

Abstract
 3-Hydroxy-4-pyrones are a class of important metal chelators with versatile medicinal applications. An efficient pathway for the preparation of new 5-amido-3-hydroxy-4-pyrone derivatives has been developed. The synthesized 5-amido-3-hydroxy-4-pyrones have been evaluated as inhibitors of matrix metalloproteinases.
3-Hydroxy-4-pyrones are a class of important metal chelators with versatile medicinal applications. An efficient pathway for the preparation of new 5-amido-3-hydroxy-4-pyrone derivatives has been developed. The synthesized 5-amido-3-hydroxy-4-pyrones have been evaluated as inhibitors of matrix metalloproteinases.
3-Hydroxy-4-pyrones (also referred to as hydroxypyrones hereafter) are an important class of biologically active compounds. They can be readily converted to analogues such as hydroxythiopyrones and hydroxypyridinones (3,4-HOPOs) (Figure 1). As charge delocalization is possible within the heterocyclic ring, the exocyclic keto group and the ortho oxyanion derived from the hydroxyl group can efficiently bind to a variety of diand trivalent metals by forming a five-membered chelate ring.1 Many reports demonstrate that hydroxypyrones and their thiopyrone and pyridinone analogues are efficient chelators for metal ions such as Fe(III),1-7 Al(III),8-11 Ga(III),3, 7, 9, 12, 13 In(III),3, 12, 13 Zn(II),14-17 Cu(II),15 Ni(II),15, 18 Pb(II),19 Ru(II),20 and [VO]2+.17, 21-25 The tight metal binding affinity of these hydroxypyrones and their analogues, as well as the high bioavailability and favorable toxicity profile suggested by maltol ((3-hydroxy-2-methyl-4-pyrones)1 and kojic acid (6-hydroxymethyl-3-hydroxyl-4-pyrone),26 has led to their exploration in medicinal inorganic chemistry. Potential medicinal applications reported in the literature include iron imbalance in anemia and iron overload disorder,4-6 aluminium removal in Alzheimer's disease,8-11 treatment of diabetes,21, 23-25 contrast agents for medical imaging,27 and regulation of metalloenzyme activity.28-30 Previous hydroxypyrone derivatives related to such investigations were synthesized by structural modification of commercially available maltol and kojic acid (Figure 1),1, 26 which are natural products and can be manufactured by biosynthetic methods from glucose. Because of the versatile coordination chemistry and potential chemotherapeutic application of hydroxypyrones and their thiopyrone and pyridinone congeners, development of new synthetic strategies to access diverse hydroxypyrone derivatives is highly desirable.
Figure 1.
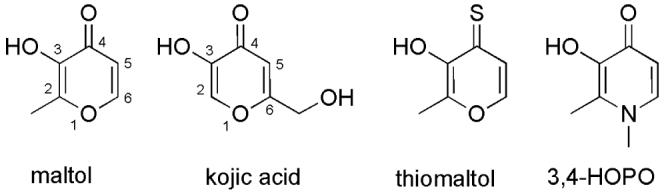
Examples of hydroxypyrone, hydroxythiopyrone, and hydroxypyridinone chelators
Our laboratory has a particular interest in the development of hydroxypyrone-based matrix metalloproteinase (MMP) inhibitors.29, 30 MMPs are a class of hydrolytic zinc-dependent enzymes catalyzing peptide bond hydrolysis. They are involved in tissue remodeling, wound healing, and growth.31 Misregulated activity of these enzymes is also implicated in a variety of diseases such as cancer, arthritis, atherosclerosis, and heart diseases.32-34 Thus, development of inhibitors for regulation of MMP activity has great therapeutic value.32-34 We have found that maltol and thiomaltol are more effective chelating inhibitors for MMP-3 (stromelysin) than the widely reported hydroxamate ligands.29, 35 Furthermore, significant improvement of inhibition potency and selectivity of such pyrone-based chelators can be achieved by introduction of a peptidomimetic backbone on the pyrone ring, which leads to enhanced interactions between the MMP subsites and the inhibitor.30 However, structural modification of maltol and kojic acid only provides access to a group of 2-, and 6-amido substituted hydroxypyrones (Scheme 1). No routes to manipulating maltol or kojic acid in the remaining 5-position have been reported in the literature.1, 5, 30, 36 As diverse substitution patterns on the pyrone ring may modulate the potency, selectivity, binding mode, and biocompatibility of the inhibitors, our interest in developing pyrone-based MMP inhibitors prompted us to synthesize 5-amido hydroxypyrones to access new potential MMP inhibitors. Analysis of the impact of substituent patterns on inhibitor activity should provide structure-activity information and guidance for further structural optimization of our inhibitor design. Herein we report the synthesis of 5-amido-3-hydroxy-4-pyrones and their activity as MMP inhibitors.
Scheme 1.
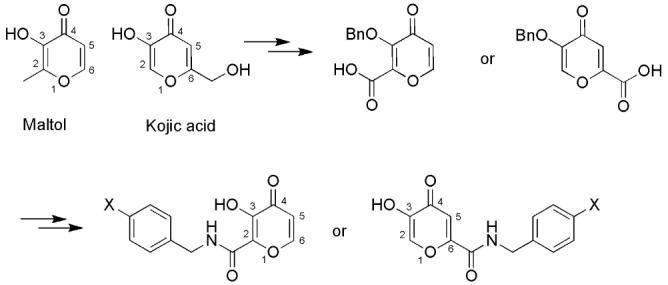
Synthetic Path for Pyrone-Based Inhibitors from Maltol and Kojic Acid
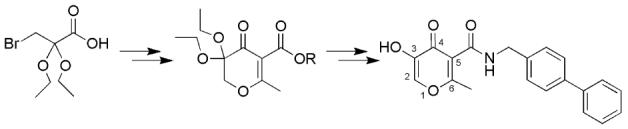
The preparation of 5-amido-3-hydroxy-4-pyrones started with commercially available 3-bromopyruvic acid 1 (Scheme 2).37 Reaction of 1 with triethyl orthoformate in the presence of concentrated H2SO4 as a catalyst provided 3-bromo-2,2-diethoxypropionic acid 2 in 85-90 % yield. The carboxylic acid intermediate 2 was then transformed to the activated ester 4-nitrophenyl 3-bromo 2,2-diethoxypropionate 3 in 60-65% yield by treatment with 4-nitrophenyl trifluoroacetate in the presence of pyridine. 5-Substituted 3,3-diethoxypyran-4-ones 4 a and 4 b were obtained by refluxing intermediate 3 with a β-keto ester anion in NaH/THF. The yields for this cyclization were in the range of 64–76%. Two 3,3-diethoxypyran-4-ones 4a (R = Et) and 4b (R = But) were prepared as they provide different choices for the subsequent hydrolysis conditions.
Scheme 2.
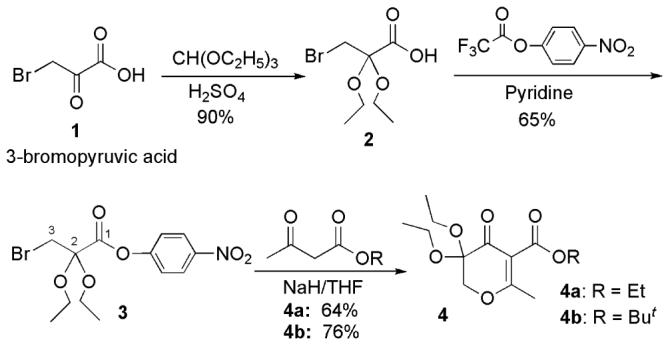
Synthesis of 5-Substituted 3,3-Diethoxypyran-4-ones
The mechanism for the formation of 3,3-diethoxypyran-4-ones 4 is rationalized in Scheme 3. Ketalization of the 2-keto group by two ethoxyl groups results in decreased nucleophilicity and a steric increase on the 3-bromo carbon of intermediate 3. Thus, the intermolecular nucleophilic reaction between compound 3 and the β-keto ester anion first takes place at the activated ester carbonyl group forming bromo intermediate I. In the presence of excess base, the α-carbon on intermediate I is further deprotonated leading to the formation of intermediate II. An intramolecular nucleophilic reaction at the bromo carbon is favorable under refluxing conditions in THF via a sixmembered intermediate III which results in the formation of 3,3-diethoxypyran-4-one 4.38
Scheme 3.
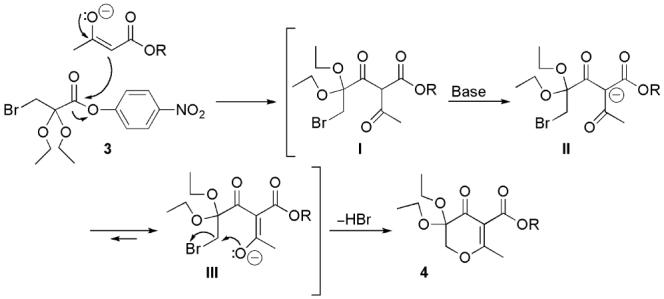
Formation of 3,3-Diethoxypyran-4-one
The deprotection of the ketal group in 4 is straightforward by treatment with formic acid or trifluoroacetic acid (TFA) in the presence of moisture (Scheme 4). For example, refluxing of 4a in THF with formic acid provided hydroxypyrone ester 5 in 67 % yield. The 5-ester and the 6-methyl groups of compound 5 provide synthetic handles for further functional extension at these positions. When compared with maltol and kojic acid (Scheme 1), 5 should be a versatile synthon for the preparation of pyrone derivatives with unprecedented substitution patterns.
Scheme 4.
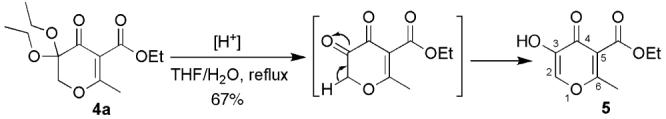
Deprotection of 3,3-Diethoxypyran-4-one
Our interest in the development of 5-amido-3-hydroxy-4-pyrone MMP inhibitors encouraged us to investigate cyclization reactions of activated bromo ester 3 with several β-keto amides 6a-c in an attempt to obtain the corresponding 5-amido 3,3-diethoxypyran-4-ones 7a-c (Scheme 5). Interestingly, the cyclization reaction did not proceed well compared to that the β-keto ester substrates (Scheme 2). It was observed that the amide substrates 6a-6c formed insoluble species under NaH/THF conditions. While reaction of N-methyl β-keto amide 6a provided the expected 5-amido dihydropyrone 7a in low yield (24%), the reactions of 3 with β-keto amide 6b and 6c only resulted in recovery of starting materials and unidentified compounds without expected product 7b and 7c.
Scheme 5.
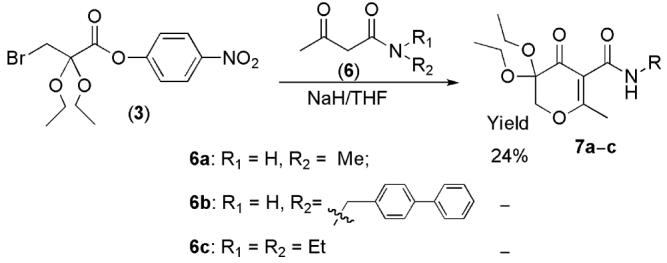
Cyclization Reactions with β-Keto Amides
Further investigations generated an improved approach for the synthesis of 5-amido-3-hydroxy-4-pyrones as shown in Scheme 6. Hydrolysis of the ester group of 4 provided carboxylic acid 8 as a versatile intermediate for the preparation of an amide. The yields for the hydrolysis of ethyl ester 4a under NaOH/H2O conditions varied substantially in the range of 30–60%. This outcome may have been due to the sensitivity of the ketal group and the α,β-unsaturated moiety of 4a under basic reaction conditions and acidic aqueous work-up. In contrast, deprotection of tert-butyl ester 4b by TFA was very efficient and selective providing carboxylic acid 8 in 95% yield. Note that the ketal protective group on the pyrone ring can be deprotected by TFA in refluxing THF/H2O, but the deprotection of tert-butyl ester by TFA in CH2Cl2 at room temperature was selective, keeping the ketal group intact. The amide backbone is introduced by coupling the carboxylic acid intermediate 8 with the corresponding amine, forming amide 7 in 63–82% yields. 5-Amido-3-hydroxy-4-pyrone 9 was obtained in 68–82% yield by deprotection of the ketal group by refluxing compound 7 in THF in the presence of formic acid or TFA. A thiopyrone analogue 10 was also prepared by thionation of the corresponding pyrone 9b with P4S10 in refluxing THF in the presence of HMDO (Scheme 7).39 Several modified conditions were pursued for this reaction, including the use of different solvents (CH2Cl2, benzene, dioxane) with or without HMDO , and alternative reagents (Lawesson's reagent); however, none of these adjustments improved the reaction yield. The low yield (16%) of this transformation may be due to a steric effect at the ketone position caused by the neighboring 3-hydroxy and 5-amido groups.18
Scheme 6.
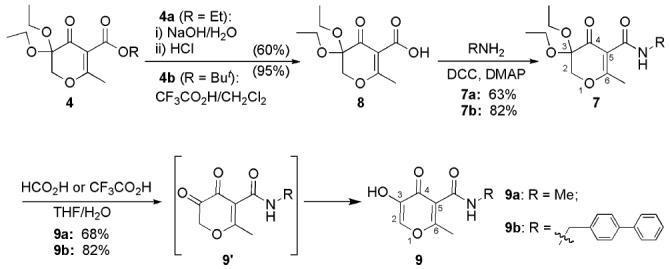
Synthesis of Compounds 9a,b
Scheme 7.
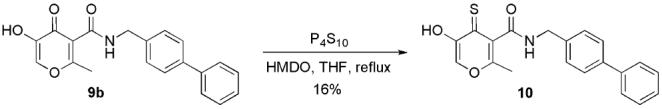
Synthesis of Inhibitor 10
The inhibitory activity of biphenyl amides 9b and 10 against MMP-1 (collagenase), MMP-2 (gelatinase A), MMP-3 (stromelysin), and MMP-9 (gelatinase B) was examined using an established fluorescence-based assay (Table 1).40 It was found that both compounds showed relatively poor inhibition against the MMPs evaluated. For hydroxypyrone 9b at 100 μM concentration, MMP activity was inhibited 21-34%. The hydroxythiopyrone 10 showed more potent inhibition than 9b. The percent activity of MMP-1, MMP-3, and MMP-9 was inhibited by 52%, 45%, and 54%, respectively, in the presence of 50 μM of 10. Furthermore, MMP-2 activity was inhibited 94 % by compound 10 at a concentration of 50 μM.
Table 1.
Inhibitory Activity of Compounds 9b and 10
Percent inhibition at 100 μM
Percent inhibition at 50 μM
IC50 = 23±2 μM
Compared with the activity of the previously reported regioisomer AM-2,30 compound 9b was several orders of magnitude less potent as an MMP inhibitor. This outcome clearly shows that the substitution pattern on the pyrone ring dramatically affects the activity of hydroxypyrone inhibitors. This suggests that the zinc-binding group and not the hydrophobic ‘backbone’ substituent are dictating the conformation of inhibitor binding in the MMP active site. This hypothesis is further supported by the lack of selectivity observed for compound 9b . AM-2 strongly inhibits MMP-2 and MMP-3, but not MMP-1; this is likely due to the large biphenyl substituent that can be accomodated in the deep S1′ pockets of MMP-2 and MMP-3, but not in the shallow S1′ pocket of MMP-1.30 In contrast, 9b shows no notable selectivity between these three enzymes, indicative of the biphenyl group no longer being directed toward the S1′ pocket. Again, this indicates that the zinc-binding group, and not the biphenyl substituent, is the dominant element dictating the mode of binding. Lastly, the significant improvement in inhibition potency of compound 10 over 9b by simply changing the chelator from an O,O to an O,S ligand is consistent with our earlier studies29 and demonstrates the substantial impact of the zinc-binding group in inhibitor activity.
In summary, the first pathway for the synthesis of 5-amido-3-hydroxy-4-pyrones has been developed. These pyrone derivatives provide new opportunities to explore the metallopharmaceutical applications of these chelators. We are currently exploring the coordination chemistry of these ligands for a variety of bioinorganic applications. These investigations, in combination with the MMP inhibition data from pyrones 9b and 10 , will provide helpful guidelines for future structural optimization of pyrone-based MMP inhibitors.
Supplementary Material
Acknowledgement
Y.-L.Y. thanks Arpita Agrawal (UCSD) and Dr. Jana A. Lewis (UCSD) for helpful discussions on the MMP activity assays. This material is based upon work supported in part by the NIH (R01 HL080049-01) and the American Heart Association (0430009N). Seth M. Cohen is a Cottrell Scholar of the Research Corporation.
Footnotes
Supporting Information Available: Synthetic procedures, characterization, and 1H NMR, 13C NMR spectra, and high-resolution mass spectra (HRMS) of all new compounds; MMP assay experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Thompson KH, Barta CA, Orvig C. Chem. Soc. Rev. 2006;35:545–556. doi: 10.1039/b416256k. [DOI] [PubMed] [Google Scholar]
- 2.Ahmet MT, Frampton CS, Silver J. J. Chem. Soc. Dalton Trans. 1988:1159. [Google Scholar]
- 3.Ellis BL, Duhme AK, Hider RC, Hossain MB, Rizvi S, van der Helm D. J. Med. Chem. 1996;39:3659–3670. doi: 10.1021/jm960220g. [DOI] [PubMed] [Google Scholar]
- 4.Liu ZD, Hider RC. Med. Res. Rev. 2002;22:26–64. doi: 10.1002/med.1027. [DOI] [PubMed] [Google Scholar]
- 5.Liu ZD, Piyamongkol S, Liu DY, Khodr HH, Lu SL, Hider RC. Bioorg. Med. Chem. 2001;9:563–573. doi: 10.1016/s0968-0896(00)00273-x. [DOI] [PubMed] [Google Scholar]
- 6.Maxton DG, Thompson RPH, Hider RC. Brit. J. Nutr. 1994;71:203–207. doi: 10.1079/bjn19940127. [DOI] [PubMed] [Google Scholar]
- 7.Santos MA, Gil M, Marques S, Gano L, Cantinho G, Chaves S. J. Inorg. Biochem. 2002;92:43–54. doi: 10.1016/s0162-0134(02)00483-x. [DOI] [PubMed] [Google Scholar]
- 8.Finnegan MM, Lutz TG, Nelson WO, Smith A, Orvig C. Inorg. Chem. 1987;26:2171–2176. [Google Scholar]
- 9.Finnegan MM, Rettig SJ, Orvig C. J. Am. Chem. Soc. 1986;108:5033–5035. [Google Scholar]
- 10.Santos MA. Coord. Chem. Rev. 2002;228:187–203. [Google Scholar]
- 11.Yokel RA. Coord. Chem. Rev. 2002;228:97–113. [Google Scholar]
- 12.Green DE, Ferreira CL, Stick RV, Patrick BO, Adam MJ, Orvig C. Bioconjugate Chem. 2005;16:1597–1609. doi: 10.1021/bc0501808. [DOI] [PubMed] [Google Scholar]
- 13.Monga V, Patrick BO, Orvig C. Inorg. Chem. 2005;44:2666–2677. doi: 10.1021/ic048693y. [DOI] [PubMed] [Google Scholar]
- 14.Emami S, Hosseinimehr SJ, Taghdisi SM, Akhlaghpoor S. Bioorg. Med. Chem. Lett. 2007;17:45–48. doi: 10.1016/j.bmcl.2006.09.097. [DOI] [PubMed] [Google Scholar]
- 15.Lewis JA, Tran BL, Puerta DT, Rumberger EM, Hendrickson DN, Cohen SM. Dalton Trans. 2005:2588–2596. doi: 10.1039/b505034k. [DOI] [PubMed] [Google Scholar]
- 16.Puerta DT, Cohen SM. Inorg. Chem. 2003;42:3423–3430. doi: 10.1021/ic026029g. [DOI] [PubMed] [Google Scholar]
- 17.Sakurai H, Kojima Y, Yoshikawa Y, Kawabe K, Yasui H. Coord. Chem. Rev. 2002;226:187–198. [Google Scholar]
- 18.Lewis JA, Puerta DT, Cohen SM. Inorg. Chem. 2003;42:7455. doi: 10.1021/ic0347135. [DOI] [PubMed] [Google Scholar]
- 19.Lewis JA, Cohen SM. Inorg. Chem. 2004;43:6534–6536. doi: 10.1021/ic0493696. [DOI] [PubMed] [Google Scholar]
- 20.Kennedy DC, Wu A, Patrick BO, James BR. Inorg. Chem. 2005;44:6529–6535. doi: 10.1021/ic050034d. [DOI] [PubMed] [Google Scholar]
- 21.McNeill JH, Yuen VG, Hoveyda HR, Orvig C. J. Med. Chem. 1992;35:1489–1491. doi: 10.1021/jm00086a020. [DOI] [PubMed] [Google Scholar]
- 22.Rangel M, Leite A, Amorim MJ, Garribba E, Micera G, Lodyga-Chruscinska E. Inorg. Chem. 2006;45:8086–8097. doi: 10.1021/ic0605571. [DOI] [PubMed] [Google Scholar]
- 23.Saatchi K, Thompson KH, Patrick BO, Pink M, Yuen VG. Inorg. Chem. 2005;44:2689–2697. doi: 10.1021/ic048186g. [DOI] [PubMed] [Google Scholar]
- 24.Song B, Saatchi K, Rawji GH, Orvig C. Inorg. Chim. Acta. 2002;339:393–399. [Google Scholar]
- 25.Thompson KH, Liboiron BD, Sun Y, Bellman KDD, Setyawati IA, Patrick BO, Karunaratne V, Rawji G, Wheeler J, Sutton K, Bhanot S, Cassidy C, McNeill JH, Yuen VG, Orvig C. J. Biol. Inorg. Chem. 2003;8:66–74. doi: 10.1007/s00775-002-0388-5. [DOI] [PubMed] [Google Scholar]
- 26.Bentley R. Nat. Prod. Rep. 2006;23:1046–1062. doi: 10.1039/b603758p. [DOI] [PubMed] [Google Scholar]
- 27.Puerta DT, Botta M, Jocher CJ, Werner EJ, Avedano S, Raymond KN, Cohen SM. J. Am. Chem. Soc. 2006;128:2222–2223. doi: 10.1021/ja057954f. [DOI] [PubMed] [Google Scholar]
- 28.Lewis JA, Mongan J, McCammon JA, Cohen SM. ChemMedChem. 2006;1:1–4. doi: 10.1002/cmdc.200600102. [DOI] [PubMed] [Google Scholar]
- 29.Puerta DT, Lewis JA, Cohen SM. J. Am. Chem. Soc. 2004;126:8388–8389. doi: 10.1021/ja0485513. [DOI] [PubMed] [Google Scholar]
- 30.Puerta DT, Mongan J, Tran BL, McCammon JA, Cohen SM. J. Am. Chem. Soc. 2005;127:14148–14149. doi: 10.1021/ja054558o. [DOI] [PubMed] [Google Scholar]
- 31.Page-McCaw A, Ewald AJ, Werb Z. Nat. Rev. Mol. Cell Biol. 2007;8:221–233. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Puerta DT, Cohen SM. Curr. Top. Med. Chem. 2004;4:1551–1573. doi: 10.2174/1568026043387368. [DOI] [PubMed] [Google Scholar]
- 33.Skiles JW, Gonnella NC, Jeng AY. Curr. Med. Chem. 2004;11:2911–2977. doi: 10.2174/0929867043364018. [DOI] [PubMed] [Google Scholar]
- 34.Whittaker M, Floyd CD, Brown P, Gearing AJH. Chem. Rev. 1999;99:2735–2776. doi: 10.1021/cr0100345. [DOI] [PubMed] [Google Scholar]
- 35.Puerta DT, Griffin MO, Lewis JA, Romero-Perez D, Garcia R, Villarreal FJ, Cohen SM. J. Biol. Inorg. Chem. 2006;11:131–138. doi: 10.1007/s00775-005-0053-x. [DOI] [PubMed] [Google Scholar]
- 36.Puerta DT. A Bioinorganic Approach to Matrix Metalloproteinse Inhibition. University of California; San Diego, San Diego: 2006. Ph.D. Thesis. [Google Scholar]
- 37.LaMattina JL, Mularski CJ. Tetrahedron Lett. 1983;24:2059–2062. [Google Scholar]
- 38.Sato K, Inoue S, Ohashi M. Bull. Chem. Soc. Japan. 1973;46:1288–1290. [Google Scholar]
- 39.Curphey TJ. J. Org. Chem. 2002;67:6461–6473. doi: 10.1021/jo0256742. [DOI] [PubMed] [Google Scholar]
- 40.Knight CG, Willenbrock F, Murphy G. FEBS. 1992;296:263–266. doi: 10.1016/0014-5793(92)80300-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.