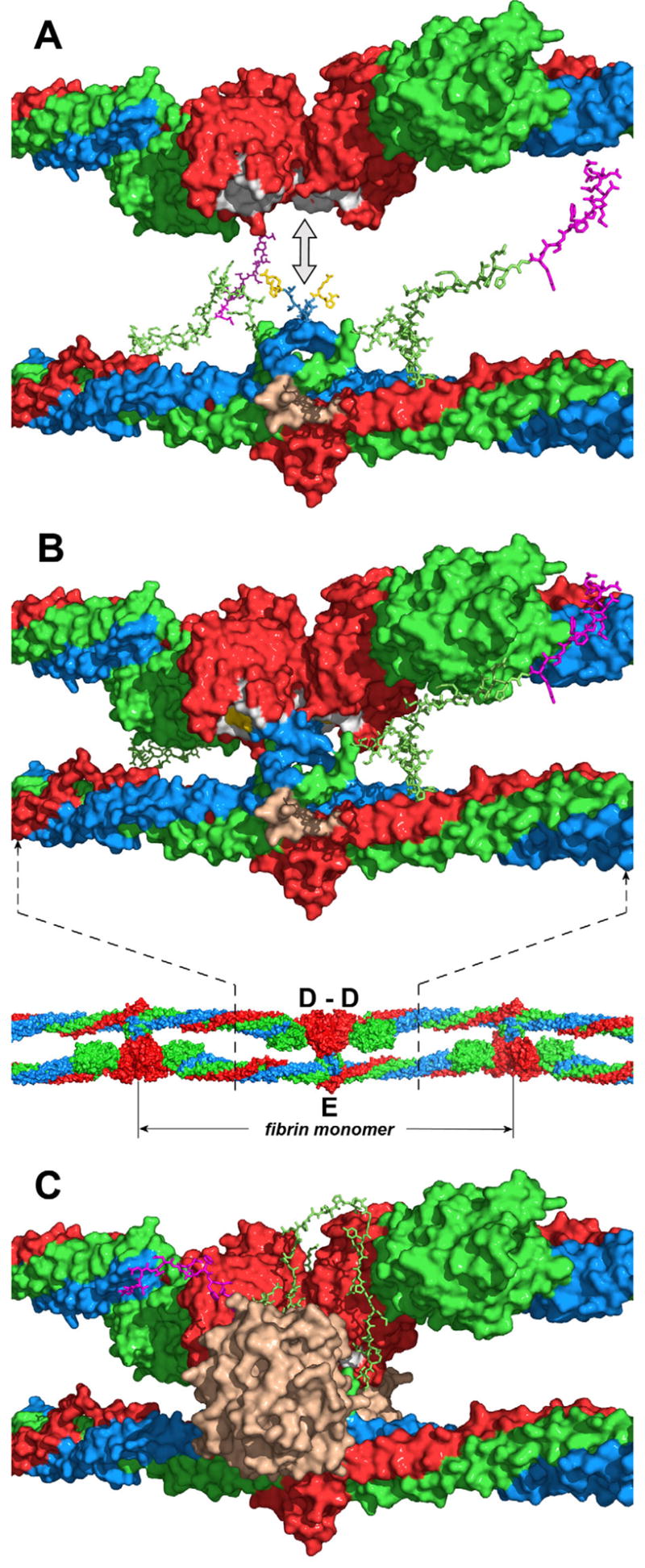
Panel A represents docking of the D dimer (top diagram) into the E region (bottom diagram) in the direction shown by the double-headed arrow to model the D:E:D interaction in a protofibril presented in panel B (top diagram). The location of the D and E regions in a protofibril is shown in the bottom diagram of panel B by dashed lines; the individual fibrin monomer in the protofibril is also denoted. The D dimer and E region are shown with solvent accessible surfaces; the randomly generated Aα17-23 and Bβ1-53 segments are represented by sticks. The Aα, Bβ and γ chains are shown in blue, green and red, respectively, fpBs in magenta, polymerization knobs “A” of the E region (Aα chain residues Gly17-Pro18-Arg19) in yellow, complementary holes “a” of the D regions in white, and the thrombin-binding site in the E region in beige. Note that although the NH2-terminal portions of the Bβ chains in panel B are shown in the same conformation as in panel A, in a protofibril they should interact with the newly formed D-D “wall” (see text). Panel C shows the same model as panel B with thrombin (in beige) bound to the E region and the NH2-terminal portion of the Bβ chain bound to the D-D “wall”. Although the exact conformation of this portion and the mode of its interaction with D-D are yet to be identified, in the model it is arranged on the D-D “wall” in a way that would facilitate its interaction with the active site cleft of bound thrombin (see text).
It should be noted that in fibrinogen variant Naples I, which is characterized by defective thrombin binding, the rate of the removal of both fibrinopeptides, fpA and fpB, is substantially reduced (40). This implies that binding of thrombin to the E region is required for effective removal of both fpA and fpB and that interaction between the DD and E regions in a protofibril should not interfere with this binding. This seems to be the case with the model of a protofibril presented in Fig. 6. Indeed, docking the structure of thrombin with that of the protofibril reveals that both thrombin binding sites in the E region are accessible to thrombin and there are no steric conflicts between bound thrombin and D-D (Fig. 6C).
Localization of the NH2-terminal Portions of the Bβ Chains in a Protofibril
Because of the length and possible conformational flexibility of the NH2-terminal portions of the Bβ chains, fpB could be quite distant from the active site cleft of thrombin bound to fibrinogen (Fig. 4B). At the same time, the well established accelerating effect of polymer formation on fpB cleavage (7–9,11,12) suggests that the structure of protofibrils should reduce conformational space for these portions in a way that promotes the binding of fpB to the active site cleft. Indeed, visual inspection of the models presented in Fig. 6 reveals that the DD regions form “a wall” separating the NH2-terminal portions of the Bβ chains from each other thus restricting conformational space for each portion to one side of a protofibril. Another possible factor restricting such space in protofibrils would be an interaction of these portions with the DD “wall”. In this regard, it has been proposed that the D-D:E1 complex is maintained by the interaction through a composite polymerization site that includes the Aα chain residues 17–19 (knob “A”) and the Bβ chain residues 20–49 (35). This implies that in protofibrils the NH2-terminal portions of the Bβ chains should interact with the DD regions through the residues located beyond fpB and knob “B”. To test this suggestion, the following experiments were performed.
We have previously prepared a recombinant dimeric (Bβ1-66)2 fragment which mimics the dimeric arrangement and properties of the NH2-terminal portions of the Bβ chains in fibrinogen (24). We have also prepared a recombinant dimeric (Bβ1-66)2 mutant, Mut-(Bβ1-66)2, with the His16Pro and Pro18Val mutations in knob “B”, and its truncated variant, Mut-(Bβ18-66)2, in which fpB and flanking knob “B” (residues 15–17) were missing (24). All three fragments were used to test the direct interactions between the NH2-terminal portions of the Bβ chains and the fibrin(ogen) D regions by surface plasmon resonance (SPR). When increasing concentrations of the wild-type fragment, (Bβ1-66)2, were added to the fibrin-derived dimeric D fragment immobilized onto a BIAcore sensor chip, a dose-dependent binding was observed (Fig. 7A). The global fitting analysis (see Experimental Procedures) of the SPR-detected binding curves obtained at various concentrations of (Bβ1-66)2 gave a Kd value of 13 μM (Table 2). The recombinant mutant fragment and the truncated variant both exhibited binding to the D dimer with Kds of 14.8 μM and 14 μM, respectively, very similar to that for the wild type fragment (Table 2). These results provide direct evidence for the interaction between the NH2-terminal portions of the Bβ chain and the dimeric D regions formed in protofibrils and fibrils. They also indicate that fpB and flanking knob “B” (residues 1–17) are not involved in this interaction.
Fig. 7. Analysis of binding of the recombinant (Bβ1-66)2 fragment to the fibrin-derived D dimer (A) and the fibrinogen-derived D1 fragment (B) by surface plasmon resonance.
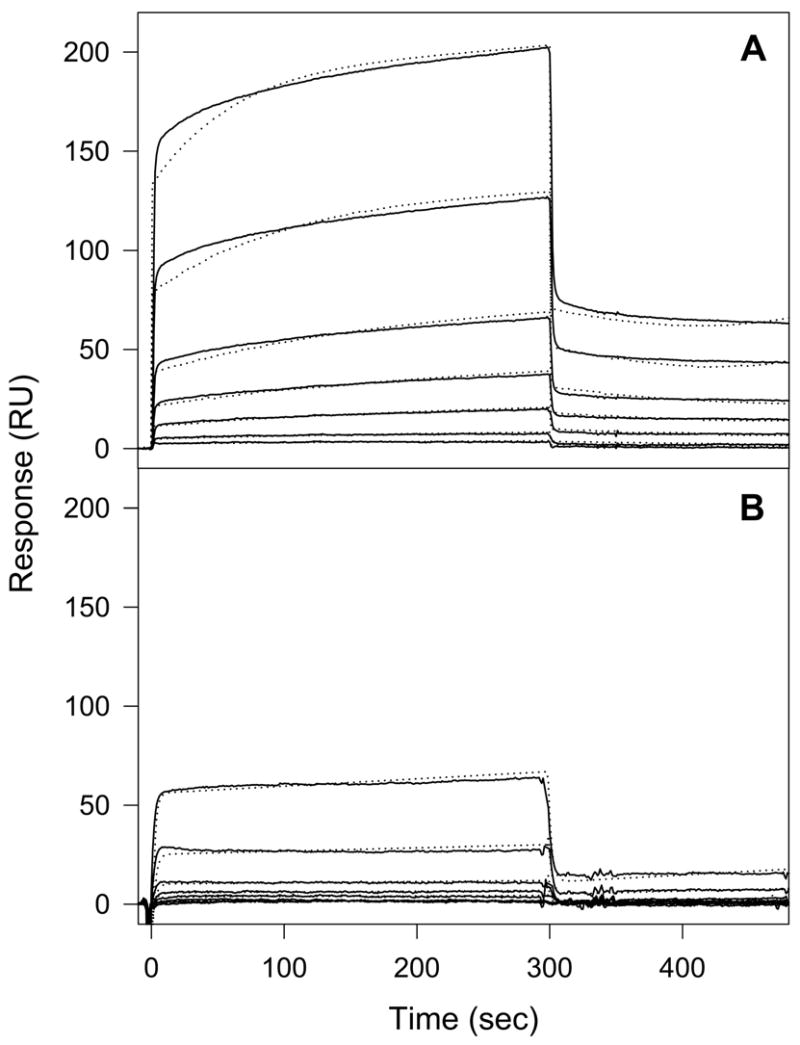
The (Bβ1-66)2 fragment was added at 0.5, 1, 2.5, 5, 10, 25 and 50 μM to the immobilized D dimer (A) or the D1 fragment (B), and its association/dissociation was monitored in real time while registering the resonance signal (response). The dotted curves represent the best fit of the binding data using global fitting analysis (see Experimental Procedures). The determined Kd values are presented in Table 2.
Table 2.
Dissociation constants (Kd) for the interaction of the recombinant fibrinogen (Bβ1-66)2 fragment and its mutant variants with the fibrinogen-derived D1 fragment and fibrin-derived D-D fragment obtained by surface plasmon resonancea
| Recombinant Fragments | Kd for interaction with D-D (μM) | Kd for interaction with D1 (μM) |
|---|---|---|
| (Bβ1-66)2 | 13.0 ± 2.0 | 153 ± 16 |
| Mut-(Bβ1-66) 2 | 14.8 ± 3.7 | 148 ± 21 |
| Mut-(β18-66)2 | 14.0 ± 0.6 | --- |
Values are the means ± S.D. of at least three independent experiments.
Next the interactions between (Bβ1-66)2 or its mutant and the immobilized fibrinogen-derived monomeric D1 fragment were tested. Both the wild type and the mutant bound to D1, however, with a much lower affinity than to the D dimer. The Kd values were found to be 153 μM and 148 μM for (Bβ1-66)2 and Mut-(Bβ1-66)2, respectively (Table 2). With such an affinity, in plasma, where the normal fibrinogen concentration is about 10 μM, the probability of interaction between individual fibrinogen molecules through the NH2-terminal portions of the Bβ chains and the D regions should be quite low. At the same time, the Kd value for the interaction of (Bβ1-66)2 with the immobilized D dimer (Kd = 13 μM) is comparable to the concentration of fibrinogen in plasma. This implies that binding of the NH2-terminal portions of the Bβ chains to the D regions occurs mainly when these regions are brought together (dimerized) in protofibrils and fibrils by the D:E:D interactions. Such binding should reduce flexibility of these portions and may “immobilize” them on the dimeric D regions in a conformation that promotes their cleavage by bound thrombin, as shown schematically in Fig. 6C. This is in agreement with the accelerating effect of polymer formation on the cleavage of fpB.
DISCUSSION
According to the current view, fibrin assembly is a highly ordered process that occurs in two stages. At the first stage thrombin cleaves a pair of fibrinopeptides A from fibrinogen to trigger the formation of protofibrils. Subsequent cleavage of a pair of fpBs at the second stage promotes lateral aggregation of protofibrils into thicker fibrils. Non-substrate interactions of thrombin with fibrin(ogen) play an important role at both stages. It is also well-established that during normal fibrin assembly fpB is mainly cleaved from assembled protofibrils and fibrils and that their formation accelerates fpB cleavage (7–9,11,12). At the same time, the mechanisms underlying these events remained to be clarified. In the previous study (16), we solved the crystal structure of thrombin in complex with a fragment, Eht, corresponding to the central region of fibrin and established the mode of the non-substrate interaction between them. In the present study, we revealed new details of the crystal structure and modeled the NH2-terminal portions of the fibrin(ogen) Aα and Bβ chains that were not identified in the complex. We also modeled their possible arrangements in protofibrils. Analyses of the resulting models established a structural basis for the preferential cleavage of fpA at the first stage of fibrin assembly and for the accelerated removal of fpB upon formation of protofibrils at the second stage of assembly.
Modeling of the fpA-containing NH2-terminal portions of the Aα chains (Aα1-25) missing in the structure of the thrombin-Eht complex was straightforward since these portions were relatively short. More importantly, the crystal structure of thrombin in complex with almost half of the portion (Aα7-19), that was available, reduced the region to be modeled to a 6-residue segment (Aα20-25). Finally, when this segment is fully extended it has practically the same length as the distance between Aα19 and Aα26. This together with the unique surface topology of Eht dictated the unambiguous accommodation of the segment on the surface of the complex, namely, in the groove between the α- and β-walls that directs this segment and the adjacent fpA towards fibrinogen-bound thrombin (Fig. 3). In contrast, the missing fpB-containing NH2-terminal portions of the Bβ chains (Bβ1-53) were too large to be modeled unambiguously. Although the size of each of these polypeptide segments is sufficient to form a thermodynamically stable domain (41), there is no experimental evidence for the presence of a folded structure in them. Therefore these portions of the molecule were generated in random conformations and located arbitrarily relative to the bulk of the molecule (Fig. 4A).
It should be noted that the conformation of the NH2-terminal portion of the Aα chain complexed with thrombin was modeled earlier by Rose and Di Cera (42). According to their model the 1–38 portion of the Aα chain envelopes thrombin making contacts with its exosite II, active site cleft, S′ groove, and exosite I. However, after the crystal structure of the thrombin-Eht complex was solved (16) and new details of the structure were revealed in this study, it became clear that the real conformation of the Aα chains is very different from the previously modeled one. Due to the antiparallel arrangement of the Aα chains in fibrinogen, thrombin interacts with the Aα33-39 portion of one Aα chain through exosite I (16), while its active site cleft is occupied by fibrinopeptide A coming from the other Aα chain. This can be seen in Fig. 2 and 3.
The model presented in Fig. 4B shows that each fpA-containing portion of the Aα chain is located in the vicinity of fibrinogen-bound thrombin and is actually directed towards its active site cleft. Furthermore, the length of each portion seems to be optimal for their fpAs to easily reach the cleft through minimal conformational adjustments. In contrast, the fpB-containing portions of the Bβ chains could be quite distant from bound thrombin due to their length and their random orientation. Although these portions should have sufficient flexibility to reach the cleft, their excessive length would prevent them from doing that as efficiently as do fpA-containing portions. Thus, the results of our modeling indicate that in fibrinogen fibrinopeptides A are “pre-positioned” for an efficient cleavage by thrombin while fibrinopeptides B are not. This provides the structural basis for the preferential removal of fpA at the first stage of fibrin assembly. It should be noted that this is not the only factor governing the preferential removal of fpA. For example, the sequence of fpB is different from that of fpA and therefore its affinity to the active site cleft of thrombin may be lower than that of fpA resulting in further reduction of the cleavage rate. In agreement, it was shown that mutation of Trp215 to Ala in the active site cleft of thrombin, which most probably affects its interaction with fpB to a lesser extend than that with fpA, results in the drop in thrombin specificity towards fibrinogen and the release of fpA and fpB with similar kinetics (43). It was also shown that substitution of a modified version of fpA (fpA′) for fpB in the fibrinogen Bβ chain increased the rate of the thrombin-catalyzed release of the former (44). However, even in such a mutant the rate of fpA′ release from the fpA′α chain was still higher than that from the fpA′β chain most probably due to the conformational restraints described above.
To clarify the structural basis for the accelerated removal of fpB upon formation of protofibrils and fibrils, we modeled the structure the D-D:E1 complex, which represents an essential portion of a protofibril, and analyzed the possible arrangement of the fpB-containing NH2-terminal portions of the Bβ chains in this structure. While modeling the complex, it was assumed that the interaction between D-D and E is not limited to the knob-to-hole contacts. As mentioned in the Results section, this assumption is in agreement with the experimental data on the D:E:D interaction-induced stabilization of the D regions and the exposure of their functional sites. It was also taken into account that the NH2-terminal portions of the Bβ chains interact with D-D. Such an interaction, which was previously suggested by Moskowitz and Budzynski (35) and confirmed by direct experiments in this study, provided a rationale for the accelerating effect of protofibril formation on the cleavage of fpB. Namely, when the D regions are brought together in a protofibril by the D:E:D interaction, the NH2-terminal portions of the Bβ chains bind to them to position their fpB segments in the vicinity of the active site cleft of bound thrombin.
It should be noted that other mechanisms of enhancing the cleavage of fpB from fibrin polymers cannot be excluded. For example, in fibrinogen the NH2-terminal portions of the Aα chains are located so close to bound thrombin that they may create a steric problem for binding of fpB to its active site cleft even after removal of their fpA. Such a problem should not exist in protofibrils and fibrils where these portions move away and become “immobilized” by the interaction with the complementary holes “a”. It should also be noted that the model presented in Fig. 6C does not provide accurate positioning of the NH2-terminal portions of the Bβ chains on the D-D “wall” as well as their exact conformations. In addition, the model does not reflect the previously proposed conformational changes in the D regions upon the D:E:D interaction (5,22). Only a crystal structure of the D-D:E1 complex would clarify such important structural details. At the same time, neither of these details is critical for the major conclusions of this study.
In summary, the results of this work clearly indicate that the conformation of the fpA-containing NH2-terminal portions of the Aα chains and their location relative to the thrombin-binding sites direct the preferential cleavage of fpA by fibrinogen-bound thrombin at the first stage of fibrin assembly. They also suggest that the accelerating effect of polymer formation on fpB cleavage is related mainly to the interaction of the fpB-containing NH2-terminal portions of the Bβ chains with the dimeric DD regions formed in protofibrils and fibrils. Finally, this and the previous study (16) highlight the involvement of non-substrate interactions of thrombin with fibrin(ogen) in governing the two-stage fibrin assembly.
Footnotes
This work was supported by National Institute of Health Grant HL-56051 to L. M.
Atomic coordinates have been deposited in the Brookhaven Protein Data Bank under accession code 2A45
fpA and fpB, fibrinopeptides A and B. Fragment Eht, thrombin-treated central region of fibrinogen prepared by digestion of human fibrinogen with proteolytic enzyme hementin (16). This fragment has the same N-terminal residues as those in fibrin, i.e. αGly17, βGly15, and γTyr1.
Certain commercial materials, instruments, and equipment are identified in this manuscript in order to specify the experimental procedure as completely as possible. In no case does such identification imply a recommendation or endorsement by the National Institute of Standards and Technology nor does it imply that the materials, instruments, or equipment identified is necessarily the best available for the purpose.
The accepted SI units of concentration, mol/L have been represented by the symbol M in order to conform to the conventions of this journal.
References
- 1.Henschen A, McDonagh J. Fibrinogen, fibrin and factor XIII. In: Zwaal RFA, Hemker HC, editors. Blood Coagulation. Elsevier Science Publishers; Amsterdam: 1986. pp. 171–241. [Google Scholar]
- 2.Blomback B. Fibrinogen and fibrin-proteins with complex roles in hemostasis and thrombosis. Thromb Res. 1996;83:1–75. doi: 10.1016/0049-3848(96)00111-9. [DOI] [PubMed] [Google Scholar]
- 3.Doolittle RF. Fibrinogen and fibrin. Annu Rev Biochem. 1984;53:195–229. doi: 10.1146/annurev.bi.53.070184.001211. [DOI] [PubMed] [Google Scholar]
- 4.Weisel JW, Medved L. The structure and function of the αC domains of fibrinogen. Ann N Y Acad Sci. 2001;936:312–327. doi: 10.1111/j.1749-6632.2001.tb03517.x. [DOI] [PubMed] [Google Scholar]
- 5.Yang Z, Mochalkin I, Doolittle RF. A model of fibrin formation based on crystal structures of fibrinogen and fibrin fragments complexed with synthetic peptides. Proc Natl Acad Sci USA. 2000;97:14156–14161. doi: 10.1073/pnas.97.26.14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blomback B, Vestermark A. Isolation of fibrinopeptides by chromatography. Arkiv Kemi. 1958;12:173–182. [Google Scholar]
- 7.Blomback B, Hessel B, Hogg D, Therkildsen L. A two-step fibrinogen-fibrin transition in blood coagulation. Nature. 1978;275:501–505. doi: 10.1038/275501a0. [DOI] [PubMed] [Google Scholar]
- 8.Martinelli RA, Scheraga HA. Steady-state kinetic study of the bovine thrombin-fibrinogen interaction. Biochemistry. 1980;19:2343–2350. doi: 10.1021/bi00552a010. [DOI] [PubMed] [Google Scholar]
- 9.Higgins DL, Lewis SD, Shafer JA. Steady state kinetic parameters for the thrombin-catalyzed conversion of human fibrinogen to fibrin. J Biol Chem. 1983;258:9276–9282. [PubMed] [Google Scholar]
- 10.Hanna LS, Scheraga HA, Francis CW, Marder VJ. Comparison of structures of various human fibrinogens and a derivative thereof by a study of the kinetics of release of fibrinopeptides. Biochemistry. 1984;23:4681–4687. doi: 10.1021/bi00315a025. [DOI] [PubMed] [Google Scholar]
- 11.Hurlet-Jensen A, Cummins HZ, Nossel HL, Liu CY. Fibrin polymerization and release of fibrinopeptide B by thrombin. Thromb Res. 1982;27:419–427. doi: 10.1016/0049-3848(82)90059-7. [DOI] [PubMed] [Google Scholar]
- 12.Ruf W, Bender A, Lane DA, Preissner KT, Selmayr E, Muller-Berghaus G. Thrombin-induced fibrinopeptide B release from normal and variant fibrinogens: influence of inhibitors of fibrin polymerization. Biochim Biophys Acta. 1988;965:169–175. doi: 10.1016/0304-4165(88)90053-0. [DOI] [PubMed] [Google Scholar]
- 13.Weisel JW, Veklich Y, Gorkun O. The sequence of cleavage of fibrinopeptides from fibrinogen is important for protofibril formation and enhancement of lateral aggregation in fibrin clots. J Mol Biol. 1993;232:285–297. doi: 10.1006/jmbi.1993.1382. [DOI] [PubMed] [Google Scholar]
- 14.Fenton JW, II, Olson TA, Zabinski MP, Wilner GD. Anion-binding exosite of human α-thrombin and fibrin(ogen) recognition. Biochemistry. 1988;27:7106–7112. doi: 10.1021/bi00418a066. [DOI] [PubMed] [Google Scholar]
- 15.Stubbs MT, Oschkinat H, Mayr I, Huber R, Angliker H, Stone SR, Bode W. The interaction of thrombin with fibrinogen. A structural basis for its specificity. Eur J Biochem. 1992;206:187–195. doi: 10.1111/j.1432-1033.1992.tb16916.x. [DOI] [PubMed] [Google Scholar]
- 16.Pechik I, Madrazo J, Mosesson MW, Hernandez I, Gilliland GL, Medved L. Crystal structure of the complex between thrombin and the central “E” region of fibrin. Proc Natl Acad Sci USA. 2004;101:2718–2723. doi: 10.1073/pnas.0303440101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McRee DE. Practical Protein Crystallography. Academic; San Diego: 1999. pp. 271–328. [Google Scholar]
- 18.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system (CNS): A new software system for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 19.Spraggon G, Everse SJ, Doolittle RF. Crystal structures of fragment D from human fibrinogen and its crosslinked counterpart from fibrin. Nature. 1997;389:455–462. doi: 10.1038/38947. [DOI] [PubMed] [Google Scholar]
- 20.Brunger AT. X-PLOR. A system for X-ray crystallography and NMR. Yale University Press; New Haven, USA: 1992. [Google Scholar]
- 21.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
- 22.Yakovlev S, Makogonenko E, Kurochkina N, Nieuwenhuizen W, Ingham K, Medved L. Conversion of fibrinogen to fibrin: mechanism of exposure of tPA- and plasminogen-binding sites. Biochemistry. 2000;39:15730–15741. doi: 10.1021/bi001847a. [DOI] [PubMed] [Google Scholar]
- 23.Olexa SA, Budzynski AZ. Primary soluble plasmic degradation product of human cross-linked fibrin. Isolation and stoichiometry of the (DD)E complex. Biochemistry. 1979;18:991–995. doi: 10.1021/bi00573a009. [DOI] [PubMed] [Google Scholar]
- 24.Gorlatov S, Medved L. Interaction of fibrin(ogen) with the endothelial cell receptor VE-cadherin: mapping of the receptor-binding site in the NH2-terminal portions of the fibrin β chains. Biochemistry. 2002;41:4107–4116. doi: 10.1021/bi0160314. [DOI] [PubMed] [Google Scholar]
- 25.Schuck P, Minton AP. Kinetic analysis of biosensor data: elementary tests for self-consistency. Trends Biochem Sci. 1996;21:458–460. doi: 10.1016/s0968-0004(96)20025-8. [DOI] [PubMed] [Google Scholar]
- 26.Yang Z, Kollman JM, Pandi L, Doolittle RF. Crystal structure of native chicken fibrinogen at 2.7 Å resolution. Biochemistry. 2001;40:12515–12523. doi: 10.1021/bi011394p. [DOI] [PubMed] [Google Scholar]
- 27.Martin PD, Robertson W, Turk D, Huber R, Bode W, Edwards BF. The structure of residues 7–16 of the Aα-chain of human fibrinogen bound to bovine thrombin at 2.3-Å resolution. J Biol Chem. 1992;267:7911–7920. [PubMed] [Google Scholar]
- 28.Martin PD, Malkowski MG, DiMaio J, Konishi Y, Ni F, Edwards BF. Bovine thrombin complexed with an uncleavable analog of residues 7–19 of fibrinogen Aα: geometry of the catalytic triad and interactions of the P1′, P2′, and P3′ substrate residues. Biochemistry. 1996;35:13030–13039. doi: 10.1021/bi960656y. [DOI] [PubMed] [Google Scholar]
- 29.Malkowski MG, Martin PD, Lord ST, Edwards BF. Crystal structure of fibrinogen-Aα peptide 1–23 (F8Y) bound to bovine thrombin explains why the mutation of Phe-8 to tyrosine strongly inhibits normal cleavage at Arg-16. Biochem J. 1997;326:811–822. doi: 10.1042/bj3260815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krishnan R, Sadler JE, Tulinsky A. Structure of the Ser195Ala mutant of human α-thrombin complexed with fibrinopeptide A(7–16): evidence for residual catalytic activity. Acta Crystallogr D. 2000;56:406–410. doi: 10.1107/s0907444900001487. [DOI] [PubMed] [Google Scholar]
- 31.Pandya BV, Gabriel JL, O’Brien J, Budzynski AZ. Polymerization site in the beta chain of fibrin: mapping of the Bβ 1–55 sequence. Biochemistry. 1991;30:162–168. doi: 10.1021/bi00215a024. [DOI] [PubMed] [Google Scholar]
- 32.Brown JH, Volkmann N, Jun G, Henschen-Edman AH, Cohen C. The crystal structure of modified bovine fibrinogen. Proc Natl Acad Sci USA. 2000;97:85–90. doi: 10.1073/pnas.97.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Z, Mochalkin I, Veerapandian L, Riley M, Doolittle RF. Crystal structure of native chicken fibrinogen at 5.5-A resolution. Proc Natl Acad Sci USA. 2000;97:3907–3912. doi: 10.1073/pnas.080065697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Madrazo J, Brown JH, Litvinovich S, Dominguez R, Yakovlev S, Medved L, Cohen C. Crystal structure of the central region of bovine fibrinogen (E5 fragment) at 1.4-Å resolution. Proc Natl Acad Sci USA. 2001;98:11967–11972. doi: 10.1073/pnas.211439798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moskowitz KA, Budzynski AZ. The (DD)E complex is maintained by a composite fibrin polymerization site. Biochemistry. 1994;33:12937–12944. doi: 10.1021/bi00248a001. [DOI] [PubMed] [Google Scholar]
- 36.Donovan JW, Mihalyi E. Clotting of fibrinogen. 1. Scanning calorimetric study of the effect of calcium. Biochemistry. 1985;24:3434–3443. doi: 10.1021/bi00335a007. [DOI] [PubMed] [Google Scholar]
- 37.Donovan JW, Mihalyi E. Clotting of fibrinogen. 2. Calorimetry of the reversal of the effect of calcium on clotting with thrombin and with ancrod. Biochemistry. 1985;24:3443–3448. doi: 10.1021/bi00335a008. [DOI] [PubMed] [Google Scholar]
- 38.Medved L, Tsurupa G, Yakovlev S. Conformational changes upon conversion of fibrinogen into fibrin. The mechanisms of exposure of cryptic sites. Ann N Y Acad Sci. 2001;936:185–204. doi: 10.1111/j.1749-6632.2001.tb03505.x. [DOI] [PubMed] [Google Scholar]
- 39.Laudano AP, Cottrell BA, Doolittle RF. Synthetic peptides modeled on fibrin polymerization sites. Ann N Y Acad Sci. 1983;408:315–329. doi: 10.1111/j.1749-6632.1983.tb23254.x. [DOI] [PubMed] [Google Scholar]
- 40.Koopman J, Haverkate F, Lord ST, Grimbergen J, Mannucci PM. Molecular basis of fibrinogen Naples associated with defective thrombin binding and thrombophilia. Homozygous substitution of Bβ 68 Ala→Thr. J Clin Invest. 1992;90:238–244. doi: 10.1172/JCI115841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Privalov PL. Stability of proteins: small globular proteins. Adv Protein Chem. 1979;33:167–241. doi: 10.1016/s0065-3233(08)60460-x. [DOI] [PubMed] [Google Scholar]
- 42.Rose T, Di Cera E. Three-dimensional modeling of thrombin-fibrinogen interaction. J Biol Chem. 2002;277:18875–18880. doi: 10.1074/jbc.M110977200. [DOI] [PubMed] [Google Scholar]
- 43.Arosio D, Ayala YM, Di Cera E. Mutation of W215 compromises thrombin cleavage of fibrinogen, but not of PAR-1 or protein C. Biochemistry. 2000;39:8095–8101. doi: 10.1021/bi0006215. [DOI] [PubMed] [Google Scholar]
- 44.Mullin JL, Gorkun OV, Binnie CG, Lord ST. Recombinant fibrinogen studies reveal that thrombin specificity dictates order of fibrinopeptide release. J Biol Chem. 2000;275:25239–25246. doi: 10.1074/jbc.M004142200. [DOI] [PubMed] [Google Scholar]