

Abstract
We report synthesis of temperature- and redox-responsive multiblock copolymers by reversible addition-fragmentation chain transfer (RAFT) polymerization. Well-defined α,ω-bis(dithioester)-functionalized poly(N-isopropylacrylamide) (PNIPAM) and poly(2-(dimethylamino) ethyl methacrylate) (PDMAEMA) were prepared using 1,4-bis(thiobenzoylthiomethyl)benzene and 1,4-bis(2-(thiobenzoylthio)prop-2-yl)benzene as RAFT agents, respectively. Dually responsive multiblock copolymers were synthesized in a single aminolysis/oxidation step from the α,ω-bis(dithioester)-terminated PNIPAM and PDMAEMA. The copolymers and their stimulus-responsive behavior were characterized by size exclusion chromatography, NMR, light scattering and atomic force microscopy. Due to the presence of redox-sensitive disulfide bonds between the blocks, the copolymers were readily reduced to the starting polymer blocks. The presence of temperature-responsive PNIPAM blocks provided the copolymers with the ability to assemble into core-shell nanostructures with hydrophobic PNIPAM as a core and cationic PDMAEMA as stabilizing shell when above the phase transition temperatures of PNIPAM. The temperature-induced assembly of the copolymers also showed substantial pH sensitivity. The phase transition temperature increased with decreasing pH, while molecular weight of the assemblies decreased.
Keywords: RAFT polymerization, reducible multiblock copolymer, 2-(dimethylamino)ethyl methacrylate, N-isopropylacrylamide
INTRODUCTION
Multiblock copolymers consist of two or more different blocks of monomers arranged in a random or alternating sequence. The interest, both theoretical and experimental, in multiblock copolymers stems from their widespread applications as adhesives, emulsifiers, barrier materials, impact modifiers, and materials for gene and drug delivery.1–10 Moreover, the experimental and theoretical studies of these systems show that multiblock copolymers often can provide more advantageous properties compared with corresponding diblock or triblock copolymers.2, 5 In particular, multiblock copolymers with combined responsiveness to different biologically relevant stimuli represent promising materials for a variety of biomedical applications. Hence, expanding the portfolio of easy methods for the synthesis of functional multiblock copolymers is of significant interest.
A general approach to synthesize multiblock copolymers is to prepare α,ω-bifunctional prepolymer and condense it with another α,ω-bifunctional prepolymer.11–18 For example, α,ω-dihalo-poly(methylphenylsilane) (PMPS) was condensed with bis-hydroxy-terminated poly(ethylene oxide) (PEO) to yield a multiblock copolymer of PMPS and PEO.11 Condensation of α,ω-dihydroxy-poly(2-vinylpyridine) (P2VP) with α,ω-dihydroxy-PEO in the presence of potassium hydroxide yielded multiblock copolymer of P2VP and PEO.18 Condensation of α,ω-diamino-polyamide with α,ω-dicarboxy-polyester led to a corresponding polyamide-polyester multiblock copolymers.12 Polymerization of bischloroformates of carboxylated polylactide (PLA) with poly(ε-caprolactone) (PCL) produced multiblock copolymers of PLA and PCL.16 Copolymerization of fluoro-terminated poly(2,5-benzophenone) and phenoxide-terminated disulfonated poly(arylene ether sulfone) formed an amphiphilic multiblock copolymer via aromatic nucleophilic substitution.10 Oxidation of thiol-terminated poly(ethylene oxide-b-propylene oxide-b-ethylene oxide) furnished thermogelling poly(ethylene oxide-b-propylene oxide-b-ethylene oxide) multiblock copolymers.19
Alternatively, multiblock copolymers can be prepared by many of the available living polymerization techniques. For example, Higaki et al. synthesized a polyurethane macroinitiator containing alkoxyamine initiating groups in the main chain and used it to synthesize multiblock copolymers of polystyrene and poly(p-tert-butoxystyrene) by nitroxide-mediated free radical polymerization.4 In another recent report, multiblock copolymers could be easily prepared by a stepwise insertion reaction of monomers into the backbone of trithiocarbonate- or dithioester-containing polymers.20 Huang et al. reported synthesis of novel ABCB-type ternary amphiphilic multiblock copolymers by stepwise insertion of monomers into the trithiocarbonate-functionalized PEO.21 Koning et al. have reported that multiblock copolymers can be synthesized by emulsion polymerization or aqueous dispersion polymerization using multifunctional RAFT agent.8, 22 Atom transfer radical polymerization and cationic polymerization have been used to prepare multiblock copolymers of methyl methacrylate, styrene, and isobutylene in a three-step synthesis.23
Redox-responsive multiblock copolymers have been proposed as promising carriers in drug and gene delivery.24–26 They utilize the redox potential gradients that exist between intracellular and extracellular environment for improved efficiency of delivery. Although the above methods provide robust tools to synthesize different types of multiblock copolymers and have been used recently to synthesize several types of reducible disulfide-containing copolymers,27–30 multiblock copolymers that respond dually to redox potential gradients and another stimulus have been seldom studied until now.
Reversible addition fragmentation chain transfer (RAFT) polymerization is an extremely versatile, controlled free radical polymerization technique for the synthesis of well-defined polymer architectures.31–36 RAFT process is applicable to a wide range of monomers and can be performed under a broad range of conditions.33, 37–39 The use of difunctional RAFT agents provides an easy route to homobifunctional dithio-polymers.31, 36, 40–42 Previously, we used oxidation of thiol-terminated peptides and polymers prepared by RAFT polymerization to form disulfide-containing reducible polymers, which could be reversibly cleaved using mild reducing agents.26, 43, 44 Here, we report an easy method to synthesize redox-responsive multiblock copolymers based on temperature-responsive N-isopropylacrylamide (NIPAM) polymer blocks and cationic 2-(dimethylamino)ethyl methacrylate (DMAEMA) polymer blocks. Different from the previously reported method of preparing reducible polymer via RAFT,40, 42, 44 the aminolysis of α,ω-dithioester-functionalized polymer and oxidization reaction were carried out in a single step without the need to isolate the α,ω-dithio-functionalized polymers. The synthesized multiblock copolymers are reduced to the original building blocks, exhibit temperature and pH dependent phase transition, and form core-shell nanoparticles with the cationic blocks as shells. These copolymers show promise as gene delivery carriers with combined responsiveness to redox potential gradients, pH and temperature.
EXPERIMENTAL SECTION
Materials
2,2′-Azobis(2-methylpropionitrile) (AIBN, Sigma-Aldrich, 98%), hexylamine (Sigma-Aldrich, 99%), 1,4-diisopropenylbenzene (TCI), sulfur (EMD, 99%), anhydrous methanol (EMD, 99.8%), sodium methoxide (Alfa, 25~30%), benzyl chloride (Alfa, 99%), carbon tetrachloride (Sigma-Aldrich, 99%), tetrahydrofuran (VWR, 99%), anhydrous diethyl ether (EMD, 99%), hexane (EMD, 98.5%), dithiothreitol (DTT, Sigma-Aldrich, >99%), phenylmagnesium bromide 1 M in THF (Sigma-Aldrich), dibromo-p-xylene (Fluka, >98%) were used as received. DMAEMA (Acros, 99%) was passed through a columnof activated basic alumina to remove the inhibitor and stored at 0 °C before use. NIPAM was recrystallized before use from a mixture of hexane/benzene 70/30. The following reagents were synthesized using previously described procedures: dithiobenzoic acid,45 1,4-bis(2-(thiobenzoylthio)prop-2-yl)benzene (BTBTPB),46 1,4-bis(thiobenzoylthiomethyl)benzene (BTBTMB).46 The reagents were characterized by 1H NMR (400 MHz) and 13C NMR (100 MHz).
Instrumentation
1H NMR and 13C NMR spectra were recorded on a Varian spectrometer (400 MHz). The number-average (Mn) and weight-average (Mw) molecular weight and polydispersity index (PDI, Mw/Mn) of the polymers were determined by size exclusion chromatography (SEC) using Shimadzu LC-10ADVP liquid chromatograph equipped with a column oven and Polymer Labs PL gel 5 μm mixed C column (300×7.5 mm, linear MW range 200–2,000,000). The system was equipped with Wyatt miniDAWN multiangle laser light scattering detector (λ = 690 nm, 30 mW) and Wyatt Optilab DSP interferometric refractometer (λ = 690 nm). N,N -Dimethylformamide (DMF) was used as an eluent at a flow rate of 1.0 mL/min and temperature of 35 °C. SEC data were analyzed using Astra 5.3.1.4 software from Wyatt Technology. Refractive index increments (ν) of the polymers were determined by the Optilab interferometric refractometer at λ = 690 nm and used in the SEC analysis.
General procedure for synthesis of α,ω-bis(dithioester)-functionalized polymers
All polymerizations were performed in ampoules under homogeneous conditions. In a typical procedure, DMAEMA (2.0 g, 1.3×10−2 mol), AIBN (0.010 mg, 6×10−5 mol), and BTBTPB RAFT agent (0.080 g, 1.71×10−4 mol) were dissolved in THF (3.0 mL) and added into an ampoule. The mixture was thoroughly deoxygenated, sealed under vacuum, and placed in a thermostated water bath at 60 °C for 60 h. The ampoule was then opened and the polymerization was quenched by lowering the temperature to 0 °C. The resulting polymer was obtained by precipitation and filtration (81% yield, Mw = 7,780, PDI = 1.18).
Synthesis of reducible multiblock copolymers
In a typical synthetic procedure, α,ω-bis(dithioester)-functionalized poly(2-(dimethylamino) ethyl methacrylate) (PDMAEMA, 50 mg) and poly(N-isopropylacrylamide) (PNIPAM, 50 mg) were added into a flask containing 2 N ammonia solution in methanol (2.0 mL), followed by an addition of a few drops of hexylamine. The reaction mixture was stirred under O2 for 24 h. The solution color changed from pink to light yellow during the reaction. After removing the solvent, the residue was dissolved in THF and precipitated into hexane (71% yield, Mw = 180,000, PDI = 5.1).
Cloud point determination
The optical transmittance of the multiblock copolymer solutions at various temperatures was measured at 360 nm with a UV-vis spectrophotometer (VARIAN CARY50). The sample cell was thermostated with a circulating water jacket from 5 to 50 °C. The temperature was gradually increased with a maximum heating rate of 0.5 °C/min. The reading was taken 5 ~10 min after each temperature increase.
Light scattering
The determination of hydrodynamic radius (RH) and ζ potential of the multiblock copolymers was performed by dynamic light scattering using Brookhaven Instruments ZetaPlus particle size and ζ potential analyzer. The measurements of particle size and ζ potential were performed at ~15 °C and 25 °C using 1.0 mg/mL solutions of the copolymers. Mean hydrodynamic radius was calculated from the size distribution by weight, assuming a log-normal distribution using the supplied algorithm, and the results are expressed as a mean of three runs. ζ potential was calculated from measured velocities using the Smoluchowski equation, and the results are expressed as a mean of 10 runs. The static light scattering measurements were performed with Wyatt DAWN EOS light scattering instrument equipped with a 30 mW linearly polarized GaAs laser (λ = 690 nm). The analysis of the copolymer solutions was conducted at an angular range θ = 22.5°–147° in 20 mL glass scintillation vials. The static light scattering data were analyzed by first- or second-order Debye fit (R(θ)/Kc vs. sin2(θ/2) to obtain apparent weight-average molecular weight (Mwapp) and z-average of the root mean square radius (radius of gyration, RG) of the copolymer assemblies. K is the optical constant which includes the square of the refractive index increment (ν); R(θ) is the Rayleigh ratio, proportional to the intensity of the light scattered from solution and the copolymer concentration c in g/mL. The refractive index increments of the copolymers were calculated as weight-average values using νPNIPAM = 0.194 mL/g and νPDMAEMA = 0.178 mL/g.47, 48
Atomic force microscopy
The copolymer was deposited on mica using dip-coating at 23 °C or 4 °C. Freshly cleaved mica was immersed in the solution for 30 min, rinsed with deionized water, and dried with N2. AFM imaging was conducted using a Nanoscope III MultiMode AFM equipped with a piezoelectric scanner type E with a maximum scan range of 10 μm (X and Y) and 2.5 μm (Z) from VEECO/Digital Instruments. Ex situ AFM imaging of samples was conducted in the Tapping Mode (oscillation frequency ~ 250 kHz) in ambient atmosphere using etched silicon probes (TESP, VEECO) with nominal radius of curvature less than 10 nm. In situ AFM imaging was conducted in aqueous solution of copolymers in the Liquid Tapping Mode (oscillation frequency ~ 8 kHz) using silicon nitride probes (NP, VEECO) with radius of curvature of 20 nm and cantilever spring constant of 0.38 N/m as provided by the manufacturer. The copolymer sample was imaged in 40 μL of water and then 10 μL of 100 mM DTT aqueous solution was directly injected into the liquid cell.
RESULTS AND DISCUSSION
Use of disulfide-containing polymers in drug and gene delivery is based on the existence of consistent redox-potential gradients between extracellular and intracellular space and an easy reversibility of disulfide bonds.49 Polyelectrolyte complexes of reducible polycations and DNA exhibit increased DNA availability for transcription and reduced cytotoxicity compared with non-reducible complexes. Evidence suggests that a combination of different functions within a single polymer can contribute to an overall improvement of carrier efficiency.24, 26 To this end, we seek to find methods to prepare synthetic reducible multiblock copolymers that would combine the redox- with another type of stimulus-responsive behavior. In this study, we describe the synthesis of multiblock copolymers responsive to redox-potential gradients and temperature.
The detailed procedure for preparing the reducible multiblock copolymers is outlined in Scheme 1. First, two difuctional RAFT agents (BTBTMB (1) and BTBTPB (2)) were synthesized and used in the AIBN-initiated RAFT polymerization of DMAEMA and NIPAM. The polymerization then yielded α,ω-bis(dithioester)-functionalized PDMAEMA (3)50 and PNIPAM (4). Treatment of the mixture of the two α,ω-bis(dithioester)-functionalized polymers with methanolic solution of ammonia and hexylamine in the presence of oxygen provided reducible multiblock copolymers of PDMAEMA and PNIPAM (5). Susceptibility of thiol-terminated polymers to oxidation makes their isolation and purification difficult. Compared with previously reported methods of preparing reducible polymers via RAFT polymerization, the method reported here eliminates the isolation of dithiol-terminated polymers due to the aminolysis and oxidization reactions being carried out in a single step.
Scheme 1.

Synthesis of reducible multiblock copolymers by reversible addition-fragmentation chain transfer polymerization.
RAFT polymerization of DMAEMA was carried out at 60 °C using BTBTPB as RAFT agent and AIBN as an initiator, producing PDMAEMA with dithioester groups at both termini as shown in Scheme 1. The synthesized α,ω-bis(dithioester)-functionalized PDMAEMA was characterized by SEC, NMR and UV spectroscopy. Figure 1 shows 1H NMR spectra of the BTBTPB RAFT agent and the synthesized PDMAEMA. The presence of the terminal phenyldithioester groups and the centrally located phenylene group in PDMAEMA is confirmed by the signals at 7.0–8.0 ppm (Figure 1). The signals at 0.8–1.3 ppm in Figure 1b are assigned to the methyl protons and the signals at 1.5–2.0 ppm are assigned to the methylene protons in polymer backbone. The living nature of the polymerization was confirmed by the data from the polymerization kinetics study (Figure 2a). The results show that the number-average molecular weight of PDMAEMA increased linearly with monomer conversion. Moreover, the polydispersity index values remain below 1.2 throughout the course of the polymerization, indicating that BTBTPB is an efficient transfer agent for the polymerization of DMAEMA. Overall, these results demonstrate that α,ω-bis(dithioester)-functionalized PDMAEMA was obtained via RAFT polymerization using BTBTPB as the RAFT agent.
Figure 1.

1H NMR spectra of 1,4-bis(2-(thiobenzoylthio)prop-2-yl)benzene (BTBTPB) in CDCl3 (a) and of α,ω-bis(dithioester)-terminated PDMAEMA in CDCl3 (b).
Figure 2.
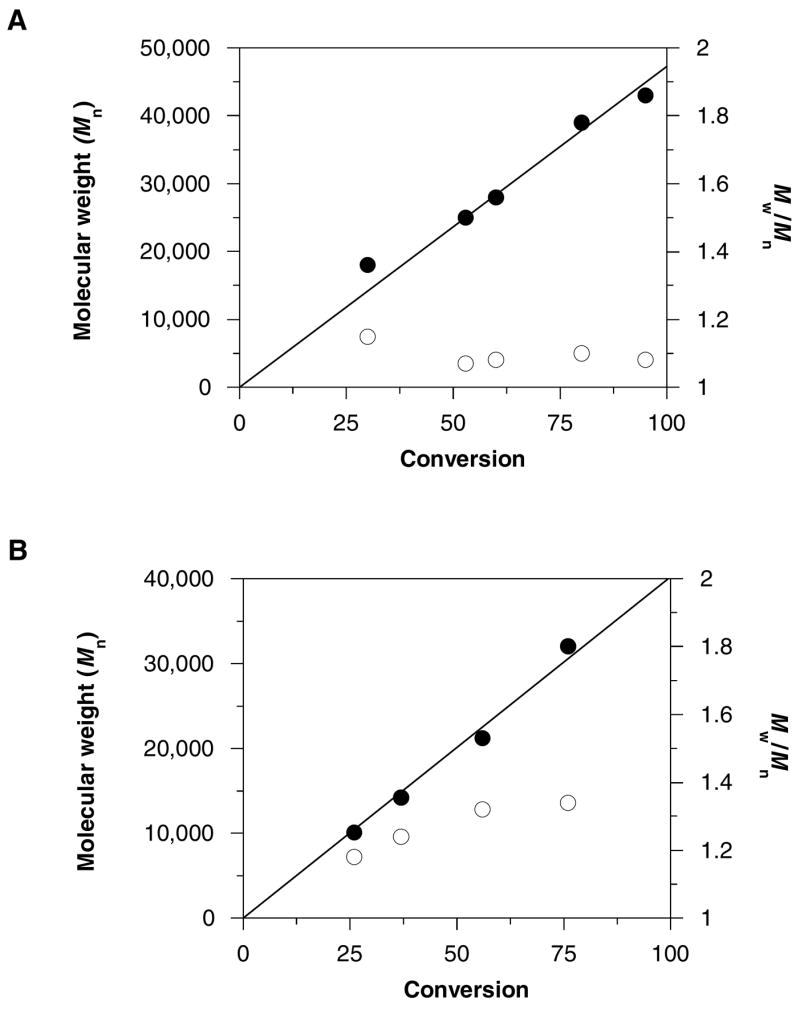
Evolution of number-average molecular weight Mn (●) and polydispersity Mw/Mn (○) (determined by size exclusion chromatography) with conversion (determined by 1H NMR) during DMAEMA (A) and NIPAM (B) polymerization.
Successful polymerization of NIPAM with difunctional RAFT agent was reported previously using trithiocarbonate compounds.41, 51 Because of the incompatibility of trithiocarbonates with a single aminolysis/oxidation step, we attempted polymerization of NIPAM with the same RAFT agent used above with DMAEMA. The polymerization of NIPAM was also carried out at 60 °C, however, no polymer was obtained even after several days of reaction, indicating that BTBTPB is not an efficient RAFT agent for the polymerization of NIPAM. We therefore synthesized BTBTMB as a possible alternative to BTBTPB. RAFT polymerization of NIPAM was carried out at 60 °C using BTBTMB as RAFT agent. Compared with RAFT polymerization of DMAEMA using BTBTPB, the rate of polymerization of NIPAM using BTBTMB was much lower and a pronounced induction period was observed at the early time points. The polymerization kinetic results, however, show that the number-average molecular weight of PNIPAM increased linearly with monomer conversion (Figure 2b). The polydispersity of the PNIPAM was generally higher than that of PDMAEMA but it was maintained below 1.4, suggesting that BTBTMB can efficiently control the RAFT polymerization of NIPAM. The NMR spectra in Figure 3 confirm the synthesis of the desired polymer by the presence of the terminal dithioester groups at 7.0–8.0 ppm (Figure 3b). The unassigned signals correspond to the methine and methylene protons of the acrylate backbone.
Figure 3.

1H NMR spectra of 1,4-bis(thiobenzoylthiomethyl)benzene (BTBTMB) in CDCl3 (a) and of α,ω-bis(dithioester)-terminated PNIPAM in CDCl3 (b).
Multiblock copolymers containing temperature-sensitive PNIPAM blocks and cationic PDMAEMA blocks connected by reducible disulfide linkages were obtained simply by treating the mixture of the synthesized α,ω-bis(dithioester)-terminated PNIPAM and PDMAEMA in ammonia methanol solution with hexylamine in the presence of oxygen. The reaction was carried out using different ratios between PNIPAM and PDMAEMA blocks and the time course of the reaction was studied for 1:1 copolymers (Table 1). High molecular weight copolymers with rather high polydispersities were obtained from all the reactions. Based on the molecular weight of each block (~8,000), we can estimate that the multiblock copolymer contain ~7–20 PDMAEMA and PNIPAM blocks. Representative 1H NMR spectrum of the multiblock copolymer in Figure 4 shows the specific signals of both PNIPAM and PDMAEMA, as well as the signal of the centrally located phenylene residues from the RAFT agents (~7.05 ppm). The absence of signals from the terminal phenyl units of the starting α,ω-bis(dithioester)-functionalized polymers clearly suggested that the aminolysis of dithioesters and oxidation of the resulting thiols have occurred successfully. The aminolysis was additionally confirmed by the disappearance of the characteristic dithioester absorption at 305 nm (Figure 5a). The SEC traces shown in Figure 5b also document the shift to higher molecular weights after oxidation. Moreover, the molecular weight of the synthesized multiblock copolymers was reduced after treatment with DTT (Figure 5b); confirming their reducible nature. All the above results demonstrate that reducible multiblock copolymers with temperature-sensitive PNIPAM blocks and cationic PDMAEMA blocks have been successfully prepared.
Table 1.
Oxidation reaction conditions and molecular parameters of the multiblock copolymers
| Sample # | Weight ratio of PNIPAM to PDMAEMA a | Reaction time (h) | Mwb (×103) | Mw/Mnb | RH above cloud point (nm) | ζ above cloud point (mV) |
|---|---|---|---|---|---|---|
| 1 | 1:1 | 4.5 | 73 | 3.0 | 55.1 | 18 |
| 2 | 1:1 | 6.0 | 180 | 5.1 | 41.7 | 40 |
| 3 | 1:1 | 7.0 | 130 | 7.0 | 50.5 | 15 |
| 4 | 1:1 | 24.0 | 125 | 3.9 | 41.8 | 44 |
| 5 | 3:7 | 7.0 | 167 | 2.8 | 53.9 | 30 |
| 6 | 7:3 | 7.0 | 80 | 5.9 | 840 | 7 |
PNIPAM (Mn = 7,860, Mw/Mn = 1.24) and PDMAEMA (Mn = 7,780, Mw/Mn = 1.18) were used
from SEC
Figure 4.

Typical 1H NMR spectrum of a multiblock copolymer of PNIPAM and PDMAEMA in CDCl3.
Figure 5.
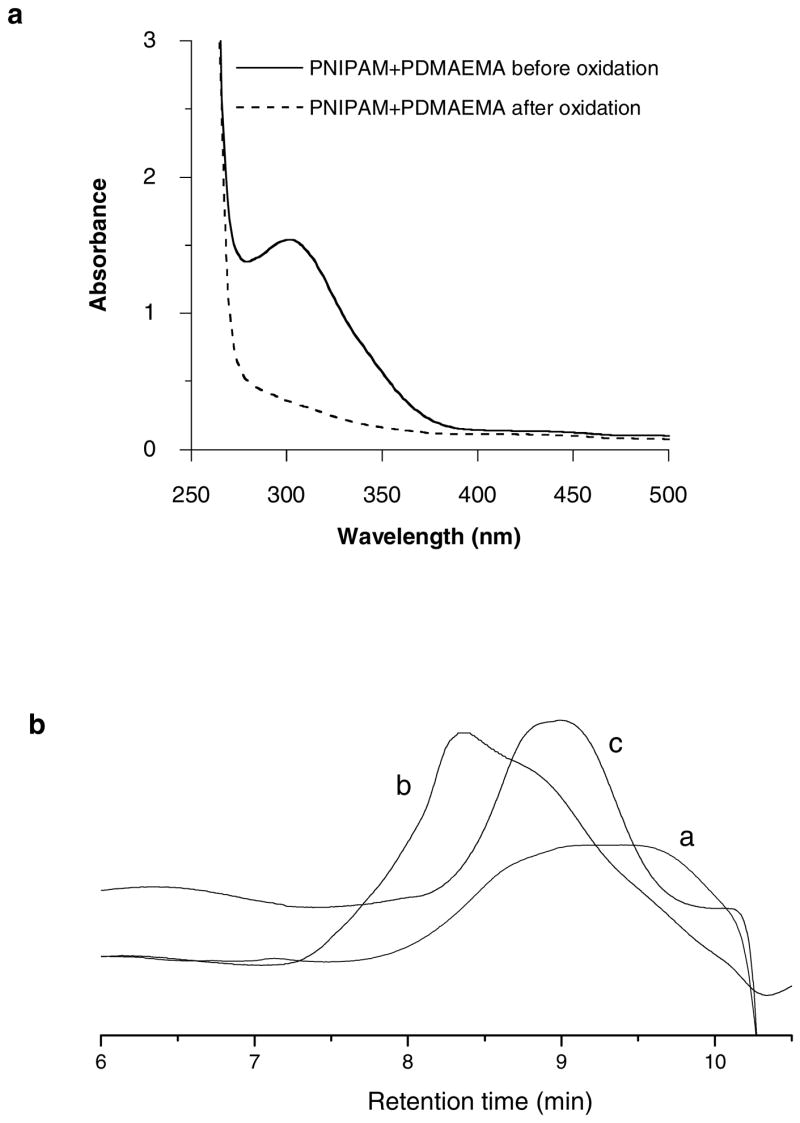
(a) UV-vis spectra of the mixture of α,ω-bis(dithioester)-terminated PDMAEMA and PNIPAM in methanol before (solid line) and after (dash line) treatment with hexylamine and oxygen. (b) SEC traces of mixture of α,ω-bis(dithioester)-functionalized PDMAEMA and PNIPAM (a) and reducible multiblock copolymer after oxidation (b) and after subsequent reduction with dithiothreitol (c).
The effect of temperature on the properties of the multiblock copolymers was investigated because of the known lower critical solution temperature (LCST) behavior of both PNIPAM and PDMAEMA. The LCST behavior of PDMAEMA is strongly dependent on the ionization of the dimethylamino groups. Here we found that under conditions employed in our studies no LCST associated with the PDMAEMA blocks was observed below 40 °C.52, 53 Thus, all the temperature-responsive behavior reported here can be ascribed to the PNIPAM blocks. At temperatures below its LCST (~32 °C), the intermolecular hydrogen bonding between the PNIPAM chains and water molecules is dominant and the PNIPAM chains are soluble in water. At temperatures above the LCST, intramolecular hydrogen bonding between C=O and N-H groups in PNIPAM results in a transition into collapsed conformations and PNIPAM insolubility in water. Preliminary experiments suggested that the multiblock copolymers have the phase transition at significantly lower temperatures than the parent PNIPAM. When measured in deionized water, the cloud point temperatures were observed around 21 °C (the cloud point of the PNIPAM blocks before aminolysis was ~15 °C). This low LCST is most likely the result of a combined effect of the hydrophobic phenylene and disulfide residues separating the short PNIPAM blocks in the copolymer structure.54
We first studied the association of the multiblock copolymers above their phase transition temperatures using dynamic light scattering in water. As illustrated in Figure 6 on the example of multiblock copolymer #2 (Table 1), the average hydrodynamic radius of the copolymer was 24.5 nm at ~15 °C. The hydrodynamic radius increased to 83.4 nm at 25 °C. Formation of relatively well-defined particles was confirmed by AFM (Figure 7). No particles were observed by AFM below the copolymer phase transition (Figure 7a), whereas particles with diameters up to 50 nm were visualized above the copolymer phase transition (Figure 7b). To evaluate the effect of reduction on the morphology of the copolymer particles, the AFM analysis was conducted in solution to visualize the particles before and after addition of 20 mM DTT (Figure 7c, d). The introduction of reducing agent to the solution resulted in particle detachment from the surface to the bulk of the solution in the AFM cell and subsequent inability to follow the morphological changes in detail. It is assumed that the reduction leads to a separation of the positively charged shell of PDMAEMA, which results in detachment of the particles from the negatively charged mica surface. It is well-known that intracellular concentrations of the most important physiological reducing agent glutathione are in mM range and that such concentrations are sufficient to break the disulfides in a variety of redox-responsive delivery systems. On the other hand, disulfide reduction proceeds inefficiently at the much lower concentrations of glutathione (μM) that are found in the extracellular space.
Figure 6.
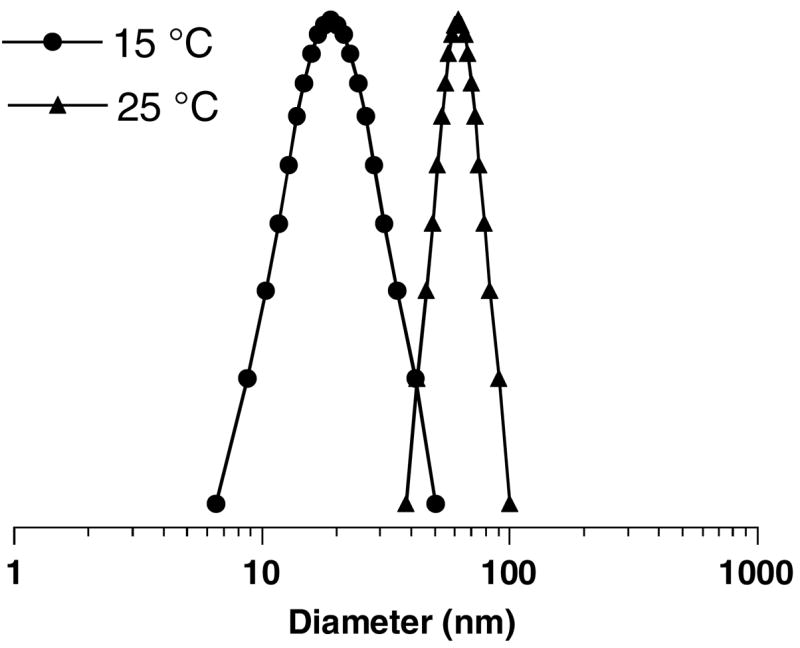
Phase transition behavior of the multiblock copolymers of PNIPAM and PDMAEMA. Hydrodynamic diameter distribution of copolymer #2 (1.0 mg/mL) in water at 15 °C and 25 °C.
Figure 7.

AFM height images of copolymer #2 (0.01 mg/mL) below phase transition (a) and above phase transition (b) acquired in dry state. Effect of reduction determined by AFM in aqueous solution of copolymer #2 in water before (c) and after the addition of 20 mM dithiothreitol. (z-range 5 nm)
As shown in Table 1, the hydrodynamic sizes of the particles formed above the copolymer phase transition were almost independent of the composition for copolymers with PNIPAM:PDMAEMA 1:1 and 3:7. Increasing the PNIPAM:PDMAEMA ratio to 7:3 resulted in a substantial aggregation and formation of large particles. We propose that PNIPAM blocks collapse to form a nanoparticle core while the cationic PDMAEMA blocks remain soluble and form a shell to stabilize the nanoparticles. This hypothesis is further supported by the ζ potential values observed for the copolymers (Table 1). For example, ζ potential of the copolymer #2 was not measurable at 15 °C due to a weak scattering of the molecularly dissolved copolymer. At 25 °C, however, ζ potential reached a highly positive values ~40 mV. This is most likely the result of the association of the multiblock copolymer blocks in which charged PDMAEMA blocks form the shell of the particles with a high surface charge density. The ζ potential values for all the other multiblock copolymers listed in Table 1 were positive. However, the multiblock copolymer with 70% PNIPAM had much lower ζ potential than others, which may be the result of a decreased PDMAEMA density on the particle surface.
We anticipated that the association of the multiblock copolymers will be affected by the ionization of PDMAEMA blocks. Transmittance of the copolymer solutions in 100 mM phosphate buffers of varying pH was measured between 10 and 50 °C to determine the cloud point temperature (Figure 8a). Increasing the pH resulted in a shift of the phase transition to lower temperatures and in sharper transitions. For example, the transition was observed at ~21 °C at pH 7.0 but it decreased at pH 8.3 to ~15 °C. On the other hand, acidification of the copolymer solution to pH 4.5 increased the transition temperature and broadened the transition. The observed pH effect on the cloud point behavior can be ascribed, to a large extent, to variations in the stabilizing effect of PDMAEMA blocks with changes of their degree of ionization. Although little change with pH was observed for RG of the copolymer particles, increasing the pH resulted in a steady increase of Mwapp above the phase transition temperature (Figure 8b).
Figure 8.
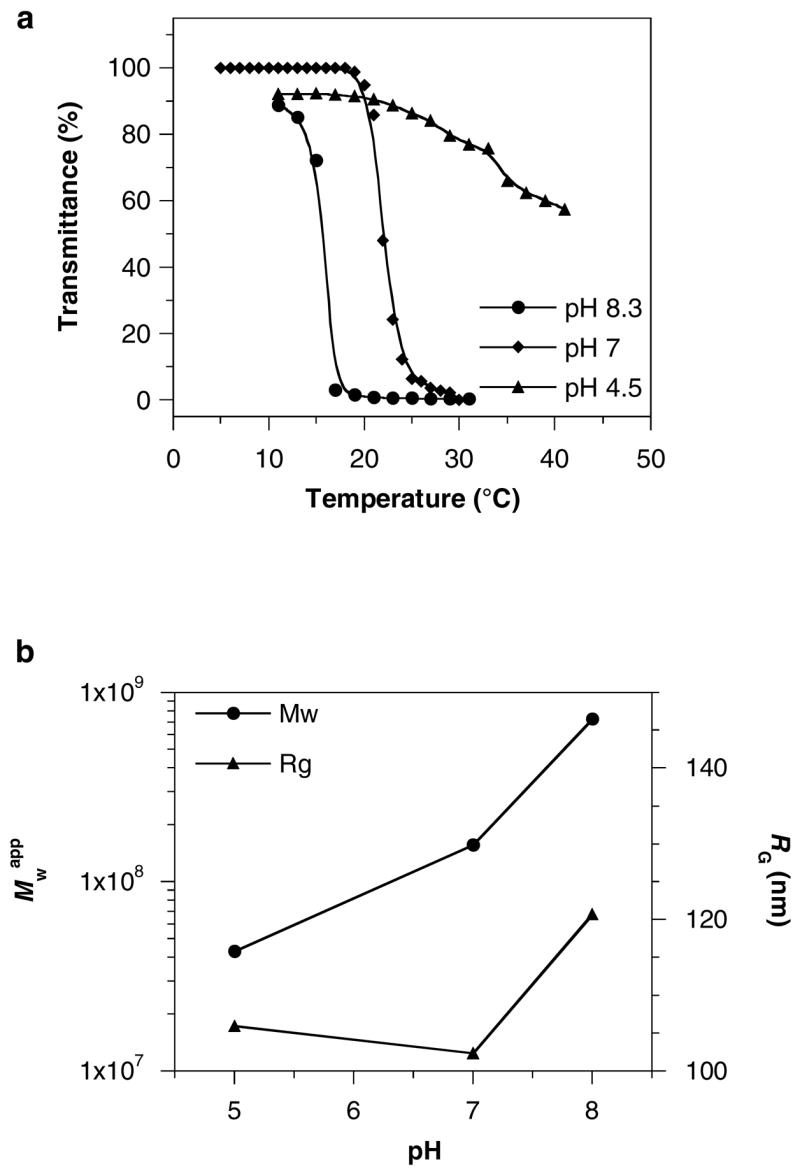
(a) Effect of pH on the cloud point of the copolymer #2 solution. (b) Effect of pH on the apparent molecular weight (Mwapp) and radius of gyration (RG) of the assemblies of copolymer #2 above the phase transition temperature in 100 mM phosphate buffers.
CONCLUSIONS
Well defined α,ω-bis(dithioester)-functionalized PDMAEMA and PNIPAM were successfully obtained by RAFT polymerization using two different RAFT agents. Multiblock copolymers containing temperature-responsive PNIPAM blocks and cationic PDMAEMA blocks connected by reducible disulfide linkages were synthesized in a single step from the α,ω-bis(dithioester) blocks. The multiblock copolymers could be easily reduced to the starting polymer blocks. The copolymers assemble into core-shell nanostructures with hydrophobic PNIPAM as the core and cationic PDMAEMA chains as the stabilizing shell. These dually responsive copolymers are attractive as gene delivery carriers because of the possibility to control their interaction with nucleic acids via two independent stimuli and because of their easy intracellular degradability.
Acknowledgments
This work was supported by NIH Grant CA109711 from the National Cancer Institute.
References
- 1.Sung BJ, Yethiraj A. J Chem Phys. 2005;123(21) doi: 10.1063/1.2131057. Art. No. 214901. [DOI] [PubMed] [Google Scholar]
- 2.Sun DC, Liang HJ. Chinese J Chem Phys. 2006;19(3):265–268. [Google Scholar]
- 3.Kavassalis TA, Whitmore MD. Macromolecules. 1991;24(19):5340–5345. [Google Scholar]
- 4.Higaki Y, Otsuka H, Takahara A. Polymer. 2003;44(23):7095–7101. [Google Scholar]
- 5.Eastwood EA, Dadmun MD. Macromolecules. 2002;35(13):5069–5077. [Google Scholar]
- 6.Eastwood EA, Dadmun MD. Macromolecules. 2001;34(4):740–747. [Google Scholar]
- 7.Cui MH, Xie HO, Guo JS. Angew Makromol Chem. 1998;257:37–42. [Google Scholar]
- 8.Bussels R, Bergman-Gottgens C, Meuldijk J, Koning C. Macromolecules. 2004;37(25):9299–9301. [Google Scholar]
- 9.Spontak RJ, Zielinski JM, Lipscomb GG. Macromolecules. 1992;25(23):6270–6276. [Google Scholar]
- 10.Ghassemi H, McGrath JE, Zawodzinski TA. Polymer. 2006;47(11):4132–4139. [Google Scholar]
- 11.Holder SJ, Hiorns RC, Sommerdijk NAJM, Williams SJ, Jones RG, Nolte RJM. Chem Commun. 1998;(14):1445–1446. [Google Scholar]
- 12.Nery L, Lefevre H, Fradet A. J Polym Sci Part A: Polym Chem. 2005;43(7):1331–1341. [Google Scholar]
- 13.Chen WN, Luo WJ, Wang SG, Bei JZ. Polym Adv Technol. 2003;14(3–5):245–253. [Google Scholar]
- 14.Cohn D, Salomon AF. Biomaterials. 2005;26(15):2297–2305. doi: 10.1016/j.biomaterials.2004.07.052. [DOI] [PubMed] [Google Scholar]
- 15.DiPasquale G, LaRosa A, Mamo A. Macromol Rapid Commun. 1997;18(4):267–272. [Google Scholar]
- 16.Jeon O, Lee SH, Kim SH, Lee YM, Kim YH. Macromolecules. 2003;36(15):5585–5592. [Google Scholar]
- 17.Petrova T, Manolova N, Rashkov I, Li SM, Vert M. Polymer Int. 1998;45(4):419–426. [Google Scholar]
- 18.Wang CY, Cui MH. J Applied Polym Sci. 2003;88(7):1632–1636. [Google Scholar]
- 19.Sun KH, Sohn YS, Jeong B. Biomacromolecules. 2006;7(10):2871–2877. doi: 10.1021/bm060512r. [DOI] [PubMed] [Google Scholar]
- 20.You YZ, Hong CY, Pan CY. Chem Commun. 2002;(23):2800–2801. doi: 10.1039/b208180f. [DOI] [PubMed] [Google Scholar]
- 21.Jia ZF, Liu C, Huang JL. Polymer. 2006;47(22):7615–7620. [Google Scholar]
- 22.Bussels R, Koning CE. Tetrahedron. 2005;61(5):1167–1174. [Google Scholar]
- 23.Toman L, Janata M, Spevacek J, Vlcek P, Latalova P, Masar B, Sikora A. J Polym Sci Part A: Polym Chem. 2004;42(23):6098–6108. [Google Scholar]
- 24.Chen CP, Kim JS, Liu D, Rettig GR, McAnuff MA, Martin ME, Rice KG. Bioconjugate Chem. 2007;18(2):371–8. doi: 10.1021/bc060229p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kwok KY, Park Y, Yang Y, McKenzie DL, Liu Y, Rice KG. J Pharm Sci. 2003;92(6):1174–85. doi: 10.1002/jps.10384. [DOI] [PubMed] [Google Scholar]
- 26.Manickam DS, Oupicky D. Bioconjugate Chem. 2006;17(6):1395–1403. doi: 10.1021/bc060104k. [DOI] [PubMed] [Google Scholar]
- 27.Tsarevsky NV, Matyjaszewski K. Macromolecules. 2002;35(24):9009–9014. [Google Scholar]
- 28.Tsarevsky NV, Matyjaszewski K. Macromolecules. 2005;38(8):3087–3092. [Google Scholar]
- 29.Li YT, Armes SP. Macromolecules. 2005;38(20):8155–8162. [Google Scholar]
- 30.Li CM, Madsen J, Armes SP, Lewis AL. Angew Chem Int Ed. 2006;45(21):3510–3513. doi: 10.1002/anie.200600324. [DOI] [PubMed] [Google Scholar]
- 31.Chiefari J, Chong YK, Ercole F, Krstina J, Jeffery J, Le TPT, Mayadunne RTA, Meijs GF, Moad CL, Moad G, Rizzardo E, Thang SH. Macromolecules. 1998;31(16):5559–5562. [Google Scholar]
- 32.Chong YK, Le TPT, Moad G, Rizzardo E, Thang SH. Macromolecules. 1999;32(6):2071–2074. [Google Scholar]
- 33.Moad G, Rizzardo E, Thang SH. Australian J Chem. 2006;59(10):669–692. [Google Scholar]
- 34.Liu J, Bulmus V, Barner-Kowollik C, Stenzel MH, Davis TP. Macromol Rapid Commun. 2007;28(3):305–314. [Google Scholar]
- 35.Quemener D, Davis TP, Barner-Kowollik C, Stenzel MH. Chem Commun. 2006;(48):5051–5053. doi: 10.1039/b611224b. [DOI] [PubMed] [Google Scholar]
- 36.Patton DL, Mullings M, Fulghum T, Advincula RC. Macromolecules. 2005;38(20):8597–8602. [Google Scholar]
- 37.Lowe AB, McCormick CL. Australian J Chem. 2002;55(6–7):367–379. [Google Scholar]
- 38.Cheng C, Khoshdel E, Wooley KL. Macromolecules. 2007;40(7):2289–2292. [Google Scholar]
- 39.Perrier S, Takolpuckdee P, Mars CA. Macromolecules. 2005;38(16):6770–6774. [Google Scholar]
- 40.Whittaker MR, Goh YK, Gemici H, Legge TM, Perrier S, Monteiro MJ. Macromolecules. 2006;39(26):9028–9034. [Google Scholar]
- 41.Qiu XP, Winnik FM. Macromolecules. 2007;40(4):872–878. [Google Scholar]
- 42.Gemici H, Legge TM, Whittaker M, Monteiro MJ, Perrier S. J Polym Sci Part A: Polym Chem. 2007;45:2334–2340. [Google Scholar]
- 43.Oupicky D, Parker AL, Seymour LW. J Am Chem Soc. 2002;124(1):8–9. doi: 10.1021/ja016440n. [DOI] [PubMed] [Google Scholar]
- 44.You YZ, Soundara Manickam D, Zhou QH, Oupicky D. Biomacromolecules. 2007;8(6):2038–2044. doi: 10.1021/bm0702049. [DOI] [PubMed] [Google Scholar]
- 45.Mitsukami Y, Donovan MS, Lowe AB, McCormick CL. Macromolecules. 2001;34(7):2248–2256. [Google Scholar]
- 46.Le TPT, Moad G, Rizzardo E, Thang SH. PCT Int Appl WO Patent 9801478. 1998 [Google Scholar]
- 47.Konak C, Reschel T, Oupicky D, Ulbrich K. Langmuir. 2002;18(21):8217–8222. [Google Scholar]
- 48.Reschel T, Konak C, Oupicky D, Seymour LW, Ulbrich K. J Controlled Rel. 2002;81(1–2):201–217. doi: 10.1016/s0168-3659(02)00045-7. [DOI] [PubMed] [Google Scholar]
- 49.Saito G, Swanson JA, Lee KD. Adv Drug Deliv Rev. 2003;55(2):199–215. doi: 10.1016/s0169-409x(02)00179-5. [DOI] [PubMed] [Google Scholar]
- 50.Krasia TC, Patrickios CS. Macromolecules. 2006;39(7):2467–2473. [Google Scholar]
- 51.Qiu XP, Winnik FM. Macromol Rapid Commun. 2006;27(19):1648–1653. [Google Scholar]
- 52.Butun V, Armes SP, Billingham NC. Polymer. 2001;42(14):5993–6008. [Google Scholar]
- 53.Triftaridou AI, Vamvakaki M, Patrickios CS. Polymer. 2002;43(10):2921–2926. [Google Scholar]
- 54.Xia Y, Burke NAD, Stover HDH. Macromolecules. 2006;39(6):2275–2283. [Google Scholar]