

Abstract
Calcium (Ca2+) is a highly versatile second messenger that controls vascular smooth muscle cell (VSMC) contraction, proliferation, and migration. By means of Ca2+ permeable channels, Ca2+ pumps and channels conducting other ions such as potassium and chloride, VSMC keep intracellular Ca2+ levels under tight control. In healthy quiescent contractile VSMC, two important components of the Ca2+ signaling pathways that regulate VSMC contraction are the plasma membrane voltage-operated Ca2+ channel of the high voltage-activated type (L-type) and the sarcoplasmic reticulum Ca2+ release channel, Ryanodine Receptor (RyR). Injury to the vessel wall is accompanied by VSMC phenotype switch from a contractile quiescent to a proliferative motile phenotype (synthetic phenotype) and by alteration of many components of VSMC Ca2+ signaling pathways. Specifically, this switch that culminates in a VSMC phenotype reminiscent of a non-excitable cell is characterized by loss of L-type channels expression and increased expression of the low voltage-activated (T-type) Ca2+ channels and the canonical transient receptor potential (TRPC) channels. The expression levels of intracellular Ca2+ release channels, pumps and Ca2+-activated proteins are also altered: the proliferative VSMC lose the RyR3 and the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase isoform 2a pump and reciprocally regulate isoforms of the ca2+/calmodulin-dependent protein kinase II. This review focuses on the changes in expression of Ca2+ signaling proteins associated with VSMC proliferation both in vitro and in vivo. The physiological implications of the altered expression of these Ca2+ signaling molecules, their contribution to VSMC dysfunction during vascular disease and their potential as targets for drug therapy will be discussed.
Keywords: Vascular smooth muscle cell proliferation, Phenotypic switching, Ca2+ signaling, Canonical transient receptor potential channels, L-type and T-type voltage-dependent Ca2+ channels, Membrane potential, Potassium channels, Ca2+/calmodulin-dependent protein kinase II (CaMKII)
Introduction
Vascular smooth muscle cells (VSMC) are one of the major cell types of the blood vessel wall and are instrumental in the maintenance of blood vessel integrity and the control of vascular tone [157]. Calcium (Ca2+) signaling plays a central role in VSMC cell function. Ca2+ can originate from two distinct sources, entry from the extracellular milieu through plasma membrane channels or release from internal stores, namely the sarcoplasmic reticulum (SR). It is widely accepted that a global rise in cytoplasmic Ca2+ concentration leads to VSMC contraction [155]. The contractile characteristics of VSMC are quite different from those of cardiac myocytes. Unlike contractions of cardiac myocytes which are rapid and of relatively short duration, VSMC are characterized by relatively slow, in some cases sustained and tonic contractions. In vivo, VSMC contractility is subject to influences from several neuronal, humoral, and endothelial factors; they are partially constricted with resting Ca2+ concentrations around 100–300 nM, several orders of magnitude lower than the extracellular space [147].
Excitation–contraction coupling
VSMC membrane potential is critical for the control of cytoplasmic Ca2+ and therefore the maintenance of the vascular tone. Depolarization of VSMC plasma membrane activates the L-type high voltage-gated Ca2+ channels (Cav1.2) at the plasma membrane leading to an increased Ca2+ entry [30]. This global rise in intracellular Ca2+ leads to the initiation of the contractile response through activation of Ca2+/calmodulin-dependent myosin light chain kinase (MLCK). In turn, Ca2+, through the process of Ca2+-induced Ca2+ release (CICR) [117], activates the Ryanodine receptor (RyR) leading to Ca2+ release from the SR [152]. Unlike cardiac muscle, where the Ca2+ entry is further amplified through CICR, this process seems to be locally restricted to few subplasmalemmal Ca2+ sparks sites in VSMC [68]. Paradoxically, Ca2+ sparks via RyR in VSMC negatively regulate contraction through activation the large conductance Ca2+-activated potassium (K2+) channels (KCa1.1; also known as BKCa) leading to outward K+ currents, subsequent hyperpolarization, decrease Ca2+ entry through L-type Ca2+ channels and VSMC relaxation [95]. Thus, the physiological effects (vasoconstriction versus vasorelaxation) of Ca2+ in VSMC are determined by the spatial distribution and the magnitude (global versus local) of the Ca2+ signal [55, 95, 152]. Reduced global Ca2+ levels and subsequent VSMC relaxation might also be achieved through extrusion or uptake of cytoplasmic Ca2+ by the plasma membrane Ca2+-ATPase (PMCA) and the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) pumps, respectively.
PLC-coupled receptors and receptor-operated channels
In addition, the contractility of VSMC in vivo is under the control of a plethora of vasoactive agonists (e.g., angiotensin II, vasopressin, endothelin) that exert their function through binding to membrane receptors that usually couple to phospholipase C (PLC) isoforms to stimulate Ca2+ release through the inositol-1,4,5-triphosphate (IP3) receptor (IP3R) [15] and concomitant Ca2+ entry via store-operated (SOC) [104, 113] and receptor-operated Ca2+ (ROC) [10, 19, 104] channels1. These channels conduct ions according to their electrochemical gradient across the plasma membrane, are operational within resting membrane potential (approximately −60 mV), and are inhibited by membrane depolarization. The exact contribution of PLC-coupled receptor activation to global Ca2+ levels during EC-coupling is still a matter of debate. However, one can imagine a scenario where discrete Ca2+ entry through ROC channels—or for that matter the generation of lipid second messengers that follow PLC-coupled receptor ligation—could affect the activation of chloride (Cl−) and/or K+ channels to alter the membrane potential and induce the activation of L-type Ca2+ channels. It has long been proposed that changes in membrane potential may occur as a result of influx of ions through ROC channels [130]. For example, activation of the non-selective receptor-activated canonical transient receptor potential 6 (TRPC6) channel (discussed in detail later in this review) in VSMC was suggested to contribute to membrane depolarization and subsequent L-type Ca2+ channel activation [128, 153]. Although nonselective cation channels such as TRPC6 conduct large sodium (Na+) currents and thus can potentially depolarize the plasma membrane, the exact mechanisms by which vasoactive factors induce depolarization remain poorly understood. Even more confusing is the suggestion that a localized Ca2+ signal can couple to either contraction or relaxation and that this coupling is, to a large extent, determined by the specific ion pumps and channels contained within the plasma membrane–SR junctional complexes [77]. For example, the Ca2+ release through the IP3R could conceptually, depending on the spatial properties of the Ca2+ signal, either cause VSMC contraction or relaxation; VSMC contraction would occur by a global IP3-mediated Ca2+ rise while spatially restricted IP3-mediated Ca2+ signals that are functionally equivalent to RyR-generated local subplasmalemmal Ca2+ sparks would cause relaxation. Studies by Bayguinov et al. have suggested such a differential regulation of VSMC contractility by PLC-coupled agonists [11, 12]. These authors showed that IP3-mediated Ca2+ signals are not only cell type dependent but that within VSMC from the same vascular bed different PLC-coupled agonists can cause either contraction or relaxation depending on their differential regulation of membrane potential through Ca2+-dependent activation of K+ channels [11, 12]. For a comprehensive review on spatially localized elementary Ca2+ signals, oscillations, and gradients and how they shape the specificity of Ca2+-mediated responses in VSMC, the reader is referred to these review articles [16, 77].
Evidently, Ca2+ dynamics during the excitation–contraction coupling (EC coupling) in VSMC are more complex with spatially and temporally coordinated events orchestrated by additional players, including but not limited to, mechano-sensitive Ca2+ entry channels, the sodium/calcium exchanger (NCX), K+ and Cl− channels. Furthermore, the impact of these Ca2+ signals extends beyond the short-term EC coupling to long-term effects through activation of transcription factors to induce profound changes in gene expression patterns, including those of contractile proteins and Ca2+ handling proteins themselves. A thorough discussion of this excitation–transcription coupling (ET coupling) is beyond the scope of this paper, and the reader is referred to the recent review articles by Owens, Lounsbury and their colleagues [9, 147].
Smooth muscle phenotypic switching
Smooth muscle provides not only structural integrity for the vessel wall but also precise regulation of vascular tone and blood pressure [101, 102]. A striking feature of smooth muscle is the considerable cellular heterogeneity apparent within the media of the vessels. The presence of phenotypically distinct VSMC populations within the vessel wall is generally hypothesized to be important in the plasticity characteristic of vascular smooth muscle [90, 101, 102]. In response to various environmental stimuli, including growth factors, cytokines, mechanical influences, and various inflammatory mediators, the resident quiescent VSMC undergo transcriptional changes affecting both the downregulation of contractile proteins and concurrent upregulation of proteins supporting a proliferative phenotype [70, 102, 147]. This plasticity is believed to have evolved as a mechanism of vascular repair during injury and/or vascular adaptation to increasing demands by enabling VSMC to “switch” to a non-contractile, proliferative and migratory phenotype. This phenotypic switch is under tight control and is often reversible but under certain circumstances can largely contribute to vascular disease states such as atherosclerosis, hypertensive microvessels, vein graft failure, and restenosis following percutaneous intervention [31, 123].
VSMC phenotypic switching during vascular disease is complex and most likely is the result of loss of humoral and endothelial factors or stimuli that sustain the differentiated state combined with acquisition of pro-proliferative and promigratory signaling pathways such as those mediated by growth factors and cytokines. Most studies of VSMC phenotypic switching have focused on identification of factors that promote this process (e.g., growths factors and cytokines, oxidative stress, mechanical stress, endothelial factors) or on transcriptional regulation of phenotype-specific genes [70, 101, 102]. These studies have mainly focused on the loss of contractile proteins with very few reports devoted to the acquisition of pro-proliferative or pro-migratory proteins such as ion channels and pumps [14, 32, 74].
It is becoming increasingly apparent that a Ca2+ signaling pathway is responsible for transcriptionally regulating its own components whereby Ca2+ entry via a specific Ca2+ channel activates gene transcription and increased expression at the plasma membrane of that channel protein, as demonstrated for TRPC6 [75]. In a similar fashion, a general model has emerged in VSMC whereby Ca2+ signals controlled by large conductance K+ channels KCa1.1, voltage-gated L-type Ca2+ channels and RyR, which are down-regulated in synthetic VSMC, couple to transcription of differentiated contractile protein markers [147]; and conversely, as will be discussed later in this review, signals resulting from activation of intermediate conductance Ca2+-activated K+ channels (KCa3.1) and TRPC channels, which are upregulated in synthetic VSMC, might couple to transcription of pro-proliferative protein markers. This review will focus on the changes in Ca2+ signaling proteins during VSMC phenotypic switch, including studies describing reciprocal regulation of CaMKII isoforms in proliferating VSMC and during vascular injury. The contributions of these changes to VSMC proliferation and the therapeutic potential of targeting components of Ca2+ signaling in the treatment of vascular disease will be discussed.
Loss of L-type and gain of T-type voltage gated Ca2+ channels
As stated earlier, L-type Ca2+ channels are important effectors in VSMC EC coupling as well as ET coupling [9, 147]. L-type Ca2+ channels are characterized by high voltage-activated currents, large single-channel conductance and slow voltage-dependent inactivation [30]. L-type Ca2+ channels are subject to regulation by protein phosphorylation through second messenger-activated kinase pathways [30]. L-type Ca2+ channels are highly expressed in differentiated contractile or growth arrested smooth muscle cells where their presence correlates with the expression of smooth-muscle specific genes such as smooth muscle α-actin (SMα-actin) and smooth muscle myosin heavy chain (SM-MHC) [48]. Studies showed that Ca2+ entry through L-type Ca2+ channels are associated with activation of expression of the smooth muscle differentiation marker genes SMα-actin, SM-MHC and myocardin [147]. The pathways controlling the activation of these differentiation markers are dependent on the RhoA-Rho kinase (ROK) cascade [146], providing a novel signaling cascade in which physiological signals that couple excitation to contraction also contribute to transcriptional regulation of VSMC differentiation marker genes. The function of the T-type Ca2+ channel (named for their transient currents) has not been fully established, though several studies suggested their involvement in potentiating smooth muscle cell proliferation [34, 118, 119].
It has been demonstrated that the number of functional L-type Ca2+ channels was significantly decreased in dedifferentiated VSMC and increased upon differentiation with retinoic acid [48]. These authors found that the expression of the L-type Ca2+ channel α1 subunit directly correlates with the expression of SMα-actin and SM-MHC [48]. Subsequent studies provided evidence for the downregulation of the L-type Ca2+ channels during VSMC proliferation and in neointimal VSMC induced after balloon injury in rat aorta [63, 114]. However, the expression of T-type Ca2+ channels was shown to increase in the VSMC synthetic phenotype [73, 141], suggesting a pro-proliferative function of T-type Ca2+ channels in VSMC. What would be the functional consequence in VSMC for replacing the L-type voltage activated Ca2+ channel by another voltage activated Ca2+ channel, the T-type? Although increased expression of T-type Ca2+ channels, which are activated by depolarization, in a proliferating cell that has lost its membrane excitability and its contractile proteins may seem paradoxical, it is worth mentioning that by comparison with L-type Ca2+ channels (threshold of activation −40 mV; full activation at 0 mV), T-type Ca2+ channels are characterized by activation at much more negative membrane potentials (20 to 40 mV more negative than the L-type) [91], rapid inactivation and small single-channel conductance [34]. Of special interest, the low voltage activation of T-type channels is particularly relevant for the synthetic VSMC, reminiscent of non-excitable cells, characterized by more hyperpolarized resting membrane potentials, at which T-type Ca2+ channels might preferentially operate. Indeed, electrophysiological recordings have determined that the resting membrane potential of VSMC in culture ranges from −45 to −70 mV, while membrane potentials of VSMC measured in vivo were −40 to −55 mV [6, 49, 91].
A different kind of switch, this time occurring within the L-type Ca2+ channel family, have been described in atherosclerotic plaques, where decreased expression of the Cav1.2α1 transcript is accompanied by the appearance of a splice variant of the same channel where exon-21 is replaced with exon-22 [133]. Ca2+ currents through the exon-22 isoform recover from inactivation significantly faster than other isoforms, suggesting increased Ca2+ current densities. Interestingly, a 15-mV shift of the exon-22 channel activation curve to more negative potentials was observed which might contribute to proliferation by the increase of Ca2+ entry in VSMC at more hyperpolarized membrane potentials. In fact, Tiwari et al. [133] showed that inhibition of cell proliferation by serum deprivation in confluent VSMC monolayers completely eliminated the exon-22 isoform of the Cav1.2α1 transcript, which was reversible upon stimulation of proliferation by addition of serum to nonconfluent cells.
Clearly, the role of L-type Ca2+ channels in vascular disease is complex, and their decreased expression during vascular injury is not common to all vascular diseases; this highlights the need of caution in extrapolating data from one vascular disease model to another. For instance, L-type Ca2+ channels are upregulated in mesenteric and skeletal arteries of spontaneously hypertensive rats where they seem to contribute to increased vascular tone [110]. By contrast, hypercholesterolemia-induced atherosclerosis is characterized by inhibition of activity (without change in expression) of L-type Ca2+ currents in the coronary circulation [24].
The idea that hyperpolarization-activated Ca2+ entry plays a role in VSMC proliferation while depolarization-activated Ca2+ channels are involved in VSMC EC-coupling is compatible with the increased expression, in the VSMC synthetic proliferative phenotype, of Ca2+ channels activated at more negative membrane potentials. This concept is supported further by the increased expression in the VSMC synthetic phenotype of TRPC channels that optimally operate at negative membrane potentials.
Increased expression of TRPC cation channels
TRPC channels are one of the major TRP families of nonselective cation channels and termed “canonical” because they are structurally the closest to the founding family member, Drosophila TRP [98, 138, 143]. The seven TRPCs share in common the property of activation through PLC-coupled receptors and were therefore proposed as components of native SOC and ROC channels in different cell types [138, 140, 143]. The TRPC family can be divided into four subfamilies: TRPC1, TRPC2, TRPC3/6/7, and TRPC4/5 (or, TRPC1 can be included with TRPC4 and -5). TRPC2 is a pseudogene in humans but encodes a functional channel in most other mammals.
The discovery of TRPC channels in the vasculature led to the hypothesis that TRPC channels were the long-sought after vascular version of SOC channels [53]. For example, all of the seven family members (TRPC1-7) are expressed in the vasculature [64, 97] and studies examining the function of TRPC1 in VSMC suggest they are linked to Ca2+ entry associated with intracellular Ca2+ store depletion [7, 13]. In no case has an expressed TRPC (which are nonselective) served to recapitulate the electrophysiological behavior of the archetypical Ca2+ selective SOC channel, CRAC found in hematopoietic cells (lymphocytes, mast cells) [104]. However, SOC current measurements in VSMC have been shown to be non selective and to present distinct electrophysiological features than those of CRAC [20]. Thus, it is conceivable that TRPC channels do function physiologically as SOC in VSMC, where they are more likely to form subunits of less Ca2+ selective SOC.
Strong evidence supports the notion that ectopically expressed TRPC channels are primarily regulated by receptor-operated mechanisms, as in most cases (but not all, TRPC1 being a notorious exception) TRPC members are activated subsequent to stimulation of receptors that activate different isoforms of PLC, in a manner independent of store depletion [138]. Under these expression conditions, it is believed that TRPC channels predominantly form homotetramers and it is clearly established that under these conditions TRPC family members 3, 6, and 7 are activated by DAG in a PKC-independent manner [56, 99, 139], while family members 1, 4, 5 are completely unresponsive to DAG activation and their exact mechanisms of activation remain elusive. It is worth mentioning that the idea that TRPC channels operate as SOC or ROC does not have to be mutually exclusive as TRPC members may form heteromultimeric channels in native tissues [57], and different subunit stoichiometry may confer the activities characteristic of either a SOC or ROC [112]. Furthermore, recent studies have reported protein–protein interactions between TRPC channels and the newly discovered Stim1 and Orai1 proteins [80, 100], which were clearly demonstrated to be respectively the ER calcium sensor and the pore forming unit of the archetypical Ca2+ selective SOC channel, CRAC [42, 81, 121, 145, 164, 165]. A role for TRPC involvement in different vascular diseases arises from extensive cell culture and whole animal studies [97]. The most commonly expressed TRPC isoforms in the vasculature are TRPC1, 4, and 6. TRPC 3, 5, and 7 isoforms have also been detected, though to a lesser extent and less frequently [64]. Only low levels of TRPC2 expression have been detected in the vasculature, and its function at this time remains unknown [25].
As stated earlier, TRPC channels (and ROC channels in general) are activated downstream of PLC, allow Ca2+ into the cells at negative membrane potentials and are inhibited by depolarization. In VSMC in culture and in models of VSMC injury as well as in other cases of vascular disease, TRPC channel expression dramatically increases. Specifically, the loss in contractile phenotype in proliferative VSMC in culture correlated with over fivefold increase in TRPC1 and TRPC6 mRNA and protein expression and increased whole cell Ca2+ currents [14]. In the same study, balloon dilatation of human internal mammary artery showed a similar increase in TRPC1 and TRPC6 mRNA expression 48 h post injury [14]. TRPC1 is upregulated following experimental vascular injury in mice and pigs, and is associated with the conversion of smooth muscle cells from a quiescent to a proliferative phenotype initiating neointimal formation [74]. Upregulation of TRPC1 was associated with enhanced Ca2+ entry and cell cycle activity. The functional relevance of TRPC1 upregulation in neointimal growth was studied in a human saphenous vein organ culture where TRPC1 function was blocked with a specific antibody targeted to its third extracellular loop (E3); this approach significantly inhibited neointimal growth in organ culture as well as calcium entry and VSMC proliferation [74].
In pulmonary artery SMC, TRPC1 and TRPC6 expression increases following serum stimulation [50, 160]. ATP-induced mitogenesis in pulmonary artery SMC was shown to be mediated through CREB-induced TRPC4 gene transcription [162]. The in vivo relevance of TRPC isoform upregulation in human pulmonary artery SMC was observed in patients with idiopathic pulmonary arterial hypertension (IPAH), a disease characterized by excessive pulmonary artery SMC proliferation [43]. In these patients, TRPC3 and TRPC6 were strongly upregulated. In pulmonary artery SMC cultured from patients with IPAH siRNA directed against TRPC6 markedly attenuated proliferation [158], suggesting TRPC channel overexpression might be important for increased proliferation during IPAH. Pulmonary hypertension caused by chronic hypoxia also involves excessive proliferation as well as contraction of pulmonary artery SMC. In a rat model of pulmonary hypertension, TRPC1 and TRPC6 upregulation was shown to require the expression of hypoxia inducible factor 1 (HIF-1) [149, 150]. TRPC channel perturbation is also proposed to be a factor involved in essential hypertension, as spontaneously hypertensive rats expressed abnormally high levels of TRPC3 compared to wild type control animals [84].
Loss of KCa1.1 and gain of KCa3.1 potassium channels
Ca2+-activated K+ channels indirectly regulate the flow of Ca2+ ions into cells by modulating the membrane potential through K+ efflux. Quiescent contractile VSMC express KCa1.1 (also called BKCa), large conductance K+ channels that are activated by intracellular Ca2+ and whose activity increases in response to membrane depolarization. Efflux of K+ through KCa1.1 causes hyperpolarization and therefore limits extracellular Ca2+ entry through the L-type voltage gated Ca2+ channels [25, 76, 95]. In this capacity, KCa1.1 serves as a negative feedback regulator that controls intracellular Ca2+ concentration and VSMC contraction.
Interestingly, KCa1.1 is downregulated when VSMC switch from a contractile to synthetic phenotype. Loss of KCa1.1 has previously been observed both in synthetic VSMC in culture and following balloon injury in the rat carotid artery [72, 96]. Reciprocally, synthetic or proliferating VSMC upregulate the intermediate conductance Ca2+-activated K+ channel protein 4 (KCa3.1), encoded by the KCNN4 gene [96]. Importantly, KCa3.1 blockade with a selective inhibitor, TRAM-34 inhibited neointimal formation by 40% after balloon-catheter induced injury in the rat, suggesting that KCa3.1 upregulation following vascular injury is functionally significant in proliferating VSMC [72]. A subsequent study showed that inhibition of KCa3.1 suppresses neointimal formation in the human saphenous vein, a vessel isolated from patients undergoing coronary artery bypass surgery and prone to failure as a result of neointimal hyperplasia [32].
As discussed above for Ca2+ channels, here again the most important difference between KCa1.1 and KCa3.1 channels appears to be their voltage dependence. Indeed, unlike KCa1.1 channels which require depolarization to optimally operate, KCa3.1 channels can open at negative membrane potentials which lead to further hyperpolarization of the cells. Thus, K+ efflux through KCa3.1 can potentially continue to hyperpolarize the cells up to a membrane potential of approximately −80 mV (value of K+ equilibrium potential) and subsequently provide additional driving force for robust Ca2+ entry through TRPC channels2. It appears therefore that KCa3.1 and TRPC (and possibly T-type Ca2+ channels) increased expression in synthetic SMC are functionally connected and represent components of the same Ca2+ signaling unit, important for increased Ca2+ entry at more hyperpolarized membrane potentials in VSMC. According to this paradigm, Ca2+ entry through hyperpolarization-activated channels might preferentially connect to downstream effectors necessary for promoting the proliferative and migratory capabilities of synthetic VSMC.
Loss of RyR3 Ca2+ release channels
RyR are Ca2+ release channels located in the membrane of the SR that are sensitive to Ca2+, caffeine and the plant alkaloid ryanodine. Opening of RyR can cause local Ca2+ transients termed Ca2+ sparks that regulate the membrane potential through activation of K+ channels [65, 66]. RyR are structurally and functionally analogous to IP3R, although they are approximately twice as large and have twice the conductance of IP3R [21]. Similar to IP3R, RyR are sensitive to cytosolic Ca2+ concentrations: nM to µM Ca2+ concentrations causing channel activation and µM to mM Ca2+ concentrations inhibiting channel function [21]. Also similar to the dual regulation of the IP3R, RyR phosphorylation by the second messenger cyclic ADP ribose (cADPR) and protein kinase A (PKA) enhance Ca2+ sensitivity while phosphorylation by protein kinase C (PKC) decreases RyR Ca2+ sensitivity [65, 83].
There are presently three molecularly distinct subtypes of RyR channels, RyR1, RyR2, and RyR3. While RyR channels are found in all muscle types, the vasculature appears to show a differential distribution of the three RyR isoforms [65, 122]. For example, rat cerebral arteries and portal vein myocytes express all three isoforms [122], while mouse myometrial smooth muscle and rat aorta express mainly the RyR3 isoform [122, 141]. It has been proposed that the RyR3 isoform is the prominent ryanodine receptor in smooth muscle [65]; however, the role of RyR3 in the vasculature is difficult to elucidate in that RyR3−/− mice do not differ in their spatial–temporal characteristics of Ca2+ sparks compared to control animals [122].
The in vitro conversion of aortic VSMC from the quiescent contractile state to the synthetic proliferating phenotype coincides with a loss in RyR3 expression and subsequent loss of the CICR mechanism [141]. The functional impact of loss of RyR3 in the intact aorta would not allow the vessel to oppose vasoconstriction and might possibly be involved in the hypersensitivity reaction to vasoactive substances observed in the pathology of hypertension and atherosclerosis. The expression levels of RyR during vascular injury have not been studied and the role of RyR in VSMC phenotypic switching and proliferation is unclear. While RyR function was not impaired in vivo in the aorta of hypercholesterolemic ApoE−/− mice in one study [142], a separate in vitro study did find that oxidized LDL induced a decrease in VSMC RyR channel density which resulted in a defect in the cells ability to regulate cytosolic Ca2+ [89]. As mentioned earlier, Ca2+ release through RyR is involved in vasodilatation of VSMC, and therefore suppression of this release pathway would alter the control of VSMC contractility. However, in vascular pathologies where VSMC lose their contractile proteins, it is reasonable to suggest that proliferation, rather than contractility, is likely responsible for the thickening of the vessel wall. Clearly, the downregulation of RyR3 represents one component (in addition to KCa1.1 and the L-type Ca2+ channels) of the Ca2+ signaling unit controlling EC-coupling that is altered during VSMC phenotypic switching. However, whether the loss of RyR3 in the synthetic VSMC is functionally connected to the promotion of the proliferative or migratory phenotype remains unknown.
Role of IP3R Ca2+ release channels
In quiescent smooth muscle, agonists that act through G protein-coupled receptors or receptor tyrosine kinases can induce intracellular Ca2+ signals through activation of PLC. PLC catalyzes the hydrolysis of phosphatidylinositol4,5-bisphosphate (PIP2) to produce the intracellular messengers IP3 and diacylglycerol (DAG). IP3 is highly diffusible in the cytoplasm and activates specific receptors (IP3R) on the sarcoplasmic reticulum membrane [15]. There are three isoforms of the IP3R: IP3R1, IP3R2, and IP3R3 with IP3R1 being the most widely expressed of these three and found in most cell types. The binding of IP3 to the IP3R changes the conformation of the IP3R causing its opening and Ca2+ release from the SR. The Ca2+ release mediated by IP3 diffuses through the cytoplasm thereby exciting neighboring receptors and providing the momentum necessary to generate a Ca2+ wave. IP3R are also sensitive to Ca2+ and in order to avoid random triggering of regenerative Ca2+ waves, Ca2+ has a biphasic effect on the receptor. As the intracellular Ca2+ concentration increases, Ca2+ exerts positive feedback and facilitate IP3R channel opening [17, 21]. However, a further increase in cytosolic Ca2+ has the opposite effect and actually inhibits the channel opening [21, 83]. Interestingly, evidence suggests that high concentrations of IP3 may prevent Ca2+-dependent inactivation of IP3R [22], an observation which is potentially important in proliferating VSMC in disease states characterized by increased pro-proliferative growth factors and endothelial factors acting through the PLC/IP3 pathway.
Increasing evidence is supporting a role for the IP3R as an important determinant of VSMC proliferation. Exposure of freshly explanted cerebral arteries to fetal bovine serum elevated IP3 and the frequency of Ca2+ waves which were subsequently required for VSMC proliferation [154]. Proliferation was inhibited by several IP3 receptor antagonists (100 µM 2-APB, 10 µM xestospongin C, or 25 µM U-73122) which dramatically reduced outgrowth cell number compared with ryanodine-treated (10 µM) arteries [154]. However, these experiments should be interpreted with caution since the effects of these pharmacological inhibitors could be partially due to inhibition of plasma membrane Ca2+ channels. In fact, 2-APB, at much lower concentrations (30 µM) is a potent inhibitor of SOC channels and is also known to partially inhibit TRPC channels [137], while U-73122 acts upstream to inhibit PLC, and would thus consequently inhibit the activation of receptor-operated channels, including all TRPC channels. Nonetheless, in serum-stimulated VSMC, proliferation is associated with a sustained increase in intracellular Ca2+ partially due to enhanced excitability of IP3R following binding of IP3. In addition, a recent report showed a sixfold increase in the expression level of IP3R1 as VSMC progressed from G0 to G1/S of the cell cycle [5]. This increase was mediated by the cell cycle-associated transcription factor c-Myb and was accompanied by increased intracellular Ca2+ at the G1/S transition [5].
Although in vivo studies have not directly examined IP3R expression/activation during vascular disease states, a recent study carried out in aortic rings of apolipoprotein E-deficient mice which had not yet developed atherosclerotic lesions, implicated IP3R as an important player during the progression of atherosclerosis. In this particular study, IP3R-mediated Ca2+ release in response to phenylephrine and ATP was significantly increased in hyperocholesteremic but plaque-free apoE−/− mice compared to wild type mice [142]; apoE−/− mice displayed an increase in basal levels of cytosolic calcium [142]. In a similar manner, endothelin-1, which induces a Ca2+ release through IP3R, produced a greater Ca2+ release in diabetic dyslipedemic swine carotid artery as compared to controls [78].
Loss of the SERCA2a isoform
SERCA maintains intracellular Ca2+ homeostasis in VSMC by pumping Ca2+ from the cytoplasm into the SR stores. The SERCA pump is encoded by a family of three genes located on separate chromosomes, SERCA1, 2, and 3 [44]. The SERCA isoform diversity is greatly increased by alternative splicing of the transcripts, resulting in more than ten different SERCA isoforms currently characterized in different tissues and developmental states [106]. SERCA isoforms differ in their affinity for Ca2+, resistance to oxidative stress and modulation by phospholamban (PLN) and Ca2+/calmodulin kinase II. The activity of SERCA2 is dependent on its interaction with PLN which is inhibitory in its dephosphorylated form. Phosphorylated PLN dissociates from SERCA2, thus activating the Ca2+ pump and SERCA2 activity is proposed to account for removal of >70% cytoplasmic Ca2+ [86].
It is clearly established that SERCA2a plays an important role in cardiac function. Earlier studies have shown that SERCA2a levels are decreased in human failing hearts and in several animal models of cardiac hypertrophy [8, 151]. The idea that SERCA2a expression might protect against cell proliferation, originated from studies where heterozygous mice with a null mutation in the SERCA2 gene develop cancers of the skin and the upper digestive tract [85], as well as cardiac hypertrophy [107]. Additional studies showed that mice in which SERCA2a has been replaced by SERCA2b develop dilated cardiac hypertrophy [144]. In contrast, cardiac-specific expression of SERCA2a in transgenic animals showed improved contractile function under basal conditions and pressure overload [94]. Furthermore, while only SERCA2b is present in proliferating BC3H muscle cells, SERCA1, SERCA2a, and SERCA2b are present in the quiescent differentiated cell variant BC3H1 [36].
Quiescent smooth muscle cells express two splice variants of the SERCA2 gene, 2a and 2b.Vallot et al., have shown that SERCA2a is repressed in vitro in VSMC during serum- and PDGF-induced proliferation [141]. Work by Lipskaia et al. showed that in proliferating VSMC, SERCA2a expression is downregulated, while SERCA2b expression remains unchanged [82]. When VSMC are growth arrested or in confluent cultures SERCA2a expression is regained, pointing to an important link between alteration in the expression of Ca2+-handling proteins and cell proliferation. Interestingly, the same authors provided evidence that adenoviral overexpression of SERCA2a inhibits VSMC proliferation and subsequent neointima formation following balloon catheter-induced injury in the rat carotid artery [82]. Specifically, overexpression of SERCA2a in serum-stimulated VSMC inhibited proliferation through inactivation of calcineurin and its target transcription factor nuclear factor of activated T-cells (NFAT) resulting in lowering of Cyclin D1 levels and cell cycle arrest at the G1 phase [82]. The authors concluded that the delivery of SERCA2a to the injured media reversed the alterations in Ca2+ handling observed in proliferating VSMC, suggesting SERCA2a expression and activity are important in maintaining the non-proliferative phenotype of smooth muscle [82, 83]. In addition, overexpression of SERCA2a in proliferating VSMC in culture reduced the amplitude and propagation of IP3-evoked Ca2+ signals [82], supporting a role of IP3-mediated Ca2+ signaling in VSMC proliferation.
Role of PMCA
The PMCA is a high-affinity Ca2+ pump dedicated to the removal of Ca2+ from the intracellular to the extracellular space. In a manner similar to SERCA, the PMCA is an ATP-driven pump which moves Ca2+ against its concentration gradient, thus functioning as an important system involved in maintaining low levels of cytoplasmic Ca2+. In mammals, four separate genes encode for the major PMCA isoforms (PMCA1 through 4) and the various gene products and splice variants which arise thereof show different patterns of expression during development as well as in different tissue beds [28, 52]. While each isoform and splice variant can be regulated by multiple kinases and protein–protein interactions, the main regulator of PMCA function is Ca2+-bound Calmodulin (CaM) [131]. Not surprisingly, it appears that the most important splicing occurs in the CaM-binding domain. Alternative splicing at this site alters the ability of CaM to bind and activate the pump. PMCA1 and PMCA4 are present in almost all cells and have therefore been proposed as housekeeping pumps, while PMCA2 and PMCA3 have a more constrained distribution and are possibly endowed with more specialized functions [27, 52, 111].
In the vasculature, medial smooth muscle cells appear to express multiple isoforms of both PMCA1 and 4 [3, 131] and cell lines from canine carotid artery or saphenous vein medial wall express PMCA1b, 4a and 4b [3]. The role of PMCA1 might be to repress VSMC proliferation as cells in the G1/S phase of the cell cycle experienced lower levels of PMCA1 mediated through repression by the transcription factor c-Myb [61, 62]. Overexpression of PMCA1 or suppression of c-Myb in these cells resulted in a reduced rate of cell proliferation assessed by a fewer number of cells entering the S phase [4, 62]. Whole animal studies in which c-Myb has been inhibited suggest that loss of PMCA1 function has important consequences on pathologic VSMC proliferation [4].
However, a study showed that upon placement in culture, carotid artery and saphenous vein VSMC showed a time-dependent upregulation of PMCA4a in a PI3-Kinase dependent manner and increased cell proliferation. Reciprocally, inhibition of PMCA4a upregulation with Wortmannin inhibited cell proliferation [3]. These results may appear incompatible with the general concept whereby decreased PMCA1 and SERCA2a, increased TRPC channels and IP3-mediated Ca2+ release contribute to the sustained increase in cytoplasmic Ca2+ levels that are necessary to drive cell proliferation. A possible explanation for this discrepancy might be that the newly synthesized PMCA4a molecules could be targeted to specific membrane subdomains and therefore not significantly affecting the pool of cytoplasmic Ca2+ associated with cell proliferation. Alternatively, heterologous expression studies of human PMCA4a and 4b showed that PMCA4a has 7- to 20-fold lower affinity for calmodulin and Ca2+ and a higher basal activity in the absence of calmodulin than PMCA4b [40]. This would suggest that PMCA4a is probably important for maintaining Ca2+ homeostasis under basal conditions and that it operates optimally to extrude Ca2+ only when very high levels of cytoplasmic Ca2+ concentrations are reached within the cells. In proliferating VSMC, PMCA4a overexpression could thus be compatible with increased cytoplasmic Ca2+ levels necessary to drive cell proliferation while, at the same time, maintaining the Ca2+ levels below a certain threshold to protect against cell death by apoptosis.
Other changes involving various ion channels
There are other ion channels that see their expression change during VSMC switching to a proliferative phenotype that seem to contribute to increased cytoplasmic Ca2+. Zhang et al. [161] demonstrated an upregulation of NCX and enhanced Ca2+ entry via NCX reverse mode in pulmonary artery SMC from patients with idiopathic arterial hypertension. These authors previously showed that cultured human pulmonary artery SMC express high levels of NCX and that enhanced NCX activity in its reverse mode produced an increase in cytoplasmic Ca2+ and promoted proliferation [163].
Quignard et al. [115] found that the tetrodotoxin-sensitive voltage gated Na+ channel was not expressed in freshly isolated myocytes from human coronary artery but both expression of these channels and large voltage gated Na+ currents were detected when the cells were grown in culture [115]. A microarray study in lung tissues from patients with idiopathic and familial pulmonary arterial hypertension showed increased mRNA expression of the voltage gated Na+ channel β1 subunit [45]. The physiological significance of increased Na+ currents in proliferating cells remains a mystery. Platoshyn et al. [109] showed evidence that increased Na+ currents does not directly influence membrane potential, intracellular Ca2+ release or proliferation in cultured human pulmonary artery smooth muscle. These authors speculated that increased Na+ currents could contribute to activation of upregulated NCX in its reverse mode, leading to an increase of the bulk of cytoplasmic Ca2+ and enhanced cell proliferation [109].
As discussed earlier, K+ channels control membrane potential which in turn regulates the flow of Ca2+ across the plasma membrane. Karkanis et al. [69] demonstrated increased inwardly rectifying K+ channels (Kir) currents in proliferative compared with contractile VSMC and showed increased mRNA and protein expression of Kir2.1 in proliferative cells, which displayed a more negative membrane potential than contractile cells [69].
The expression profile of voltage gated K+ (Kv) channels is also altered during the transition of VSMC from the contractile to the proliferating phenotypes. While Kv1 channel members were the main contributors to the Kv currents in the human uterine contractile phenotype, proliferating VSMC upregulated Kv3.4 expression [92]; Kv3.4 is characterized by a rapid inactivation. Interestingly, selective blockade of Kv3.4 channels decreased VSMC proliferation [92]. Another study showed that pulmonary artery SMC proliferation is associated with a decrease in Kv1.5 currents [159]. Early studies by DeCoursey et al. [37] have proposed voltage gated K+ channels as important players in T-cell mitogenesis. Subsequent studies implicated many isoforms of K+ channels in normal and/or pathological cell proliferation in different cell types [103]. The exact mechanisms by which voltage gated K+ channel might regulate proliferation in VSMC that have lost their electrical excitability (or for that matter in electrically non-excitable cells in general) remain poorly understood and may well involve regulation of membrane potential and/or cell volume.
Cl− channels which are expressed in both the plasma membrane and intracellular organelles of cells play an important role in various cell functions, including ion homeostasis, cell volume regulation, transepithelial transport, and regulation of electrical excitability [67]. There is also evidence implicating ClC-3, a voltage gated Cl− channels, in VSMC proliferation; different Cl− channel blockers were shown to inhibit the proliferation of VSMC induced by endothelin-1 (ET-1) [156], and transient transfection of rat aortic VSMC with antisense oligonucleotide specific to ClC-3 caused an inhibition in ET-1-induced expression of ClC-3 protein and cell proliferation of VSMC [148]. ClC-3 expression was upregulated in pulmonary artery smooth muscle cells and cardiac myocytes from hypertensive mice and in response to inflammatory mediators such as ET-1, platelet-derived growth factor (PDGF), IL-1β and TNFα; ClC-3 increase was shown to confer increased viability and enhanced protection against cell death induced by reactive oxygen species [35]. Voltage gated Cl− channels can induce VSMC depolarization; the Cl− equilibrium potential in VSMC is approximately −26 mV, a range at which L-type Ca2+ channels can be effectively activated. However, how exactly increased ClC-3 expression might functionally affect the proliferation of VSMC that has lost L-type Ca2+ channels expression is not known. As pointed out by Guan et al., medial hypertrophy, a key feature involved in arterial remodeling during hypertension, is caused mainly by the enhancement of VSMC proliferation in response to vascular injury. Because cell proliferation must sometimes lead to increased cell volume, it is therefore not surprising that ClC-3 function is associated with VSMC proliferation during vascular disease [51]. However, the involvement of ClC-3 as a volume-regulated channel remains controversial, and findings from several groups, unable to consistently measure plasma membrane ClC-3 currents, suggested that ClC-3 can reach the plasma membrane only under some conditions of overexpression and provided evidence for the expression of ClC-3 in intracellular vesicles (see [67] and references therein). Whether ClC-3 increased expression in proliferating VSMC is associated with increased ClC-3 proteins at the plasma membrane or in intracellular vesicles and how this pertain to VSMC proliferation remains a mystery.
Decrease of CaMKIIγ and increase of CaMK IIδ2
The Ca2+/calmodulin (Ca2+/CaM)-dependent protein kinase II (CaMKII) is a ubiquitously expressed and multifunctional serine/threonine kinase that mediate many of the downstream effects of Ca2+ signaling. CaMKII isoforms can associate to form either homo- or hetero-multimeric holoenzyme composed of 10–14 subunits. In mammals, CaMKII isoforms are encoded by four different genes (α, β, γ, δ) which share extensive sequence homology and structure [135]. CaMKII transcripts are in turn subject to extensive alternative splicing, resulting in kinases with variable domains and specific cellular functions. Although ubiquitously expressed, CaMKII isoforms have a distinct pattern of expression in different tissue beds. Distribution of three distinct α and seven distinct β splice variants are mainly limited to the central nervous system where they are estimated to comprise up to 1% to 2% of the total protein in the forebrain and hippocampus, respectively [41, 135]. In the brain, CaMKII is critical for regulating synaptic and behavioral plasticity and possibly memory [33]. To date, there are 13 recognized CaMKIIγ splice variants. Similarly, 15 splice variants have been identified for the δ gene [29]. Both CaMKIIγ and CaMKIIδ are ubiquitously expressed with the highest levels observed in peripheral tissue [134]. Many studies have particularly focused on CaMKII function in heart and vascular smooth muscle and implicated the involvement of CaMKII in various functions including the following: mediation of Ca2+-dependent gene transcription [116], regulation of protein interactions involved in contractility and cell motility [105, 108, 120, 132], and control of intracellular Ca2+ dynamics [87].
In the vasculature, differentiated VSMC express CaMKII γ and δ variants [124, 125]. In these cells, CaMKII γ isoforms have been implicated in the control of the differentiated contractile function [71, 88, 120]. However, cultured VSMC predominantly express the δ2 (also referred to as δC) isoform [124]. CaMKII δ isoforms couple to ERK1/2 signaling [1, 47], and the δ2 isoform was shown to specifically regulate VSMC migration [105, 108]. Upregulation of CaMKII δ isoforms was shown to occur in models of cardiac hypertrophy and heart failure [23, 166], as well as in regenerating skeletal muscle after injury [2].
Recently, House et al. have showed that following dispersion and primary culture of VSMC, a significant increase in CaMKII δ2 protein and a significant decrease in CaMKII γ protein within 30 h of dispersion which coincided with VSMC phenotypic switching including increased DNA synthesis and cell proliferation [58]. Interestingly, this reciprocal regulation of CaMKII isoforms during phenotypic switching occurred even when cells were dispersed under low serum concentrations (0.4%) which do not support proliferation, suggesting that the CaMKII isoform switch (and possibly changes in expression of other molecular players) occurs irrespective of VSMC proliferation [58]. Attenuation of the initial upregulation of the CaMKII δ2 isoform in primary cultured cells using small-interfering RNA resulted in decreased serum-stimulated DNA synthesis and cell proliferation [58], arguing for a specific role of CaMKII δ2 in driving VSMC proliferation. In a follow up study, similar results were obtained with rat carotid arteries subjected to balloon angioplasty where a significant increase of CaMKII δ2 and a significant decrease of CaMKII γ were observed within 3 days post-injury in both medial smooth muscle and adventitial fibroblasts [59]. In this model, the neointimal VSMC expressed primarily CaMKII δ2; incubation of the injured vessels with adenovirus encoding siRNA against CaMKII δ isoforms prevented upregulation of CaMKII δ2 in the media and adventitia, inhibited cell migration and proliferation and neointima formation [59].
What is the physiological significance of the switch in CaMKII isoform expression? CaMKII isoforms are capable of autoregulation through autophosphorylation which confers to the kinase the unique ability to maintain catalytic activity without bound Ca2+/CaM [60, 129]. This is particularly important for the memory of the kinase allowing continuous activation after intracellular levels of Ca2+ has returned to their basal state [126]. It is increasingly acknowledged that different sources, frequencies, amplitudes, and spatiotemporal distributions of Ca2+ signals may result in activation of different downstream effectors, different patterns of gene expression and thus lead to different physiological outcomes [154]. CaMKII isoforms are capable of decoding different Ca2+ signals and responding to Ca2+ transients in a frequency-dependent manner [60]. Although the physiological significance of this remain largely unexplored, the CaMKII δ2 isoform might functionally connect to Ca2+ sources generated by growth factors receptors through IP3-mediated Ca2+ release and Ca2+ entry via TRPC channels. Indeed, the CaMKII δ2 isoform was shown to be activated in VSMC by growth factors acting on G protein-coupled receptors and receptor tyrosine kinase [46, 47]. As argued earlier for other Ca2+ signaling molecules, the acquisition of the CaMKII δ2 isoform by VSMC may be one component of a functional molecular unit that is acquired during phenotypic switching and is necessary to initiate and/or sustain cell proliferation and migration.
What triggers the molecular switch in Ca2+ signaling pathways?
Normal VSMC in the vessel wall are quiescent and differentiated but can undergo remarkable and reversible changes into a proliferative phenotype that is likely of fundamental importance during vascular development, regeneration, and normal vascular physiology. It is intuitive to think that it is the maintenance of the proliferative phenotype that contributes to the development of atherosclerotic plaques and neointimal hyperplasia during occlusive vascular disease. Though the molecular mechanisms controlling the phenotypic switch of VSMC have been intensely studied, they remain incompletely understood. The vast majority of studies in the literature have focused on the transcriptional regulation of the contractile genes [70, 102]. Most of the regulatory aspects of VSMC contractile genes identified to date have been shown to depend on CArG regulatory sequences found in their promoters and on the binding of the transcription factor serum response factor (SRF) [102, 147]. In recent years, increasing number of studies have focused on the ion channels and pumps acquired or lost during VSMC switch to the synthetic phenotype and their role in proliferation [14, 32, 74, 82]. The transcription factors that control the gene regulation of these proteins are starting to emerge [32, 75, 149, 150, 160, 162]. However, the pro-proliferative downstream signaling pathways that they regulate remain poorly understood.
What triggers the phenotypic switch? For good reasons, most studies have focused on the identification of growth factors, cytokines, and endothelium-derived factors as initiators of VSMC proliferation [70, 102]. The expression levels of TRPC channels appear to be under the control of growth factors. Indeed, studies from one laboratory showed that TRPC6 cation channel was upregulated through the c-Jun/STAT-3 pathway during PDGF-induced pulmonary VSMC proliferation [160], and that TRPC4 was upregulated in ATP-induced mitogenesis through cAMP response element-binding protein (CREB) activation in the same cells [162], providing a mechanism for upregulation of these channels during cell proliferation. Furthermore, SERCA2a expression (or loss thereof) in proliferative VSMC seems to depend on a proliferative signal because expression of SERCA2a can be regained when VSMC are growth arrested [82]. However, the modulation of smooth muscle differentiation markers [54] as well as the reciprocal regulation of the CAMKII isoforms [58] occurs in the absence of VSMC proliferation. In this case, proliferative factors alone cannot account for the change in expression of these proteins and future investigations should consider alternative hypotheses such as loss of endothelial contact/factors, change in matrix interactions, secretion of lipid mediators, mechanical stimuli and oxidative stress and the subsequent signaling pathways they activate. Whether the regulation of the ion channels and pumps during VSMC phenotypic switching can also occur in the absence of serum mitogens under growth arrest conditions remain to be demonstrated. It is tempting to speculate that change in expression of a group of proteins might represent the trigger for the molecular switch in VSMC that occur in a mitogen-independent manner while the maintenance of this switch would require mitogen-induced expression (or repression) of another group of signaling molecules.
The aforementioned idea leads to important questions that remain unexplored: what are the time course and the specific temporal sequence of the molecular changes of Ca2+ handling proteins during phenotypic switching of VSMC? What are the transcriptional mechanisms that control change in expression during vascular injury? Insight into the latter question as it pertains to the K+ channel KCa3.1 expression was gained from a recent study showing that repressor element 1 silencing transcription factor (REST) downregulation is a critical event enabling KCa3.1 expression and proliferation of VSMC [32]. A genome wide analysis using conservative sequences of REST target genes, identified 1892 REST sites in the human genome corresponding to genes transcriptionally repressed by REST and encoding among others: ion channels, growth factors, cytoskeleton proteins, intracellular signaling molecules, and transcription factors [26]. From this study, additional questions arise: What are the ion channels and pumps whose expression is affected by REST downregulation during VSMC phenotypic switching? What causes REST downregulation in the first place? And what pathway ends this retrogression? Clearly, what happens during VSMC phenotypic switching is likely complex, involves more than one pathway and crosstalk between multiple molecular players that are individually capable of autoregulation through feedback loops.
Ca2+ is involved in a wide array of biological functions and the fact that Ca2+ regulates the cell cycle is well established. Ca2+ has the capability of acting directly on transcription factors (such as DREAM) or indirectly through protein phosphatases (calcineurin/NFAT) or protein kinases (CaMKII or CaMKIV/CREB; PKC/ NF-κB) to induce the activation of numerous target genes. The universality of Ca2+ as a second messenger and the ubiquitous requirement for Ca2+ signals as activators of many signaling pathways should not be interpreted to mean a lack of specificity. Quite the opposite, Ca2+ signals show an impressive variation in their modes of transport, localization, amplitude and frequency and thus their action on downstream transcription factors is remarkably specific. For instance, transcription factors such as NF-κB and NFAT were shown to be activated by different Ca2+ signals that vary in their amplitude, frequency and duration; the former is activated by a large and transient Ca2+ rise, whereas the latter is activated by low, sustained CA2+ signals [39]. The altered expression of Ca2+ signaling molecules in proliferating VSMC should instead be interpreted within this paradigm where the source of the Ca2+ signal, its amplitude, frequency, and duration are important determinants in VSMC phenotypic switching.
The simplistic view would involve the shift in resting membrane potential to more negative values, the increased expression and subsequent activation (constitutive or agonist-induced) of ROC channels such as TRPC channels that optimally operate at these membrane potential values and the loss of Ca2+ pumps responsible for Ca2+ clearance would presumably lead to increased basal cytoplasmic Ca2+ levels that might be necessary to drive VSMC proliferation. As such, the adaptive changes of VSMC to a non-excitable mode by altering the levels of Ca2+ channels, pumps, and Ca2+-activated enzymes appear to be necessary for proliferation. Clearly, future studies on the correlation between the dynamic changes in Ca2+ signaling proteins expression during VSMC proliferation and their specific cellular localization in regions of the plasma membrane or in microdomains of the sub-sarcolemmal space are needed, so the precise mechanisms by which these Ca2+ signaling molecules promote VSMC proliferation and migration can be fully elucidated.
Conclusions
There is growing evidence that alteration in Ca2+ signaling molecules (ion channels, pumps, Ca2+-activated enzymes) expression is closely associated with VSMC phenotypic modulation from a contractile quiescent to a synthetic proliferative phenotype. Figure 1 summarizes the changes in expression of channels and pumps that occur during VSMC phenotypic switching. Here, we argued that these changes in expression are interconnected and each acquired Ca2+ signaling molecule represents a component of a pro-proliferative module functioning as a single unit. The functional consequence of these changes is the acquisition by VSMC of a non-excitable-like phenotype, an increased and sustained cytoplasmic Ca2+ at more negative membrane potentials and subsequent proliferation. Alternatively, the shift in the source of Ca2+ entry from voltage-gated channels to receptor-operated channels would lead to Ca2+ entry that connects to pro-proliferative downstream signaling pathways. Interestingly, and regardless of the mechanisms by which this Ca2+ signaling switch promotes VSMC proliferation, in most studies where an increased expression of Ca2+ signaling molecules have been reported in proliferative VSMC, silencing of these Ca2+ signaling molecules have invariably produced an inhibition of proliferation. This highlights the importance of these proteins in VSMC proliferation and their potential use as targets for novel therapeutic strategies of vascular disease.
Fig. 1.
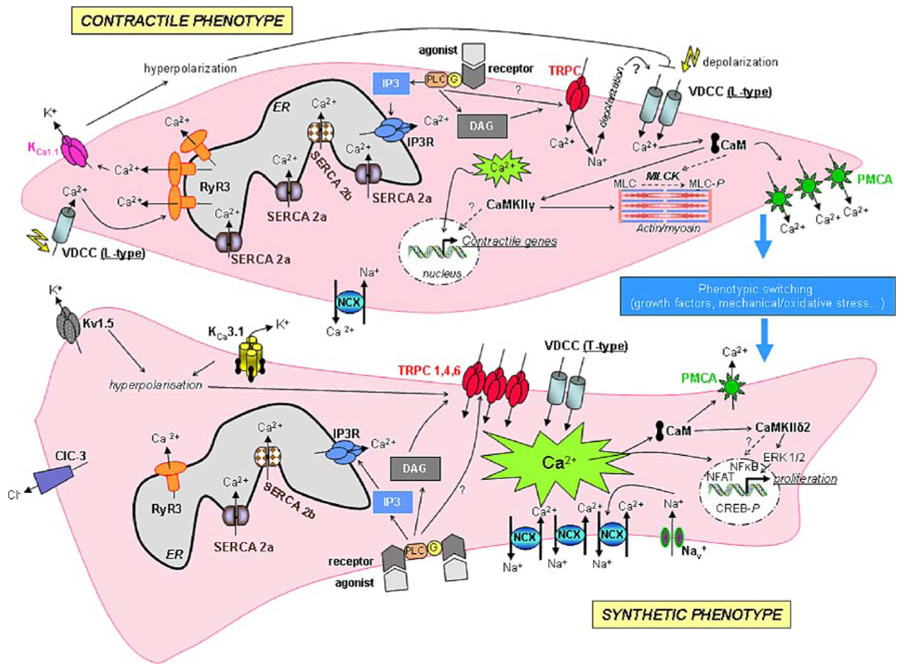
Ion channel modulation during phenotypic switching of VSMC. In contractile VSMC, extracellular Ca2+ enters the cell mainly through L-type Ca2+ channels and Ca2+ release occurs through RyR. Vasoactive agonists, by binding to their receptors, mediate Ca2+ release from the ER through IP3R and Ca2+ entry via TRPC. Intracellular Ca2+ maintains precise control of its own homeostasis through regulation of K+, Cl−, and Ca2+ release channels and Ca2+ pumps. Ca2+ regulates VSMC contraction through activation of myosin light chain kinase (MLCK) which leads to myosin light chain (MLC) phosphorylation (MLC-P). Vascular tissue injury is associated with a modulation of ion channels, pumps and Ca2+-binding proteins and phenotype modulation to a proliferative synthetic phenotype. Nav voltage-dependent Na+ channel; Kv1.5 voltage-dependent K+ channel; ClC3 voltage dependent Cl− channel; VDCC voltage-dependent Ca2+ channel; CaM calmodulin
The remarkable complexity and variability of Ca2+ channels (and ion channels in general) makes them prime targets for drug therapy of disease. For over half a century, pharmacological blockers of the L-type voltage gated Ca2+ channels in VSMC have been used in the treatment of cardiovascular diseases, including hypertension and angina [79]. Perhaps the limited therapeutic efficacy of drugs against L-type Ca2+ channels is not surprising if some of the vascular pathologies treated with these drugs are characterized by the loss of VSMC L-type Ca2+ channel expression, as it is the case during vascular injury. Therefore, it makes implicit sense to focus drug targeting efforts on ion channels that see their expression increased in vascular disease. Among such ion channels, arguably the most attractive current candidates for drug targeting are the TRP superfamily members of channels; this is because of the important role they play in the vasculature and their differential expression among cell types [79, 97, 136]. While increased TRP channel expression of members of the Melastatin- (TRPM) and Vanilloid (TRPV)-families have been reported in several forms of human cancers ([93, 97] and references therein), their involvement in VSMC proliferation remains unexplored. Nonetheless, in the light of canonical TRPC members increased expression in proliferating VSMC in various disease models, these ion channels could represent realistic targets for therapy of VSMC proliferation in cardiovascular diseases. Similarly, other ion channels (KCa3.1, T-type channels), transporters (NCX) and Ca2+ signaling molecules (CaMKIIδ2) that see their expression increased during VSMC proliferation could also represent promising targets for future drug therapy.
Acknowledgments
Funding for the authors work was supported by NIH grants to M.T (K22ES014729) and to H.A.S (R01 HL-4942612).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures The authors have no conflict of interest to disclose.
SOC channels can be considered a type of ROC channels. SOC channels are physiologically activated by the depletion of internal Ca2+ stores through IP3-mediated Ca2+ release, and also by pharmacological agents that deplete the internal Ca2+ stores without increasing the levels of IP3 such as the SERCA pump inhibitor thapsigargin. ROC channels include channels that are activated by signaling mechanisms downstream of membrane receptors, including second messengers such as IP3, diacylglycerol (DAG) and arachidonic acid (SMOC channels), Ca2+, cyclic nucleotides etc. In fact, the use of the ROC nomenclature which lumps together diverse ionic conductances is a reflection of our ignorance of the molecular identity and the exact mechanisms of activation of these ion channels.
Some TRPC channels such as TRPC3 and TRPC7 are known to possess considerable constitutive activity [38], and it was clearly demonstrated that growth factors such as the epidermal growth factor (EGF) can increase the membrane expression of constitutively active TRPC channels through increased vesicular trafficking [18, 127]. Thus, it is reasonable to assume that strong hyperpolarization in VSMC would lead to increased constitutive activity of TRPC channels, and increased basal levels of cytoplasmic Ca2+.
References
- 1.Abraham ST, Benscoter HA, Schworer CM, Singer HA. A role for Ca2+/calmodulin-dependent protein kinase II in the mitogen-activated protein kinase signaling cascade of cultured rat aortic vascular smooth muscle cells. Circ Res. 1997;81:575–584. doi: 10.1161/01.res.81.4.575. [DOI] [PubMed] [Google Scholar]
- 2.Abraham ST, Shaw C. Increased expression of deltaCaMKII isoforms in skeletal muscle regeneration: Implications in dystrophic muscle disease. J Cell Biochem. 2006;97:621–632. doi: 10.1002/jcb.20669. [DOI] [PubMed] [Google Scholar]
- 3.Abramowitz J, Aydemir-Koksoy A, Helgason T, Jemelka S, Odebunmi T, Seidel CL, Allen JC. Expression of plasma membrane calcium ATPases in phenotypically distinct canine vascular smooth muscle cells. J Mol Cell Cardiol. 2000;32:777–789. doi: 10.1006/jmcc.2000.1120. [DOI] [PubMed] [Google Scholar]
- 4.Afroze T, Husain M. Cell cycle dependent regulation of intracellular calcium concentration in vascular smooth muscle cells: a potential target for drug therapy. Current Drug Targets. 2001;1:23–40. doi: 10.2174/1568006013338060. [DOI] [PubMed] [Google Scholar]
- 5.Afroze T, Sadi AM, Momen MA, Gu S, Heximer S, Husain M. c-Myb-dependent inositol 1,4,5-trisphosphate receptor type-1 expression in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2007;27:1305–1311. doi: 10.1161/ATVBAHA.107.142059. [DOI] [PubMed] [Google Scholar]
- 6.Akata T. Cellular and molecular mechanisms regulating vascular tone. Part 1: basic mechanisms controlling cytosolic Ca2+ concentration and the Ca2+-dependent regulation of vascular tone. Journal of anesthesia. 2007;21:220–231. doi: 10.1007/s00540-006-0487-5. [DOI] [PubMed] [Google Scholar]
- 7.Albert AP, Saleh SN, Peppiatt-Wildman CM, Large WA. Multiple activation mechanisms of store-operated TRPC channels in smooth muscle cells. J Physiol. 2007;583:25–36. doi: 10.1113/jphysiol.2007.137802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arai M, Matsui H, Periasamy M. Sarcoplasmic reticulum gene expression in cardiac hypertrophy and heart failure. Circ Res. 1994:555–564. doi: 10.1161/01.res.74.4.555. [DOI] [PubMed] [Google Scholar]
- 9.Barlow CA, Rose P, Pulver-Kaste RA, Lounsbury KM. Excitation-transcription coupling in smooth muscle. J Physiol. 2006;570:59–64. doi: 10.1113/jphysiol.2005.098426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barritt GJ. Receptor-activated Ca2+ inflow in animal cells: a variety of pathways tailored to meet different intracellular Ca2+ signalling requirements. Biochem J. 1999;337(Pt 2):153–169. [PMC free article] [PubMed] [Google Scholar]
- 11.Bayguinov O, Hagen B, Bonev AD, Nelson MT, Sanders KM. Intracellular calcium events activated by ATP in murine colonic myocytes. Am J Physiol Cell Physiol. 2000;279:C126–C135. doi: 10.1152/ajpcell.2000.279.1.C126. [DOI] [PubMed] [Google Scholar]
- 12.Bayguinov O, Hagen B, Sanders KM. Muscarinic stimulation increases basal Ca(2+) and inhibits spontaneous Ca (2+) transients in murine colonic myocytes. Am J Physiol Cell Physiol. 2001;280:C689–C700. doi: 10.1152/ajpcell.2001.280.3.C689. [DOI] [PubMed] [Google Scholar]
- 13.Beech DJ. TRPC1: store-operated channel and more. Pflugers Arch. 2005;451:53–60. doi: 10.1007/s00424-005-1441-3. [DOI] [PubMed] [Google Scholar]
- 14.Bergdahl A, Gomez MF, Wihlborg AK, Erlinge D, Eyjolfson A, Xu SZ, Beech DJ, Dreja K, Hellstrand P. Plasticity of TRPC expression in arterial smooth muscle: correlation with store-operated Ca2+ entry. Am J Physiol Cell Physiol. 2005;288:C872–C880. doi: 10.1152/ajpcell.00334.2004. [DOI] [PubMed] [Google Scholar]
- 15.Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 16.Berridge MJ. Calcium microdomains: organization and function. Cell Calcium. 2006;40:405–412. doi: 10.1016/j.ceca.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 17.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 18.Bezzerides VJ, Ramsey IS, Kotecha S, Greka A, Clapham DE. Rapid vesicular translocation and insertion of TRP channels. Nat Cell Biol. 2004;6:709–720. doi: 10.1038/ncb1150. [DOI] [PubMed] [Google Scholar]
- 19.Bird GS, Aziz O, Lievremont JP, Wedel BJ, Trebak M, Vazquez G, Putney JW., Jr Mechanisms of phospholipase Cregulated calcium entry. Curr Mol Med. 2004;4:291–301. doi: 10.2174/1566524043360681. [DOI] [PubMed] [Google Scholar]
- 20.Bolotina VM. Store-operated channels: diversity and activation mechanisms. Sci STKE. 2004;2004:pe34. doi: 10.1126/stke.2432004pe34. [DOI] [PubMed] [Google Scholar]
- 21.Bootman MD, Collins TJ, Peppiatt CM, Prothero LS, MacKenzie L, De Smet P, Travers M, Tovey SC, Seo JT, Berridge MJ, Ciccolini F, Lipp P. Calcium signalling–an overview. Semin Cell Dev Biol. 2001;12:3–10. doi: 10.1006/scdb.2000.0211. [DOI] [PubMed] [Google Scholar]
- 22.Bootman MD, Lipp P. Ringing changes to the ‘bell-shaped curve’. Curr Biol. 1999;9:R876–R878. doi: 10.1016/s0960-9822(00)80072-x. [DOI] [PubMed] [Google Scholar]
- 23.Bossuyt J, Helmstadter K, Wu X, Clements-Jewery H, Haworth RS, Avkiran M, Martin JL, Pogwizd SM, Bers DM. Ca2+/Calmodulin-Dependent Protein Kinase II{delta} and Protein Kinase D Overexpression Reinforce the Histone Deacetylase 5 Redistribution in Heart Failure. Circulation research. 2008 doi: 10.1161/CIRCRESAHA.107.169755. [DOI] [PubMed] [Google Scholar]
- 24.Bowles DK, Heaps CL, Turk JR, Maddali KK, Price EM. Hypercholesterolemia inhibits L-type calcium current in coronary macro-, not microcirculation. J Appl Physiol. 2004;96:2240–2248. doi: 10.1152/japplphysiol.01229.2003. [DOI] [PubMed] [Google Scholar]
- 25.Brenner R, Perez GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW. Vaso-regulation by the beta1 subunit of the calcium-activated potassium channel. Nature. 2000;407:870–876. doi: 10.1038/35038011. [DOI] [PubMed] [Google Scholar]
- 26.Bruce AW, Donaldson IJ, Wood IC, Yerbury SA, Sadowski MI, Chapman M, Gottgens B, Buckley NJ. Genome-wide analysis of repressor element 1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) target genes. Proc Natl Acad Sci USA. 2004;101:10458–10463. doi: 10.1073/pnas.0401827101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carafoli E. Biogenesis: plasma membrane calcium ATPase: 15 years of work on the purified enzyme. Faseb J. 1994;8:993–1002. [PubMed] [Google Scholar]
- 28.Carafoli E, Stauffer T. The plasma membrane calcium pump: functional domains, regulation of the activity, and tissue specificity of isoform expression. J Neurobiol. 1994;25:312–324. doi: 10.1002/neu.480250311. [DOI] [PubMed] [Google Scholar]
- 29.Caran N, Johnson LD, Jenkins KJ, Tombes RM. Cytosolic targeting domains of gamma and delta calmodulin-dependent protein kinase II. J Biol Chem. 2001;276:42514–42519. doi: 10.1074/jbc.M103013200. [DOI] [PubMed] [Google Scholar]
- 30.Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- 31.Chamley-Campbell J, Campbell GR, Ross R. The smooth muscle cell in culture. Physiol Rev. 1979;59:1–61. doi: 10.1152/physrev.1979.59.1.1. [DOI] [PubMed] [Google Scholar]
- 32.Cheong A, Bingham AJ, Li J, Kumar B, Sukumar P, Munsch C, Buckley NJ, Neylon CB, Porter KE, Beech DJ, Wood IC. Downregulated REST transcription factor is a switch enabling critical potassium channel expression and cell proliferation. Mol Cell. 2005;20:45–52. doi: 10.1016/j.molcel.2005.08.030. [DOI] [PubMed] [Google Scholar]
- 33.Colbran RJ, Brown AM. Calcium/calmodulin-dependent protein kinase II and synaptic plasticity. Curr Opin Neurobiol. 2004;14:318–327. doi: 10.1016/j.conb.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 34.Cribbs LL. T-type Ca2+ channels in vascular smooth muscle: multiple functions. Cell Calcium. 2006;40:221–230. doi: 10.1016/j.ceca.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 35.Dai YP, Bongalon S, Hatton WJ, Hume JR, Yamboliev IA. ClC-3 chloride channel is upregulated by hypertrophy and inflammation in rat and canine pulmonary artery. Br J Pharmacol. 2005;145:5–14. doi: 10.1038/sj.bjp.0706135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Jaegere S, Wuytack F, De Smedt H, Van den Bosch L, Casteels R. Alternative processing of the gene transcripts encoding a plasma-membrane and a sarco/endoplasmic reticulum Ca2+ pump during differentiation of BC3H1 muscle cells. Biochim Biophys Acta. 1993;1173:188–194. doi: 10.1016/0167-4781(93)90180-l. [DOI] [PubMed] [Google Scholar]
- 37.DeCoursey TE, Chandy KG, Gupta S, Cahalan MD. Voltage-gated K+ channels in human T lymphocytes: a role in mitogenesis? Nature. 1984;307:465–468. doi: 10.1038/307465a0. [DOI] [PubMed] [Google Scholar]
- 38.Dietrich A, Mederos y Schnitzler M, Emmel J, Kalwa H, Hofmann T, Gudermann T. N-linked protein glycosylation is a major determinant for basal TRPC3 and TRPC6 channel activity. J Biol Chem. 2003;278:47842–47852. doi: 10.1074/jbc.M302983200. [DOI] [PubMed] [Google Scholar]
- 39.Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 1997;386:855–858. doi: 10.1038/386855a0. [DOI] [PubMed] [Google Scholar]
- 40.Enyedi A, Verma AK, Heim R, Adamo HP, Filoteo AG, Strehler EE, Penniston JT. The Ca2+ affinity of the plasma membrane Ca2+ pump is controlled by alternative splicing. J Biol Chem. 1994;269:41–43. [PubMed] [Google Scholar]
- 41.Erondu NE, Kennedy MB. Regional distribution of type II Ca2+/calmodulin-dependent protein kinase in rat brain. J Neurosci. 1985;5:3270–3277. doi: 10.1523/JNEUROSCI.05-12-03270.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feske S, Prakriya M, Rao A, Lewis RS. A severe defect in CRAC Ca2+ channel activation and altered K+ channel gating in T cells from immunodeficient patients. J Exp Med. 2005;202:651–662. doi: 10.1084/jem.20050687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Firth AL, Remillard CV, Yuan JX. TRP channels in hypertension. Biochim Biophys Acta. 2007;1772:895–906. doi: 10.1016/j.bbadis.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Floyd R, Wray S. Calcium transporters and signalling in smooth muscles. Cell Calcium. 2007;42:467–476. doi: 10.1016/j.ceca.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 45.Geraci MW, Moore M, Gesell T, Yeager ME, Alger L, Golpon H, Gao B, Loyd JE, Tuder RM, Voelkel NF. Gene expression patterns in the lungs of patients with primary pulmonary hypertension: a gene microarray analysis. Circ Res. 2001;88:555–562. doi: 10.1161/01.res.88.6.555. [DOI] [PubMed] [Google Scholar]
- 46.Ginnan R, Pfleiderer PJ, Pumiglia K, Singer HA. PKC-delta and CaMKII-delta 2 mediate ATP-dependent activation of ERK1/2 in vascular smooth muscle. Am J Physiol Cell Physiol. 2004;286:C1281–C1289. doi: 10.1152/ajpcell.00202.2003. [DOI] [PubMed] [Google Scholar]
- 47.Ginnan R, Singer HA. CaM kinase II-dependent activation of tyrosine kinases and ERK1/2 in vascular smooth muscle. Am J Physiol Cell Physiol. 2002;282:C754–C761. doi: 10.1152/ajpcell.00335.2001. [DOI] [PubMed] [Google Scholar]
- 48.Gollasch M, Haase H, Ried C, Lindschau C, Morano I, Luft FC, Haller H. L-type calcium channel expression depends on the differentiated state of vascular smooth muscle cells. Faseb J. 1998;12:593–601. doi: 10.1096/fasebj.12.7.593. [DOI] [PubMed] [Google Scholar]
- 49.Gollasch M, Nelson MT. Voltage-dependent Ca2+ channels in arterial smooth muscle cells. Kidney Blood Press Res. 1997;20:355–371. doi: 10.1159/000174250. [DOI] [PubMed] [Google Scholar]
- 50.Golovina VA, Platoshyn O, Bailey CL, Wang J, Limsuwan A, Sweeney M, Rubin LJ, Yuan JX. Upregulated TRP and enhanced capacitative Ca(2+) entry in human pulmonary artery myocytes during proliferation. Am J Physiol. 2001;280:H746–H755. doi: 10.1152/ajpheart.2001.280.2.H746. [DOI] [PubMed] [Google Scholar]
- 51.Guan YY, Wang GL, Zhou JG. The ClC-3 Cl- channel in cell volume regulation, proliferation and apoptosis in vascular smooth muscle cells. Trends Pharmacol Sci. 2006;27:290–296. doi: 10.1016/j.tips.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 52.Guerini D. The significance of the isoforms of plasma membrane calcium ATPase. Cell Tissue Res. 1998;292:191–197. doi: 10.1007/s004410051050. [DOI] [PubMed] [Google Scholar]
- 53.Hardie RC, Minke B. Novel Ca2+ channels underlying transduction in Drosophila photoreceptors: implications for phosphoinositide-mediated Ca2+ mobilization. Trends Neurosci. 1993;16:371–376. doi: 10.1016/0166-2236(93)90095-4. [DOI] [PubMed] [Google Scholar]
- 54.Hedin U, Sjolund M, Hultgardh-Nilsson A, Thyberg J. Changes in expression and organization of smooth-muscle-specific alpha-actin during fibronectin-mediated modulation of arterial smooth muscle cell phenotype. Differentiation; research in biological diversity. 1990;44:222–231. doi: 10.1111/j.1432-0436.1990.tb00621.x. [DOI] [PubMed] [Google Scholar]
- 55.Herrera GM, Nelson MT. Sarcoplasmic reticulum and membrane currents. Novartis Found Symp. 2002;246:189–203. discussion 203–187, 221–187. [PubMed] [Google Scholar]
- 56.Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- 57.Hofmann T, Schaefer M, Schultz G, Gudermann T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci USA. 2002;99:7461–7466. doi: 10.1073/pnas.102596199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.House SJ, Ginnan RG, Armstrong SE, Singer HA. Calcium/calmodulin-dependent protein kinase II-delta isoform regulation of vascular smooth muscle cell proliferation. Am J Physiol Cell Physiol. 2007;292:C2276–C2287. doi: 10.1152/ajpcell.00606.2006. [DOI] [PubMed] [Google Scholar]
- 59.House SJ, Singer HA. CaMKII-{delta} Isoform Regulation of Neointima Formation After Vascular Injury. Arterioscler Thromb Vasc Biol. 2008;28:441. doi: 10.1161/ATVBAHA.107.156810. [DOI] [PubMed] [Google Scholar]
- 60.Hudmon A, Schulman H. Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem J. 2002;364:593–611. doi: 10.1042/BJ20020228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Husain M, Bein K, Jiang L, Alper SL, Simons M, Rosenberg RD. c-Myb-dependent cell cycle progression and Ca2+ storage in cultured vascular smooth muscle cells. Circ Res. 1997;80:617–626. doi: 10.1161/01.res.80.5.617. [DOI] [PubMed] [Google Scholar]
- 62.Husain M, Jiang L, See V, Bein K, Simons M, Alper SL, Rosenberg RD. Regulation of vascular smooth muscle cell proliferation by plasma membrane Ca(2+)-ATPase. Am J Physiol. 1997;272:C1947–C1959. doi: 10.1152/ajpcell.1997.272.6.C1947. [DOI] [PubMed] [Google Scholar]
- 63.Ihara E, Hirano K, Hirano M, Nishimura J, Nawata H, Kanaide H. Mechanism of down-regulation of L-type Ca(2+) channel in the proliferating smooth muscle cells of rat aorta. J Cell Biochem. 2002;87:242–251. doi: 10.1002/jcb.10295. [DOI] [PubMed] [Google Scholar]
- 64.Inoue R, Jensen LJ, Shi J, Morita H, Nishida M, Honda A, Ito Y. Transient receptor potential channels in cardiovascular function and disease. Circ Res. 2006;99:119–131. doi: 10.1161/01.RES.0000233356.10630.8a. [DOI] [PubMed] [Google Scholar]
- 65.Jaggar JH, Porter VA, Lederer WJ, Nelson MT. Calcium sparks in smooth muscle. Am J Physiol Cell Physiol. 2000;278:C235–C256. doi: 10.1152/ajpcell.2000.278.2.C235. [DOI] [PubMed] [Google Scholar]
- 66.Jaggar JH, Wellman GC, Heppner TJ, Porter VA, Perez GJ, Gollasch M, Kleppisch T, Rubart M, Stevenson AS, Lederer WJ, Knot HJ, Bonev AD, Nelson MT. Ca2+ channels, ryanodine receptors and Ca(2+)-activated K+ channels: a functional unit for regulating arterial tone. Acta Physiol Scand. 1998;164:577–587. doi: 10.1046/j.1365-201X.1998.00462.x. [DOI] [PubMed] [Google Scholar]
- 67.Jentsch TJ, Stein V, Weinreich F, Zdebik AA. Molecular structure and physiological function of chloride channels. Physiol Rev. 2002;82:503–568. doi: 10.1152/physrev.00029.2001. [DOI] [PubMed] [Google Scholar]
- 68.Kamishima T, Quayle JM. Ca2+-induced Ca2+ release in cardiac and smooth muscle cells. Biochem Soc Trans. 2003;31:943–946. doi: 10.1042/bst0310943. [DOI] [PubMed] [Google Scholar]
- 69.Karkanis T, Li S, Pickering JG, Sims SM. Plasticity of KIR channels in human smooth muscle cells from internal thoracic artery. Am J Physiol. 2003;284:H2325–H2334. doi: 10.1152/ajpheart.00559.2002. [DOI] [PubMed] [Google Scholar]
- 70.Kawai-Kowase K, Owens GK. Multiple repressor pathways contribute to phenotypic switching of vascular smooth muscle cells. Am J Physiol Cell Physiol. 2007;292:C59–C69. doi: 10.1152/ajpcell.00394.2006. [DOI] [PubMed] [Google Scholar]
- 71.Kim I, Je HD, Gallant C, Zhan Q, Riper DV, Badwey JA, Singer HA, Morgan KG. Ca2+-calmodulin-dependent protein kinase II-dependent activation of contractility in ferret aorta. J Physiol. 2000;526(Pt 2):367–374. doi: 10.1111/j.1469-7793.2000.00367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kohler R, Wulff H, Eichler I, Kneifel M, Neumann D, Knorr A, Grgic I, Kampfe D, Si H, Wibawa J, Real R, Borner K, Brakemeier S, Orzechowski HD, Reusch HP, Paul M, Chandy KG, Hoyer J. Blockade of the intermediate-conductance calcium-activated potassium channel as a new therapeutic strategy for restenosis. Circulation. 2003;108:1119–1125. doi: 10.1161/01.CIR.0000086464.04719.DD. [DOI] [PubMed] [Google Scholar]
- 73.Kuga T, Kobayashi S, Hirakawa Y, Kanaide H, Takeshita A. Cell cycle–dependent expression of L- and T-type Ca2+ currents in rat aortic smooth muscle cells in primary culture. Circ Res. 1996;79:14–19. doi: 10.1161/01.res.79.1.14. [DOI] [PubMed] [Google Scholar]
- 74.Kumar B, Dreja K, Shah SS, Cheong A, Xu SZ, Sukumar P, Naylor J, Forte A, Cipollaro M, McHugh D, Kingston PA, Heagerty AM, Munsch CM, Bergdahl A, Hultgardh-Nilsson A, Gomez MF, Porter KE, Hellstrand P, Beech DJ. Upregulated TRPC1 channel in vascular injury in vivo and its role in human neointimal hyperplasia. Circ Res. 2006;98:557–563. doi: 10.1161/01.RES.0000204724.29685.db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kuwahara K, Wang Y, McAnally J, Richardson JA, Bassel-Duby R, Hill JA, Olson EN. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest. 2006;116:3114–3126. doi: 10.1172/JCI27702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ledoux J, Werner ME, Brayden JE, Nelson MT. Calcium-activated potassium channels and the regulation of vascular tone. Physiology (Bethesda, Md) 2006;21:69–78. doi: 10.1152/physiol.00040.2005. [DOI] [PubMed] [Google Scholar]
- 77.Lee CH, Poburko D, Kuo KH, Seow CY, van Breemen C. Ca(2+) oscillations, gradients, and homeostasis in vascular smooth muscle. Am J Physiol. 2002;282:H1571–H1583. doi: 10.1152/ajpheart.01035.2001. [DOI] [PubMed] [Google Scholar]
- 78.Lee DL, Wamhoff BR, Katwa LC, Reddy HK, Voelker DJ, Dixon JL, Sturek M. Increased endothelin-induced Ca2+ signaling, tyrosine phosphorylation, and coronary artery disease in diabetic dyslipidemic Swine are prevented by atorvastatin. J Pharm Exp Ther. 2003;306:132–140. doi: 10.1124/jpet.103.049577. [DOI] [PubMed] [Google Scholar]
- 79.Li S, Westwick J, Cox B, Poll CT. TRP channels as drug targets. Novartis Found Symp. 2004;258:204–213. discussion 213–221, 263–206. [PubMed] [Google Scholar]
- 80.Liao Y, Erxleben C, Yildirim E, Abramowitz J, Armstrong DL, Birnbaumer L. Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc Natl Acad Sci USA. 2007;104:4682–4687. doi: 10.1073/pnas.0611692104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lipskaia L, del Monte F, Capiod T, Yacoubi S, Hadri L, Hours M, Hajjar RJ, Lompre AM. Sarco/endoplasmic reticulum Ca2+-ATPase gene transfer reduces vascular smooth muscle cell proliferation and neointima formation in the rat. Circ Res. 2005;97:488–495. doi: 10.1161/01.RES.0000180663.42594.aa. [DOI] [PubMed] [Google Scholar]
- 83.Lipskaia L, Lompre AM. Alteration in temporal kinetics of Ca2+ signaling and control of growth and proliferation. Biology of the cell/under the auspices of the European Cell Biology Organization. 2004;96:55–68. doi: 10.1016/j.biolcel.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 84.Liu D, Scholze A, Zhu Z, Kreutz R, Wehland-von-Trebra M, Zidek W, Tepel M. Increased transient receptor potential channel TRPC3 expression in spontaneously hypertensive rats. Am J Hypertens. 2005;18:1503–1507. doi: 10.1016/j.amjhyper.2005.05.033. [DOI] [PubMed] [Google Scholar]
- 85.Liu LH, Boivin GP, Prasad V, Periasamy M, Shull GE. Squamous cell tumors in mice heterozygous for a null allele of Atp2a2, encoding the sarco(endo)plasmic reticulum Ca2+-ATPase isoform 2 Ca2+ pump. J Biol Chem. 2001;276:26737–26740. doi: 10.1074/jbc.C100275200. [DOI] [PubMed] [Google Scholar]
- 86.MacLennan DH, Asahi M, Tupling AR. The regulation of SERCA-type pumps by phospholamban and sarcolipin. Ann NY Acad Sci. 2003;986:472–480. doi: 10.1111/j.1749-6632.2003.tb07231.x. [DOI] [PubMed] [Google Scholar]
- 87.Maier LS, Zhang T, Chen L, DeSantiago J, Brown JH, Bers DM. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ Res. 2003;92:904–911. doi: 10.1161/01.RES.0000069685.20258.F1. [DOI] [PubMed] [Google Scholar]
- 88.Marganski WA, Gangopadhyay SS, Je HD, Gallant C, Morgan KG. Targeting of a novel Ca+2/calmodulin-dependent protein kinase II is essential for extracellular signal-regulated kinase-mediated signaling in differentiated smooth muscle cells. Circ Res. 2005;97:541–549. doi: 10.1161/01.RES.0000182630.29093.0d. [DOI] [PubMed] [Google Scholar]
- 89.Massaeli H, Austria JA, Pierce GN. Lesions in ryanodine channels in smooth muscle cells exposed to oxidized low density lipoprotein. Arterioscler Thromb Vasc Biol. 2000;20:328–334. doi: 10.1161/01.atv.20.2.328. [DOI] [PubMed] [Google Scholar]
- 90.McDonald OG, Owens GK. Programming smooth muscle plasticity with chromatin dynamics. Circ Res. 2007;100:1428–1441. doi: 10.1161/01.RES.0000266448.30370.a0. [DOI] [PubMed] [Google Scholar]
- 91.McDonald TF, Pelzer S, Trautwein W, Pelzer DJ. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiol Rev. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- 92.Miguel-Velado E, Moreno-Dominguez A, Colinas O, Cidad P, Heras M, Perez-Garcia MT, Lopez-Lopez JR. Contribution of Kv channels to phenotypic remodeling of human uterine artery smooth muscle cells. Circ Res. 2005;97:1280–1287. doi: 10.1161/01.RES.0000194322.91255.13. [DOI] [PubMed] [Google Scholar]
- 93.Monteith GR, McAndrew D, Faddy HM, Roberts-Thomson SJ. Calcium and cancer: targeting Ca2+ transport. Nat Rev Cancer. 2007;7:519–530. doi: 10.1038/nrc2171. [DOI] [PubMed] [Google Scholar]
- 94.Muller OJ, Lange M, Rattunde H, Lorenzen HP, Muller M, Frey N, Bittner C, Simonides W, Katus HA, Franz WM. Transgenic rat hearts overexpressing SERCA2a show improved contractility under baseline conditions and pressure overload. Cardiovasc Res. 2003;59:380–389. doi: 10.1016/s0008-6363(03)00429-2. [DOI] [PubMed] [Google Scholar]
- 95.Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- 96.Neylon CB, Lang RJ, Fu Y, Bobik A, Reinhart PH. Molecular cloning and characterization of the intermediate-conductance Ca(2+)-activated K(+) channel in vascular smooth muscle: relationship between K(Ca) channel diversity and smooth muscle cell function. Circ Res. 1999;85:e33–e43. doi: 10.1161/01.res.85.9.e33. [DOI] [PubMed] [Google Scholar]
- 97.Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev. 2007;87:165–217. doi: 10.1152/physrev.00021.2006. [DOI] [PubMed] [Google Scholar]
- 98.Nilius B, Voets T. TRP channels: a TR(I)P through a world of multifunctional cation channels. Pflugers Arch. 2005;451:1–10. doi: 10.1007/s00424-005-1462-y. [DOI] [PubMed] [Google Scholar]
- 99.Okada T, Inoue R, Yamazaki K, Maeda A, Kurosaki T, Yamakuni T, Tanaka I, Shimizu S, Ikenaka K, Imoto K, Mori Y. Molecular and functional characterization of a novel mouse transient receptor potential protein homologue TRP7. Ca (2+)-permeable cation channel that is constitutively activated and enhanced by stimulation of G protein-coupled receptor. J Biol Chem. 1999;274:27359–27370. doi: 10.1074/jbc.274.39.27359. [DOI] [PubMed] [Google Scholar]
- 100.Ong HL, Cheng KT, Liu X, Bandyopadhyay BC, Paria BC, Soboloff J, Pani B, Gwack Y, Srikanth S, Singh BB, Gill D, Ambudkar IS. Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J Biol Chem. 2007;282:9105–9116. doi: 10.1074/jbc.M608942200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiol Rev. 1995;75:487–517. doi: 10.1152/physrev.1995.75.3.487. [DOI] [PubMed] [Google Scholar]
- 102.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 103.Pardo LA. Voltage-gated potassium channels in cell proliferation. Physiology (Bethesda, Md) 2004;19:285–292. doi: 10.1152/physiol.00011.2004. [DOI] [PubMed] [Google Scholar]
- 104.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 105.Pauly RR, Bilato C, Sollott SJ, Monticone R, Kelly PT, Lakatta EG, Crow MT. Role of calcium/calmodulin-dependent protein kinase II in the regulation of vascular smooth muscle cell migration. Circulation. 1995;91:1107–1115. doi: 10.1161/01.cir.91.4.1107. [DOI] [PubMed] [Google Scholar]
- 106.Periasamy M, Kalyanasundaram A. SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve. 2007;35:430–442. doi: 10.1002/mus.20745. [DOI] [PubMed] [Google Scholar]
- 107.Periasamy M, Reed TD, Liu LH, Ji Y, Loukianov E, Paul RJ, Nieman ML, Riddle T, Duffy JJ, Doetschman T, Lorenz JN, Shull GE. Impaired cardiac performance in heterozygous mice with a null mutation in the sarco(endo)plasmic reticulum Ca2+-ATPase isoform 2 (SERCA2) gene. J Biol Chem. 1999;274:2556–2562. doi: 10.1074/jbc.274.4.2556. [DOI] [PubMed] [Google Scholar]
- 108.Pfleiderer PJ, Lu KK, Crow MT, Keller RS, Singer HA. Modulation of vascular smooth muscle cell migration by calcium/ calmodulin-dependent protein kinase II-delta 2. Am J Physiol Cell Physiol. 2004;286:C1238–C1245. doi: 10.1152/ajpcell.00536.2003. [DOI] [PubMed] [Google Scholar]
- 109.Platoshyn O, Remillard CV, Fantozzi I, Sison T, Yuan JX. Identification of functional voltage-gated Na(+) channels in cultured human pulmonary artery smooth muscle cells. Pflugers Arch. 2005;451:380–387. doi: 10.1007/s00424-005-1478-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pratt PF, Bonnet S, Ludwig LM, Bonnet P, Rusch NJ. Upregulation of L-type Ca2+ channels in mesenteric and skeletal arteries of SHR. Hypertension. 2002;40:214–219. doi: 10.1161/01.hyp.0000025877.23309.36. [DOI] [PubMed] [Google Scholar]
- 111.Preiano BS, Guerini D, Carafoli E. Expression and functional characterization of isoforms 4 of the plasma membrane calcium pump. Biochemistry. 1996;35:7946–7953. doi: 10.1021/bi9527404. [DOI] [PubMed] [Google Scholar]
- 112.Putney JW. Physiological mechanisms of TRPC activation. Pflugers Arch. 2005;451:29–34. doi: 10.1007/s00424-005-1416-4. [DOI] [PubMed] [Google Scholar]
- 113.Putney JW., Jr A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 114.Quignard JF, Harricane MC, Menard C, Lory P, Nargeot J, Capron L, Mornet D, Richard S. Transient down-regulation of L-type Ca(2+) channel and dystrophin expression after balloon injury in rat aortic cells. Cardiovasc Res. 2001;49:177–188. doi: 10.1016/s0008-6363(00)00210-8. [DOI] [PubMed] [Google Scholar]
- 115.Quignard JF, Ryckwaert F, Albat B, Nargeot J, Richard S. A novel tetrodotoxin-sensitive Na+ current in cultured human coronary myocytes. Circ Res. 1997;80:377–382. [PubMed] [Google Scholar]
- 116.Ramirez MT, Zhao XL, Schulman H, Brown JH. The nuclear deltaB isoform of Ca2+/calmodulin-dependent protein kinase II regulates atrial natriuretic factor gene expression in ventricular myocytes. J Biol Chem. 1997;272:31203–31208. doi: 10.1074/jbc.272.49.31203. [DOI] [PubMed] [Google Scholar]
- 117.Roderick HL, Berridge MJ, Bootman MD. Calcium-induced calcium release. Curr Biol. 2003;13:R425. doi: 10.1016/s0960-9822(03)00358-0. [DOI] [PubMed] [Google Scholar]
- 118.Rodman DM, Harral J, Wu S, West J, Hoedt-Miller M, Reese KA, Fagan K. The low-voltage-activated calcium channel CAV3.1 controls proliferation of human pulmonary artery myocytes. Chest. 2005;128:581S–582S. doi: 10.1378/chest.128.6_suppl.581S. [DOI] [PubMed] [Google Scholar]
- 119.Rodman DM, Reese K, Harral J, Fouty B, Wu S, West J, Hoedt-Miller M, Tada Y, Li KX, Cool C, Fagan K, Cribbs L. Low-voltage-activated (T-type) calcium channels control proliferation of human pulmonary artery myocytes. Circ Res. 2005;96:864–872. doi: 10.1161/01.RES.0000163066.07472.ff. [DOI] [PubMed] [Google Scholar]
- 120.Rokolya A, Singer HA. Inhibition of CaM kinase II activation and force maintenance by KN-93 in arterial smooth muscle. Am J Physiol Cell Physiol. 2000;278:C537–C545. doi: 10.1152/ajpcell.2000.278.3.C537. [DOI] [PubMed] [Google Scholar]
- 121.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rossi D, Sorrentino V. Molecular genetics of ryanodine receptors Ca2+-release channels. Cell Calcium. 2002;32:307–319. doi: 10.1016/s0143416002001987. [DOI] [PubMed] [Google Scholar]
- 123.Schwartz SM, Campbell GR, Campbell JH. Replication of smooth muscle cells in vascular disease. Circ Res. 1986;58:427–444. doi: 10.1161/01.res.58.4.427. [DOI] [PubMed] [Google Scholar]
- 124.Schworer CM, Rothblum LI, Thekkumkara TJ, Singer HA. Identification of novel isoforms of the delta subunit of Ca2+/calmodulin-dependent protein kinase II. Differential expression in rat brain and aorta. J Biol Chem. 1993;268:14443–14449. [PubMed] [Google Scholar]
- 125.Singer HA, Benscoter HA, Schworer CM. Novel Ca2+/calmodulin-dependent protein kinase II gamma-subunit variants expressed in vascular smooth muscle, brain, and cardiomyocytes. J Biol Chem. 1997;272:9393–9400. doi: 10.1074/jbc.272.14.9393. [DOI] [PubMed] [Google Scholar]
- 126.Singla SI, Hudmon A, Goldberg JM, Smith JL, Schulman H. Molecular characterization of calmodulin trapping by calcium/calmodulin-dependent protein kinase II. J Biol Chem. 2001;276:29353–29360. doi: 10.1074/jbc.M101744200. [DOI] [PubMed] [Google Scholar]
- 127.Smyth JT, Lemonnier L, Vazquez G, Bird GS, Putney JW., Jr Dissociation of regulated trafficking of TRPC3 channels to the plasma membrane from their activation by phospholipase C. J Biol Chem. 2006;281:11712–11720. doi: 10.1074/jbc.M510541200. [DOI] [PubMed] [Google Scholar]
- 128.Soboloff J, Spassova M, Xu W, He LP, Cuesta N, Gill DL. Role of endogenous TRPC6 channels in Ca2+ signal generation in A7r5 smooth muscle cells. J Biol Chem. 2005;280:39786–39794. doi: 10.1074/jbc.M506064200. [DOI] [PubMed] [Google Scholar]
- 129.Soderling TR, Chang B, Brickey D. Cellular signaling through multifunctional Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 2001;276:3719–3722. doi: 10.1074/jbc.R000013200. [DOI] [PubMed] [Google Scholar]
- 130.Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–236. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- 131.Strehler EE, Caride AJ, Filoteo AG, Xiong Y, Penniston JT, Enyedi A. Plasma membrane Ca2+ ATPases as dynamic regulators of cellular calcium handling. Ann NY Acad Sci. 2007;1099:226–236. doi: 10.1196/annals.1387.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tansey MG, Word RA, Hidaka H, Singer HA, Schworer CM, Kamm KE, Stull JT. Phosphorylation of myosin light chain kinase by the multifunctional calmodulin-dependent protein kinase II in smooth muscle cells. J Biol Chem. 1992;267:12511–12516. [PubMed] [Google Scholar]
- 133.Tiwari S, Zhang Y, Heller J, Abernethy DR, Soldatov NM. Atherosclerosis-related molecular alteration of the human CaV1.2 calcium channel alpha1C subunit. Proc Natl Acad Sci USA. 2006;103:17024–17029. doi: 10.1073/pnas.0606539103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tobimatsu T, Fujisawa H. Tissue-specific expression of four types of rat calmodulin-dependent protein kinase II mRNAs. J Biol Chem. 1989;264:17907–17912. [PubMed] [Google Scholar]
- 135.Tombes RM, Faison MO, Turbeville JM. Organization and evolution of multifunctional Ca(2+)/CaM-dependent protein kinase genes. Gene. 2003;322:17–31. doi: 10.1016/j.gene.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 136.Trebak M. Canonical transient receptor potential channels in disease: targets for novel drug therapy? Drug Discov Today. 2006;11:924–930. doi: 10.1016/j.drudis.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 137.Trebak M, Bird GS, McKay RR, Putney JW., Jr Comparison of human TRPC3 channels in receptor-activated and store-operated modes. Differential sensitivity to channel blockers suggests fundamental differences in channel composition. J Biol Chem. 2002;277:21617–21623. doi: 10.1074/jbc.M202549200. [DOI] [PubMed] [Google Scholar]
- 138.Trebak M, Lemonnier L, Smyth JT, Vazquez G, Putney JW., Jr Phospholipase C-coupled receptors and activation of TRPC channels. Handb Exp Pharmacol. 2007:593–614. doi: 10.1007/978-3-540-34891-7_35. [DOI] [PubMed] [Google Scholar]
- 139.Trebak M, St JBG, McKay RR, Birnbaumer L, Putney JW., Jr Signaling mechanism for receptor-activated canonical transient receptor potential 3 (TRPC3) channels. J Biol Chem. 2003;278:16244–16252. doi: 10.1074/jbc.M300544200. [DOI] [PubMed] [Google Scholar]
- 140.Trebak M, Vazquez G, Bird GS, Putney JW., Jr The TRPC3/6/7 subfamily of cation channels. Cell Calcium. 2003;33:451–461. doi: 10.1016/s0143-4160(03)00056-3. [DOI] [PubMed] [Google Scholar]
- 141.Vallot O, Combettes L, Jourdon P, Inamo J, Marty I, Claret M, Lompre AM. Intracellular Ca(2+) handling in vascular smooth muscle cells is affected by proliferation. Arterioscler Thromb Vasc Biol. 2000;20:1225–1235. doi: 10.1161/01.atv.20.5.1225. [DOI] [PubMed] [Google Scholar]
- 142.Van Assche T, Fransen P, Guns PJ, Herman AG, Bult H. Altered Ca2+ handling of smooth muscle cells in aorta of apolipoprotein E-deficient mice before development of atherosclerotic lesions. Cell Calcium. 2007;41:295–302. doi: 10.1016/j.ceca.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 143.Vazquez G, Wedel BJ, Aziz O, Trebak M, Putney JW., Jr The mammalian TRPC cation channels. Biochim Biophys Acta. 2004;1742:21–36. doi: 10.1016/j.bbamcr.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 144.Ver Heyen M, Heymans S, Antoons G, Reed T, Periasamy M, Awede B, Lebacq J, Vangheluwe P, Dewerchin M, Collen D, Sipido K, Carmeliet P, Wuytack F. Replacement of the muscle-specific sarcoplasmic reticulum Ca(2+)-ATPase isoform SERCA2a by the nonmuscle SERCA2b homologue causes mild concentric hypertrophy and impairs contraction-relaxation of the heart. Circ Res. 2001;89:838–846. doi: 10.1161/hh2101.098466. [DOI] [PubMed] [Google Scholar]
- 145.Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, Kinet JP. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wamhoff BR, Bowles DK, McDonald OG, Sinha S, Somlyo AP, Somlyo AV, Owens GK. L-type voltage-gated Ca2+ channels modulate expression of smooth muscle differentiation marker genes via a rho kinase/myocardin/SRF-dependent mechanism. Circ Res. 2004;95:406–414. doi: 10.1161/01.RES.0000138582.36921.9e. [DOI] [PubMed] [Google Scholar]
- 147.Wamhoff BR, Bowles DK, Owens GK. Excitation-transcription coupling in arterial smooth muscle. Circ Res. 2006;98:868–878. doi: 10.1161/01.RES.0000216596.73005.3c. [DOI] [PubMed] [Google Scholar]
- 148.Wang GL, Wang XR, Lin MJ, He H, Lan XJ, Guan YY. Deficiency in ClC-3 chloride channels prevents rat aortic smooth muscle cell proliferation. Circ Res. 2002;91:E28–E32. doi: 10.1161/01.res.0000042062.69653.e4. [DOI] [PubMed] [Google Scholar]
- 149.Wang J, Shimoda LA, Weigand L, Wang W, Sun D, Sylvester JT. Acute hypoxia increases intracellular [Ca2+] in pulmonary arterial smooth muscle by enhancing capacitative Ca2+ entry. Am J Physiol Lung Cell Mol Physiol. 2005;288:L1059–L1069. doi: 10.1152/ajplung.00448.2004. [DOI] [PubMed] [Google Scholar]
- 150.Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL, Shimoda LA. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res. 2006;98:1528–1537. doi: 10.1161/01.RES.0000227551.68124.98. [DOI] [PubMed] [Google Scholar]
- 151.Wankerl M, Schwartz K. Calcium transport proteins in the nonfailing and failing heart: gene expression and function. J Mol Med (Berlin, Germany) 1995;73:487–496. doi: 10.1007/BF00198900. [DOI] [PubMed] [Google Scholar]
- 152.Wellman GC, Nelson MT. Signaling between SR and plasmalemma in smooth muscle: sparks and the activation of Ca2+-sensitive ion channels. Cell Calcium. 2003;34:211–229. doi: 10.1016/s0143-4160(03)00124-6. [DOI] [PubMed] [Google Scholar]
- 153.Welsh DG, Morielli AD, Nelson MT, Brayden JE. Transient receptor potential channels regulate myogenic tone of resistance arteries. Circ Res. 2002;90:248–250. doi: 10.1161/hh0302.105662. [DOI] [PubMed] [Google Scholar]
- 154.Wilkerson MK, Heppner TJ, Bonev AD, Nelson MT. Inositol trisphosphate receptor calcium release is required for cerebral artery smooth muscle cell proliferation. Am J Physiol. 2006;290:H240–H247. doi: 10.1152/ajpheart.01191.2004. [DOI] [PubMed] [Google Scholar]
- 155.Wray S, Burdyga T, Noble K. Calcium signalling in smooth muscle. Cell Calcium. 2005;38:397–407. doi: 10.1016/j.ceca.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 156.Xiao GN, Guan YY, He H. Effects of Cl- channel blockers on endothelin-1-induced proliferation of rat vascular smooth muscle cells. Life Sci. 2002;70:2233–2241. doi: 10.1016/s0024-3205(02)01508-4. [DOI] [PubMed] [Google Scholar]
- 157.Yoshida T, Owens GK. Molecular determinants of vascular smooth muscle cell diversity. Circ Res. 2005;96:280–291. doi: 10.1161/01.RES.0000155951.62152.2e. [DOI] [PubMed] [Google Scholar]
- 158.Yu Y, Fantozzi I, Remillard CV, Landsberg JW, Kunichika N, Platoshyn O, Tigno DD, Thistlethwaite PA, Rubin LJ, Yuan JX. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci USA. 2004;101:13861–13866. doi: 10.1073/pnas.0405908101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Yu Y, Platoshyn O, Zhang J, Krick S, Zhao Y, Rubin LJ, Rothman A, Yuan JX. c-Jun decreases voltage-gated K(+) channel activity in pulmonary artery smooth muscle cells. Circulation. 2001;104:1557–1563. doi: 10.1161/hc3801.095662. [DOI] [PubMed] [Google Scholar]
- 160.Yu Y, Sweeney M, Zhang S, Platoshyn O, Landsberg J, Rothman A, Yuan JX. PDGF stimulates pulmonary vascular smooth muscle cell proliferation by upregulating TRPC6 expression. Am J Physiol Cell Physiol. 2003;284:C316–C330. doi: 10.1152/ajpcell.00125.2002. [DOI] [PubMed] [Google Scholar]
- 161.Zhang S, Dong H, Rubin LJ, Yuan JX. Upregulation of Na+/Ca2+ exchanger contributes to the enhanced Ca2+ entry in pulmonary artery smooth muscle cells from patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol. 2007;292:C2297–C2305. doi: 10.1152/ajpcell.00383.2006. [DOI] [PubMed] [Google Scholar]
- 162.Zhang S, Remillard CV, Fantozzi I, Yuan JX. ATP-induced mitogenesis is mediated by cyclic AMP response element-binding protein-enhanced TRPC4 expression and activity in human pulmonary artery smooth muscle cells. Am J Physiol Cell Physiol. 2004;287:C1192–C1201. doi: 10.1152/ajpcell.00158.2004. [DOI] [PubMed] [Google Scholar]
- 163.Zhang S, Yuan JX, Barrett KE, Dong H. Role of Na+/Ca2+ exchange in regulating cytosolic Ca2+ in cultured human pulmonary artery smooth muscle cells. Am J Physiol Cell Physiol. 2005;288:C245–C252. doi: 10.1152/ajpcell.00411.2004. [DOI] [PubMed] [Google Scholar]
- 164.Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc Natl Acad Sci USA. 2006;103:9357–9362. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zhang T, Brown JH. Role of Ca2+/calmodulin-dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovasc Res. 2004;63:476–486. doi: 10.1016/j.cardiores.2004.04.026. [DOI] [PubMed] [Google Scholar]