

Summary
Endothelial cells in growing tumors express activated Akt, which when modeled by transgenic endothelial expression of myrAkt1 was sufficient to recapitulate the abnormal structural and functional features of tumor blood vessels in nontumor tissues. Sustained endothelial Akt activation caused increased blood vessel size and generalized edema from chronic vascular permeability, while acute permeability in response to VEGF-A was unaffected. These changes were reversible, demonstrating an ongoing requirement for Akt signaling for the maintenance of these phenotypes. Furthermore, rapamycin inhibited endothelial Akt signaling, vascular changes from myrAkt1, tumor growth, and tumor vascular permeability. Akt signaling in the tumor vascular stroma was sensitive to rapamycin, suggesting that rapamycin may affect tumor growth in part by acting as a vascular Akt inhibitor.
Introduction
The development and maturation of blood vessels result from a complex interplay of pro- and antiangiogenic regulators. Dys-regulation of the balance between these factors is thought to result in the formation of pathological blood vessels, such as those found in tumors (Bergers and Benjamin, 2003). Tumor blood vessels are distinctly abnormal in structure and function. While heterogeneous, many tumor blood vessels are enlarged and tortuous, and compared to normal vessels of similar sizes, they are poorly invested by pericytes and have defective basement membranes (Baluk et al., 2003; Carmeliet and Jain, 2000; Morikawa et al., 2002). Tumor blood vessels are also hyper-permeable to circulating macromolecules, partly due to the overproduction of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF-A). This results in extravasation of plasma fluid and proteins (Dvorak, 1990; Dvorak et al., 1999). Several possible pathways by which macromolecules and other plasma solutes cross vascular endothelium involves intercellular gaps, fenestrae, trans-endothelial cell pores, and vesiculo-vacuolar organelles (VVOs) (Baluk and McDonald, 1994; Feng et al., 1999,2000b; McDonald and Baluk, 2002).
VEGF-A is one of the major cytokines involved in tumor angiogenesis, and its expression is regulated not only by tumor hypoxia but also by oncogenes and tumor suppressor genes (Harris, 2002; Pal et al., 2001; Rak et al., 1995; Rak and Kerbel, 2001; Shweiki et al., 1992). Overexpression of VEGF-A by tumor cells leads to the formation of new blood vessels with exaggerated size, tortuosity, and permeability that have prolonged dependence on VEGF-A for survival (Benjamin and Keshet, 1997). In some tumors, vessels that acquire pericyte/smooth muscle investment are more resistant to VEGF-A deprivation (Benjamin et al., 1999). VEGF-A overexpression has also been shown to induce tumor-like blood vessels in normal tissues in the absence of tumor cells (Feng et al., 2000a; Pettersson et al., 2000; Sundberg et al., 2001).
The PI3K/Akt signaling pathway functions downstream of VEGF-A to promote endothelial cell survival (Dimmeler and Zeiher, 2000; Fujio and Walsh, 1999; Gerber et al., 1998; Liu et al., 2000). Recent investigation of different Akt isoforms has demonstrated that Akt1 expression is required for pathological angiogenesis in a hindlimb ischemia model and in response to VEGF-A-induced acute permeability (Ackah et al., 2005). VEGF-A-induced acute permeability is dependent on Akt signaling and downstream activation of endothelial nitric oxide synthase (eNOS) (Dimmeler et al., 1999; Fukumura et al., 2001; Fulton et al., 1999; Six et al., 2002). Another downstream pathway that has an impact on angiogenesis is the mammalian target of rapamycin (mTOR) (Guba et al., 2002). Recent studies have shown that the rapamycin-insensitive rictor-mTOR complex (mTORC2) is the one that directly phosphorylates Akt at S473 (Sarbassov dos et al., 2005). Despite the straightforward prediction that rapamycin action on the raptor containing rapamycin-sensitive complex (mTORC1) would result in increased Akt activation due to compensatory higher levels of mTORC2, in fact, long-term treatment of rapamycin on cells in culture decreased activation of Akt (Edinger et al., 2003; O’Reilly et al., 2006). Investigation of the effects of rapamycin on mTORC1 and mTORC2 complexes revealed that prolonged treatment with rapamycin interferes with the formation of mTORC2 (Sarbassov dos et al., 2006).
We have observed that tumor endothelial cells have chronic Akt activation. To explore the function of such activation, we have used transgenic mice that allow us to induce or repress constitutively active myristoylated Akt1 (myrAkt1) in endothelial cells of normal tissues to form a surrogate tumor stroma without the complications of tumor cell contributions. We show that sustained Akt activation induces the formation of enlarged, hyper-permeable blood vessels that recapitulate the structural and functional abnormalities of tumor vessels and mimic the effects of VEGF-A-induced angiogenesis. Both the abnormal structure and function were blocked by rapamycin and correlate to rapamycin-induced downregulation of Akt phosphorylation in endothelial cells and normal mouse tissues. Rapamycin inhibited tumor growth and tumor vascular permeability, which we hypothesize are in part due to effects of rapamycin on endothelial Akt activation.
SIGNIFICANCE.
Most of the common alterations in structure and function associated with tumor blood vessels can be recapitulated by overexpression of VEGF-A. This study demonstrates that the Akt pathway, a key signaling pathway activated in response to VEGF-A, induces similar structural and functional alterations. Sustained endothelial activation of Akt in otherwise healthy tissue induces vascular malformations and systemic vascular permeability. Moreover, the activation of Akt and the associated pathological features of the blood vessels can be blocked by rapamycin. These findings indicate a possible clinical utility of rapamycin as an angiogenesis inhibitor and support a pathway for rapamycin action via Akt inhibition.
Results
Increased phosphorylated Akt in tumor blood vessels
We have previously shown that VEGF-A functions as a survival factor for newly formed blood vessels and that abrupt withdrawal of VEGF-A results in regression of tumor vessels in rat C6 glioma cells (Benjamin and Keshet, 1997). We hypothesize that the vasculature in tumors is abnormal in part due to chronic activation of endothelial Akt signaling pathways as a result of high expression of angiogenic cytokines intrinsic within the tumor. Should this be correct, we would expect to see high levels of phosphorylated Akt (pAkt) in endothelial cells lining these tumor blood vessels. Immunofluorescence staining of xenografts of rat C6 glioma cells overexpressing VEGF-A showed that many tumor endothelial cells expressed pAkt (Figure 1). Many tumor cells, and especially those surrounding blood vessels, also expressed pAkt. Such perivascular cuffs of tumor cells with activated Akt may reflect increased Akt survival signaling in cells receiving increased oxygen and nutrients due to their close proximity to the blood vessels. Expression of pAkt is strong in tumor tissue but not in adjacent normal skin, and similarly, quiescent vessels in normal skin do not show increased activation of Akt (Figure S1 in the Supplemental Data available with this article online).
Figure 1. Akt activation in tumor vessels.
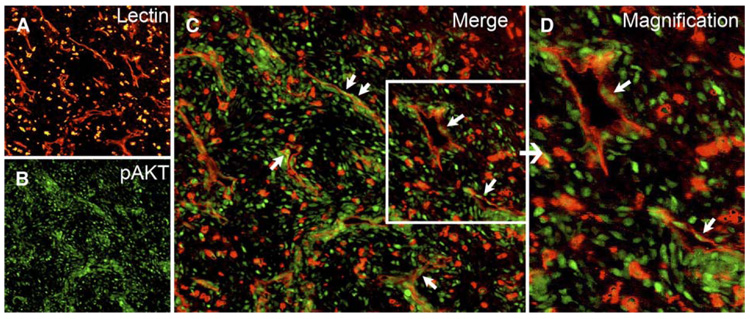
A–D: VEGF-A-expressing rat C6 glioma cells grown subcutaneously in Nu/Nu mice were double stained with TRITC-lectin (A, red) to label blood vessels and anti-phosphorylated Akt antibody (B, green). Merged images are shown in C, with arrows indicating endothelial cell nuclei positive for phosphorylated Akt. Phosphorylated Akt is also seen in tumor cells. A higher magnification of the blood vessels within the boxed area is shown in D.
Sustained myrAkt1 activation in endothelial cells induces surrogate tumor blood vessels in nontumor tissue
To mimic chronically activated Akt in tumor blood vessels in an otherwise normal microenvironment, we used a transgenic model of tetracycline-repressible endothelial-specific Akt activation (Sun et al., 2005). Overexpression of myrAkt1 in endothelial cells results in embryonic lethality but is overcome by maintaining mothers and their offspring on tetracycline. When the mice were 6–8 weeks of age, tetracycline was withdrawn for various lengths of time to induce myrAkt1 expression. Endothelial cell-specific expression of the transgene was also demonstrated by crossing the VE-cadherin:tTA line with a reporter strain that has the tetracycline-responsive promoter TET driving lacZ gene expression (TET:lacZ). VE-cadherin:tTA × TET:lacZ double transgenic mice expressed β-galactosidase in endothelial cells in the skin in adult mice (Figure 2A). For these studies we have used two lines of VE-cadherin:tTA, the D5 line with higher expression and the D4 line with lower expression of tTA. In combination with the same responder line carrying the TET:myrAkt1 transgene, D5 animals develop an overt vascular phenotype in 8–12 days after tetracycline withdrawal, while D4 animals require 6–7 weeks to produce a similar phenotype. We examined the skin blood vessels of normal and double transgenic mice for structural alterations and Akt signaling.
Figure 2. Pathological blood vessel formation in double transgenic myrAkt1 mice.
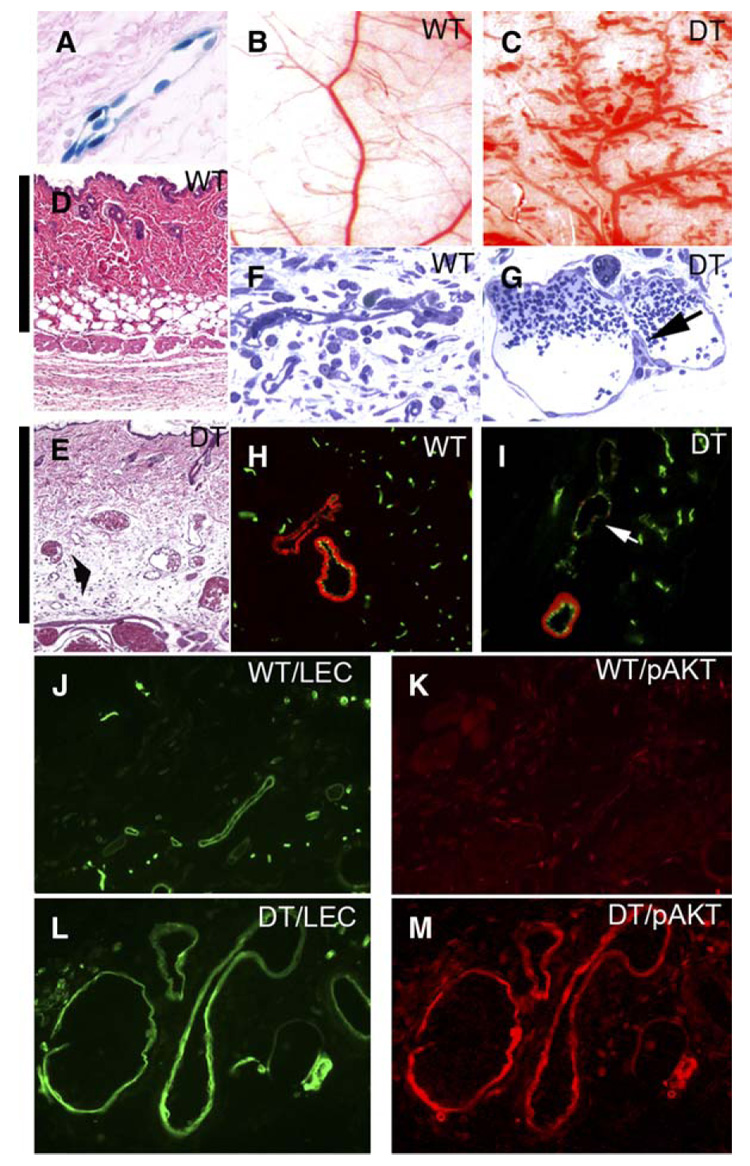
A: VE-Cadherin:†TA line drives expression of TET:lacZ reporter gene in adult vasculature as shown by histology of the skin expressing β-galactosidase activity (blue stain) in endothelial cells.
B–E: Whole tissue view of the flank skin of wild-type (WT) mice (B) and double transgenic (DT) littermates (C). Corresponding histology sections of wild-type (D) and double transgenic (E) skin taken at the same magnification showing enlarged vessels in double transgenic skin. Cutaneous edema separating dermal collagen is shown (E, arrow). Note the increase in thickness of double transgenic dermis from edema as indicated by a bar on the left side of each panel.
F and G: One micrometer sections of blood vessels in wild-type (F) and double transgenic (G) mice. MyrAkt1 expression induced enlarged vessels with elongated endothelial cells and transluminal bridges (arrow).
H and I: Skin sections from wild-type (H) and double transgenic mice (I) were analyzed for the presence of smooth muscle cells/pericytes by double immunofluorescent staining with anti-SMA antibody (red) and BS-1 lectin (green), which labels endothelial cells. Arrow in I shows enlarged vessel with deficient SMA staining.
J–M: Increased levels of activated Akt in the vasculature in double transgenic myrAkt1 mice. Wild-type skin stained with FITC-lectin (J, green) to label blood vessels, and anti-phosphorylated Akt antibody (K, red). The same magnification of myrAkt1 double transgenic skin labeled with FITC-lectin (L, green) and anti-phosphorylated Akt antibody (M, red).
Necropsy of double transgenic mice after myrAkt1 induction demonstrated marked changes in the vasculature in most organs, most notably in the skin and subcutaneous fat, mammary fat pads, visceral fat, retina, lungs, uterus, testes, and skeletal muscle. Many vessels in these mice became abnormally enlarged and irregular with multifocal areas of “aneurysm” or “balloon-like” enlargement (Figures 2B and 2C). The percentage of the skin surface covered by blood vessels in double transgenic mice was approximately twice that of wild-type control littermates (37.029% ± 8.092% versus 17.809% ± 4.652%, respectively; p < 0.01; controls n = 11; double transgenics n = 14).
The dermis and subcutaneous tissue were significantly thickened as the result of edema in double transgenic mice (note vertical bar covering the distance from the epidermis to the subcutaneous muscle layer) (Figures 2D and 2E). Edema was also evident as separation of dermal collagen bundles (Figure 2E). Edema was also present in other tissues, such as the lungs, resulting in increased wet lung weight in double transgenic mice compared to control mice (0.26 ± 0.08 g versus 0.17 ± 0.04 g, respectively; n = 4 animals per group). There was substantial accumulation of fluid in the pleural cavity in double transgenic mice compared to controls (1.1 ± 0.2 ml versus 0 ml, respectively; n = 4 animals per group). Enlarged blood vessels were lined with a single layer of elongated endothelial cells (Figures 2F and 2G). Some of these vessels contained structures previously described as “transluminal bridges” seen in “mother vessels” (Dvorak, 2002; Pettersson et al., 2000) in response to VEGF-A stimulation (Figure 2G, arrow). These bridges represent endothelial cell cytoplasmic processes that project into and across the vessel lumens and are thought to divide blood flow into smaller-sized channels as “mother vessels” evolve into “daughter vessels.” Similar to the reported features of deficient mural support in tumor blood vessels, many of these enlarged vessels were partially deficient in mural cell coverage (i.e., pericytes and smooth muscle cells). Figure 2H shows wild-type skin with many small capillaries and a pair of larger vessels, both of which were covered by a complete layer of smooth muscle actin (SMA)-positive cells. In double transgenic mice, some large SMA-positive vessels persisted, but there were also enlarged vessels deficient in mural cell coverage (Figure 2I, arrow). Tissues from wild-type and double transgenic littermates were stained with BS-1 lectin to highlight blood vessels and with antibodies to pAkt. Skin vessels from wild-type mice were smaller in size and had little activated Akt (Figures 2J and 2K). In contrast, pAkt was strongly expressed in endothelial cells of enlarged blood vessels in double transgenic mice (Figures 2L and 2M).
Sustained endothelial Akt activation leads to increased blood volume and leaky blood vessels
We determined the changes in blood volume and vascular permeability in the whole animal in double transgenic mice and control littermates by MRI following intravenous (i.v.) injection of the contrast agent biotin-BSA-Gadolinium-DTPA (biotin-BSA-Gd-DTPA). Intravascular contrast enhancement, as detected immediately after administration of biotin-BSA-Gd-DTPA, was significantly lower in the double transgenic mice relative to control mice (Figure 3A). The lower enhancement in double transgenic mice corresponded to a significantly increased total blood volume over which the same amount of contrast material distributed (p = 0.007) (Figure 3B). Blood volume in various tissues (indicated by blood volume fraction [fBV]) was obtained from the ratio of the contrast agent concentration in the tissue at time zero to the concentration of the contrast agent in the vena cava blood at time zero. The fBV in myrAkt1 double transgenic mice was elevated in all tissues examined, including brain, hind-limb, liver, and kidney, with statistically significant differences seen in the brain and hindlimb of myrAkt1 mice compared to control animals (brain, p = 0.005; limb, p = 0.003) (Figure 3C). These findings are consistent with the increase in microvessel diameter. The changes in fBV in the liver and the kidney were not statistically significant due to large variance in measurements. The changes in intravascular contrast material concentration over time were used as a way to assess vascular permeability. As for most other macromolecules, the leakage rate of biotin-BSA-Gd-DTPA across normal vessels was relatively low. Hyperpermeable vessels would be expected to leak contrast agent at a higher rate. The extravasation rate (indicated by permeability surface area product [PS]) was calculated as the rate of contrast accumulation in the tissues as a function of time. Although a trend of higher permeability was seen in many organs in double transgenic mice, only the liver showed statistically significant higher permeability over the course of 15 min (p = 0.01) (Figure 3D). All tissues were harvested, fixed, and examined histologically. In some organs, such as skeletal muscle, even though quantitation of vascular leak in the relatively short time period assayed by MRI was not dramatic (Figure 3D), clear evidence of edema was easily observed by separation in myofibers in hematoxylin and eosin-stained sections (Figures 3E and 3F).
Figure 3. MRI studies of the effects of endothelial myrAkt1 on blood volume and vascular permeability.
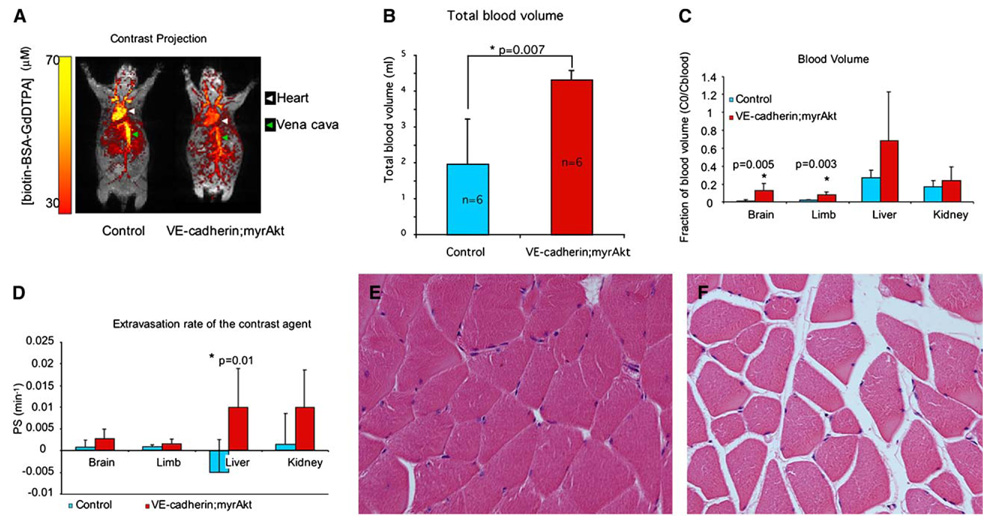
A: Mice were taken off tetracycline for 7 days, then imaged by dynamic contrast enhanced MRI using intravenously administered biotin-BSA-Gd-DTPA. The intravascular distribution of contrast agent in vivo is seen in overlay of maximal intensity projections maps from images that were taken prior to contrast injection (gray) and immediately after contrast injection (color).
B: Total blood volume was significantly higher in myrAkt1 double transgenic mice than in control mice (p = 0.007; n = 6 mice per group).
C: Significant changes in blood volume fraction (fBV) were observed in the brain and hindlimb of double transgenicmice as compared to control mice (brain, p = 0.005; hindlimb, p = 0.003; n = 6 mice per group).
D: Increased permeability surface area product (PS) was observed in the brain, limb, liver, and kidney, with statistically significant differences seen in the liver (p = 0.01; n = 6 mice per group).
E and F: Edema is seen in skeletal muscles in double transgenic mice as shown by separation of muscle fibers in these mice (F) as compared to controlmice (E). Bars in graphs represent means ± standard deviations.
We also employed additional methods to explore vascular permeability in the vessels of double transgenic mice and control littermates. Intravenous injection of the macromolecular tracer dextran (70 kDa) conjugated to the fluorophore FITC allows one to visualize alterations in vascular structure and vascular leak after 2 hr. Abnormal blood vessels in double transgenic animals were enlarged and irregularly shaped, and the extravasated FITC-dextran left a perivascular fluorescent haze not observed around normal vessels in wild-type mice (Figures 4A and 4B).
Figure 4. Sustained endothelial Akt activation leads to structural and functional changes.
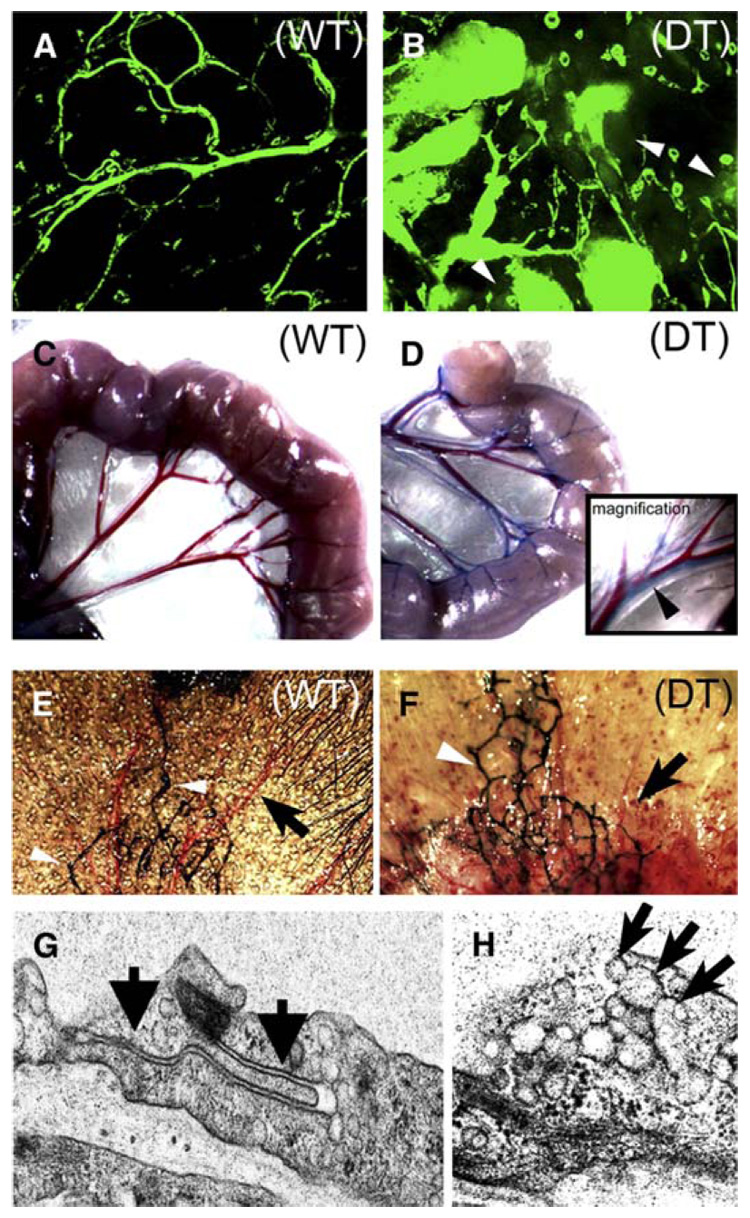
A and B: Vascular permeability was assessed in wild-type (WT) and double transgenic (DT) littermates. Confocal microscopic images of skin from wild-type (A) and double transgenic (B) mice after FITC-dextran perfusion for 2 hr. Arrows indicate extravasated FITC-dextran into the interstitial space.
C–F: Lymphatic structure and function were assessed in control and double transgenic animals. Lymphatic function was demonstrated by Evans blue dye extravasation and lymphatic uptake in wild-type (C) and double transgenic (D)mesentery. Inset in D shows higher magnification of mesenteric lymphatic (blue, arrow)and adjacent blood vessels (red). Intralymphatic injection of colloidal carbon (India ink) in the mouse ear highlights normal lymphatic network (white arrows) in the ear skin in both wild-type (E) and double transgenic (F) mice. The blood vessels in double transgenic mice are enlarged compared to the normal vascular pattern in control mice (black arrows).
G and H: Electron microscopic studies of double transgenic endothelial cells demonstrate intact and closed intercellular junctions (G, arrows) and many VVOs (H, arrows).
Edema can result from increased vascular permeability and from defective lymphatic drainage. Therefore, we investigated the functional status of lymphatics in double transgenic mice. Evans blue dye, which binds albumin and allows us to measure the extravasation of plasma proteins, was injected i.v. Uptake of the dye in the intestine lymphatic network was evident after 30 min in double transgenic mice, indicating that blood vessels had leaked dye and lymphatic vessels were functioning to reabsorb it (Figures 4C and 4D). We visualized the structure of the lymphatics following injection of colloidal carbon in the terminal lymphatics in the ears of wild-type and double transgenic mice. Colloidal carbon entered the lymphatics normally and revealed a normal pattern of lymphatic valves and branching networks in both control and double transgenic mice (Figures 4E and 4F). Electron microscopy was used to investigate the structure of the enlarged blood vessels in double transgenic mice. Examination of many endothelial cell junctions in multiple skin sections from several double transgenic and control animals did not reveal evidence of junctional disassembly (Figure 4G). VVOs, structures that provide one pathway of transport of macromolecules across endothelial cell membranes (Feng et al., 1996, 2000b), were normally present in the endothelial cells lining these vessels (Figure 4H). The VVOs in double transgenic mice and wild-type littermates were similar in number and ultrastructural morphology. Although these findings do not conclusively determine the cellular mechanism of vascular leak, they suggest that VVOs are present and may to contribute to the edema observed.
Acute versus chronic permeability in Akt1 null and myrAkt1 transgenic mice
Vascular permeability can be quantified by measuring the amount of Evans blue dye that leaks from otherwise equal vascular beds into the surrounding tissue following a stimulus. However, there is a basal level of transvascular flux of fluid and protein that can be similarly quantified with longer measurements than those needed to see permeability in response to a stimulus. However, when the vascular beds are unequal, other factors contribute to the final quantity of Evans blue accumulation in the tissue besides “permeability” of the endothelium, including vascular density and overall endothelial surface area. We will suggest that the molecular pathways for the rapid extravasation of Evans blue to stimuli such as VEGF-A and the more gradual leakage that occurs over longer periods of time are different. For the sake of our discussion, we are distinguishing these by the terms “acute” versus “chronic” permeability. The acute vascular permeability response to VEGF-A is typically measured in 15–30 min; however, if one waits long enough even normal blood vessels allow Evans blue to extravasate. By 2–3 hr the skin of the myrAkt1 transgenic mice was visibly more blue, and by 24 hr even the wild-type mice were blue. To measure this chronic leak of Evans blue, we quantified extravasation at 8 hr. Because we wished to minimize alternative explanations for the increased extravasation, such as differences in endothelial surface area, we used animals that had been removed from tetracycline for 5 days and had very little change to the vessel structure and number in order to attempt to separate between the structural alterations in the blood vessels and the signaling that could impact these measurements (Figure 5A). Even before dramatic alterations in vessel structure and number were evident, we observed significantly increased extravasation of Evans blue in myrAkt1 double transgenic mice, which we interpreted as a result of chronic vascular permeability (Figure 5B). The Evans blue accumulation in the lungs is shown prior to extraction (Figure 5C). To assess acute permeability in response to VEGF-A, a Miles assay was performed following intradermal injection of VEGF-A (Figure 5D). Extravasation of Evans blue dye in double transgenic mice was not statistically different from control animals (Figure 5E). Recent reports have demonstrated that acute permeability to VEGF-A in a Miles assay was blunted in the Akt1−/− mice (Ackah et al., 2005). We could not find significant differences in permeability to Evans blue or to alterations in the vascular structure of the Akt+/+, Akt1+/−, or Akt1−/− mice (Figures 5F–5H). Taken together these data suggest that Akt1 is required for acute permeability in response to VEGF-A but that chronic Akt1 activation leads to low-level chronic vascular leak accompanied by edema. Despite our attempts to measure chronic permeability before the vasculature had become significantly abnormal, we cannot rule out the possibility that this chronic permeability is secondary to Akt-induced morphological changes including factors such as reduced pericyte-endothelial interactions. The overall health and normal vascular structure of the Akt1 null mice in combination with these data contributes to our emerging hypothesis that Akt1 is critical for pathological vascular responses but not for those regulated in a physiological manner in healthy adult vascular homeostasis.
Figure 5. Baseline vascular permeability is increased in double transgenic myrAkt1 mice but not in Akt1 null mice.
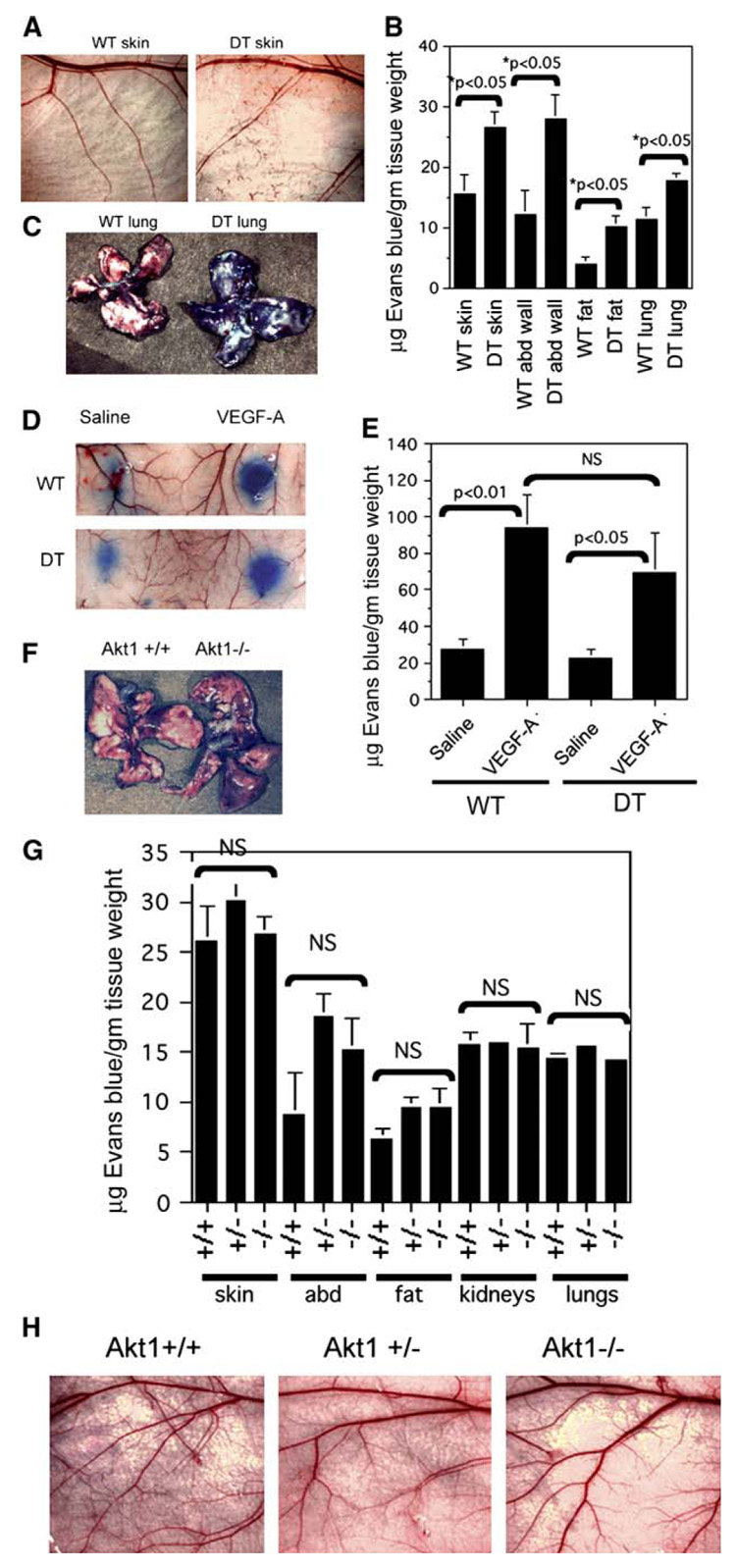
A–C: Wild-type (WT) and double transgenic (DT) mice were taken off tetracycline for 5 days prior to analysis of baseline permeability. Evans blue dye (50 mg/kg) was injected i.v. and allowed to circulate for 8 hr, at which time the animals were sacrificed and various organs were collected for analysis. Relatively normal skin vasculature in wild-type and double transgenic mice is shown in A. Evans blue dye was extracted from various organs, and the amount of extracted dye was measured by spectrophotometric absorbance at 620 nm (B). Lungs from WT and DT mice 8 hr after Evans blue injection (C).
D and E: Miles assay in WT and DT mice that were off tetracycline for 5 days. Evans blue dye was injected in the tail vein. Saline or VEGF-A was immediately injected intradermally in the back skin (D). Quantification of Evans blue dye extracted from the skin (E).
F–H: Baseline vascular permeability in Akt1−/− mice and control littermates was determined as described in A. F: Whole lungs taken from Akt1+/+ and Akt1−/− mice 8 hr after Evans blue injection. G: Quantitation of extravasated Evans blue in various organs from Akt1+/+, Akt1+/−, and Akt1−/− mice. H: Normal vasculature is seen in the flank skin in these mice. Data from all the experiments above represented four mice per group and were calculated as micrograms of dye per gram tissue weight (mean ± SEM). p value < 0.05 was considered statistically significant. NS is not statistically significant.
Plasticity of the adult vasculature in response to induction followed by suppression of sustained endothelial Akt signaling
In order to determine whether the vascular abnormalities induced by myrAkt1 could be reversed, we first removed tetracycline from the drinking water of double transgenic mice to induce myrAkt1 expression, allowed the vascular phenotype to become fully developed, and then restored tetracycline. These animals without tetracycline became sickly, with marked edema and fluid accumulation making the animals appear fat, with ruffled fur, nearly closed eyelids, and vascular anomalies in the ears (Figure 6B) and flank skin (Figure 6F). Even in our lower expresser D4 line, in which the phenotype developed slowly over 6–7 weeks, the vascular abnormalities were still reversible. Within a matter of days following readministration of tetracycline, a general resolution of fluid and healthy appearance returned. The same mouse shown in Figures 6A and 6B now appears healthy, with a normal-appearing slim body, smooth hair, and open eyes, although the vascular anomalies in the ears were still apparent (Figure 6C). After 4 weeks back on tetracycline, the vascular anomalies had largely disappeared (Figures 6D and 6G). These results demonstrate the enormous “plasticity” of the microvasculature in the adult animal in response to the induction or suppression of endothelial Akt signaling.
Figure 6. Reversibility of the vascular phenotype in double transgenic myrAkt1 mice.
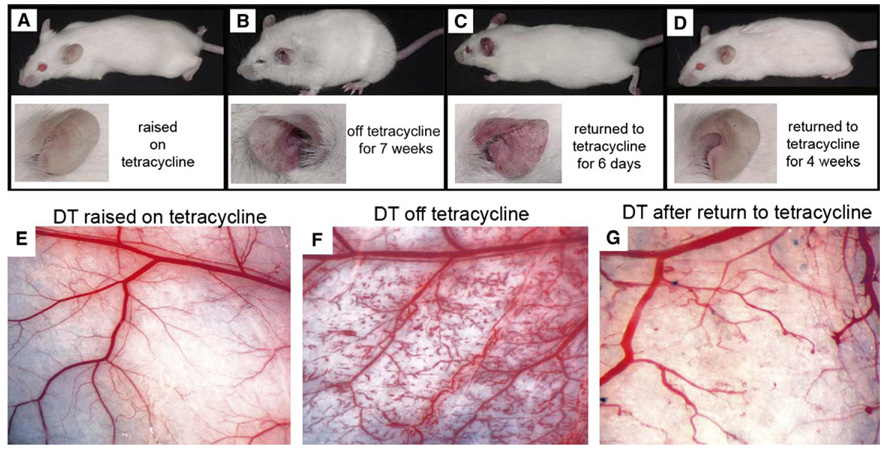
A–D: The same double transgenic mouse was followed over a time course of tetracycline administration. Images of the whole body and ear are shown for the mouse raised to maturity on tetracycline (A), followed by tetracycline withdrawal for 7 weeks (B), then back on tetracycline for 6 days (C) and 4 weeks (D).
E–G: The normal cutaneous vasculature in similarly treated representative double transgenic mice (E) was dramatically altered in response to tetracycline withdrawal (F). Restoration of a near-normal vascular pattern was observed when these animals were placed back on tetracycline for 4 weeks (G).
Induction of myrAkt1 expression in endothelial cells leads to activation of Akt and its canonical downstream effectors
To investigate the signaling pathways downstream of Akt in our animal model, we isolated primary endothelial cells from double transgenic animals. Culture purity was assessed by uptake of DiI-acetylated LDL and immunoreactivity to CD31 (Figures S2A–S2C). There were enhanced levels of pAkt in the absence of tetracycline but no changes in the levels of other signaling pathways also activated by VEGF, such as phosphorylated Erk-1/2 (Figure S2D). Moreover, myrAkt1 expression was associated with activation of downstream signaling pathways, as evidenced by increased phosphorylation of mTOR, GSK-3β, and eNOS (Figure S2E). Total levels of these proteins were unchanged by tetracycline conditions and myrAkt1 expression. Taken together, these data demonstrate the specificity of myrAkt1 in inducing Akt signaling pathways in double transgenic endothelial cells.
Investigation of eNOS in VEGF-A-induced acute permeability and myrAkt1-induced chronic permeability
We investigated the contribution of eNOS activation to the morphological and functional changes in double transgenic animals. VEGF-A-induced vascular permeability in endothelial cells has been shown to be dependent on eNOS activity. However, a correlation to vascular morphology has not been reported. To explore the potential role of eNOS in mediating the effects of myrAkt1, we treated double transgenic mice and wild-type littermates with antennapedia-caveolin-1 peptide (AP-Cav), an inhibitor of eNOS that does not block nNOS, iNOS, or Src phosphorylation (Bucci et al., 2000; Gratton et al., 2003). Daily dosing of the AP-Cav peptide reduces tumor-associated eNOS activity and tumor progression (Gratton et al., 2003). We confirmed that administration of AP-Cav blocked VEGF-A-induced permeability using the Miles assay as previously reported (Gratton et al., 2003). We used a dose of AP-Cav that was shown to be sufficient to block VEGF-A-induced vascular permeability and tumor growth in vivo. In the Miles assay, mice were pretreated with either AP-Cav peptide or control an-tennapedia peptide without the active caveolin-1 domain (AP) for 45 min prior to intravenous administration of the tracer Evans blue dye. To induce vascular permeability, either saline or VEGF-A was injected intradermally in the mouse ear. After 30 min, there was increased extravasation of Evans blue dye in response to VEGF-A in mice pretreated with control AP peptide (Figure S3A). In contrast, VEGF-A did not increase Evans blue extravasation in mice pretreated with AP-Cav, demonstrating that AP-Cav was effective in blocking VEGF-A-induced vascular permeability. The differences in Evans blue extravasation between AP- and AP-Cav-treated animals were statistically significant (Figure S3B).
To determine whether eNOS mediates myrAkt1-induced chronic permeability, wild-type and double transgenic myrAkt1 littermates were taken off tetracycline and injected intraperitoneally (i.p.) with AP-Cav or AP peptides each day for 7 days using a protocol that was efficacious in reducing tumor growth and permeability (Gratton et al., 2003). On day 8 of the study, the mice were injected intravenously with the macromolecular tracer FITC-dextran to perfuse the vasculature. After 2 hr, the flank skin was harvested and analyzed by confocal microscopy for extravasation of FITC-dextran. AP-Cav, at concentrations that effectively inhibit eNOS in vivo (Gratton et al., 2003), did not block the development of pathological blood vessel morphology or vascular permeability in myrAkt1-expressing mice (Figures S3D and S3H compared to Figures S3E and S3I). Similar results were obtained when the dose of AP-Cav was doubled, or when N-nitro-L-arginine methyl ester (L-NAME), a structurally different eNOS inhibitor, was used (Figure S3F). Thus, we cannot find an indication of eNOS requirement in myrAkt1-induced structural alterations or chronic permeability.
Rapamycin blocks pathological blood vessel formation and vascular permeability
Rapamycin is a highly specific inhibitor of mTOR that is also an immunosuppressant that is under investigation as an anticancer therapy (Bjornsti and Houghton, 2004). Studies using rapamycin have suggested that it may be antiangiogenic (Guba et al., 2002). To determine whether the mTOR inhibitor rapamycin inhibits vascular permeability induced by VEGF-A, we pretreated Nu/Nu mice with rapamycin or control solvent for 24 hr prior to performing a Miles assay. We observed increased extravasation of Evans blue dye in response to VEGF-A in mice pretreated with control solvent but not in mice pretreated with rapamycin (Figures 7A–7C). This demonstrated that rapamycin was effective in blocking VEGF-A-induced acute vascular permeability in a normal vasculature. To determine whether rapamycin inhibits the formation of pathological blood vessels in double transgenic myrAkt1 mice, the mice were taken off tetracycline and on the same day were injected i.p. with rapamycin or vehicle for 7 days. Treatment with rapamycin almost completely blocked the formation of pathological blood vessels in myrAkt1 transgenic mice (Figures 7D–7F). Rapamycin also blocked the extravasation of FITC-dextran (Figures 7G–7I). Necropsy examination of rapamycin-treated mice showed that these animals did not develop tissue edema or pleural fluid.
Figure 7. Effects of rapamycin on pathological vessel formation in double transgenic myrAkt1 mice.
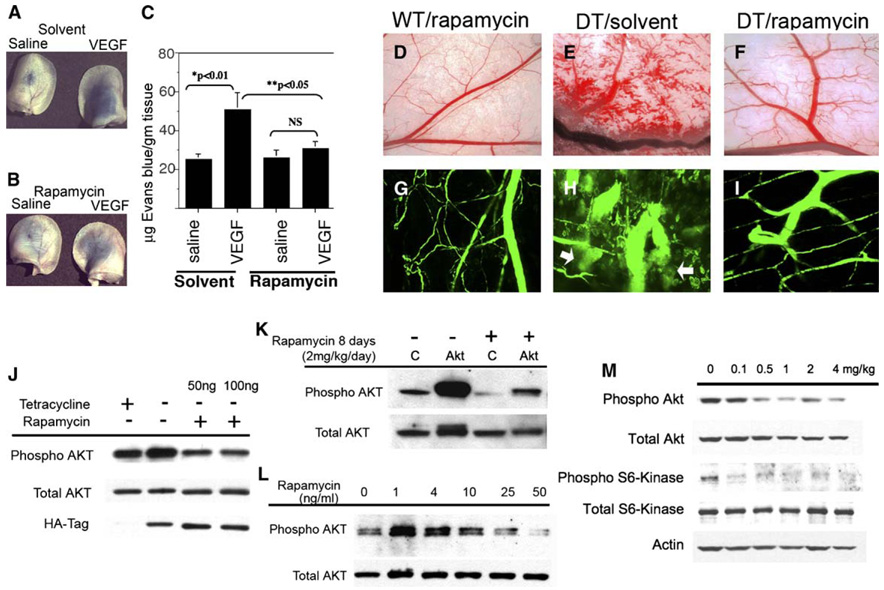
A–C: Effects of rapamycin on VEGF-A-induced vascular permeability. Athymic Nu/Nu mice were pretreated with either solvent (A) or rapamycin (4 mg/kg, intraperitoneal) (B) for 24 hr prior to intravenous injection of Evans blue dye. Immediately following Evans blue injection, saline or VEGF-A was injected intradermally in the ears and back skin. Photographs of extravasated Evans blue in the ears of treated animals are shown. Quantitation of Evans blue dye extracted from the ears and skin (C). Data represent five to six mice per rapamycin- or solvent-treated group, three sites per saline or VEGF-A treatment, and were calculated as micrograms of dye per gram tissue weight (mean ± SEM). p value < 0.05 was considered statistically significant. NS, not statistically significant.
D–I: Wild-type (WT) and double transgenic (DT) littermates were taken off tetracycline to induce myrAkt1 expression. Starting on the same day, they were given daily intraperitoneal injections of rapamycin or solvent for 7 days. On day 8, the mice were perfused intravenously with FITC-dextran for 60 min, and the flank skin was examined (D–F) (whole tissue view, magnification ×2.5) and harvested for confocal microscopy (G–I) (magnification x200). Extravasation of FITC-dextran from the blood vessels in solvent-treated DT mice is shown in H (arrows).
J: Double transgenic endothelial cells were treated for 48 hr ± tetracycline ± rapamycin and analyzed by Western blot for phosphorylated Akt and expression of HA-tagged myrAkt1 transgene.
K: Control (C) and double transgenic myrAkt1 mice were taken off tetracycline and starting on the same day injected intraperitoneally with rapamycin for 4 days. The animals were sacrificed and the kidneys were harvested and analyzed by Western blot for phosphorylated and total Akt.
L: Primary human dermal microvascular endothelial cells were treated with rapamycin (1–50 ng/ml) for 3 days and analyzed by Western blot for phosphorylated and total Akt.
M: Inbred FVBmice were injected intraperitoneally with rapamycin (0.1–4 mg/kg/day) for 4 days. The animals were sacrificed, and the kidneys were harvested and analyzed by Western blot.
Surprisingly, in myrAkt1-expressing endothelial cells derived from double transgenic mice, rapamycin reduced pAkt levels (Figure 7J). Similarly, treatment with rapamycin was also found to inhibit pAkt levels in kidneys of both wild-type and double transgenic mice (Figure 7K). To determine whether the anti-pAkt effects of rapamycin were independent of myrAkt1, we performed dose response studies in primary endothelial cells isolated from human skin. After 3 days we observed an increase in pAkt levels with 1–4 ng/ml rapamycin, followed by a dose-dependent decrease from 10 to 50 ng/ml (Figure 7L). To determine whether the effects of rapamycin on pAkt were within the range of rapamycin given to patients, we performed a dose response study on wild-type mice and correlated the blood concentrations of rapamycin to the effects on phosphorylation of Akt and S6 Kinase, a downstream molecule activated by mTOR. Mouse blood was analyzed for rapamycin levels by our clinical lab, where similar assays are performed on blood samples from patients treated with rapamycin. We found that treatment of mice with rapamycin in the dose range of 0.5 mg/kg (7.1 ± 0.1 ng/ml trough blood levels) to 1 mg/kg (16.9 ± 6.2 ng/ml trough blood levels) matched the optimal trough blood levels of 6–15 ng/ml in humans (Meier-Kriesche and Kaplan, 2000). At this dose we have decreased pS6 Kinase and pAkt levels in whole kidney lysates after 4 days (Figure 7M). Our findings suggest that long-term treatment with rapamycin may be an effective “Akt inhibitor” of endothelial cells, and perhaps of other normal cells in vitro and in vivo.
Rapamycin reduces tumor growth and tumor vascular permeability
Rapamycin is known to inhibit tumor growth and tumor angiogenesis (Guba et al., 2002). We investigated whether rapamycin treatment also affects tumor vessel permeability. VEGF-A-expressing C6 rat glioma cells were inoculated subcutaneously into Nu/Nu mice, allowed to grow to approximately 0.1 cm3, and then treated with either control solvent or rapamycin for 4, 9, and 12 days. Treatment of the animals with rapamycin did not influence normal bodily functions and behavior such as weight gain, appetite, or grooming behavior. Similar to the published studies, rapamycin significantly reduced tumor growth after 7 days in Nu/Nu mice (Figure 8A). The final tumor volumes at day 12 were 1.49 ± 0.49 cm3 (solvent) and 0.40 ± 0.18 cm3 (rapamycin). Statistically significant reductions in C6 tumor growth were also observed after 8 days of drug treatment at much lower doses of rapamycin, for example, compared to solvent, which had tumor volumes of 1.10 ± 0.82 cm3, at 0.5 mg/kg/day, the volumes were 0.37 ± 0.23 cm3, and at 1 mg/kg/day they were 0.32 ± 0.15 cm3 (n = 6 tumors/group). Tumor vascular permeability was assayed by i.v. injection of Evans blue dye as previously reported (Gratton et al., 2003). Evans blue in the tumor tissue was significantly reduced after 12 days of rapamycin treatment (Figure 8B). In contrast to our observations of normal kidneys and primary human endothelial cells, Western blot of total tumor tissue lysates indicated that rapamycin effectively inhibited mTOR signaling (indicated by decreased S6 Kinase phosphorylation) but increased pAkt levels in the total tumor lysates (Figure 8C). We attribute this increase in pAkt levels to the effects of rapamycin in increasing pAkt in tumor cells rather than stromal cells, as tumor cells make up most of the cellular mass of this tumor. This interpretation was further supported by immunohistochemistry with anti-CD31 antibodies to label endothelial cells (green), anti-pAkt antibodies (red), and Hoechst dye to label nuclei (blue) (Figures 8D and 8E). In solvent-treated animals, pAkt was detected at high levels in both tumor cells and blood vessels, but in the rapamycin-treated animals, pAkt levels in tumor blood vessels were clearly reduced in comparison to the surrounding tumor. While overall pAkt levels from sample to sample cannot be accurately assessed by this method, we can conclude that the response of the vascular stroma compared to the adjacent tumor was a dramatic relative decrease in endothelial pAkt levels. These data suggest that clinically relevant doses of rapamycin decrease tumor growth, decrease Akt signaling in the vascular stroma, but at least in C6 glioma increase pAkt signaling in the tumor. Additional studies are needed to determine whether decreases in tumor growth are strictly due to antiangiogenic effects, or to both antiangiogenic and antitumor effects that result from loss of downstream mTOR signaling irrespective of pAkt signaling. Similarly, more tumor studies are needed to determine if the increase in pAkt levels in C6 tumors following rapamycin treatment is representative of all tumor types or varies from tumor to tumor.
Figure 8. Effects of rapamycin on C6 rat glioma tumor.
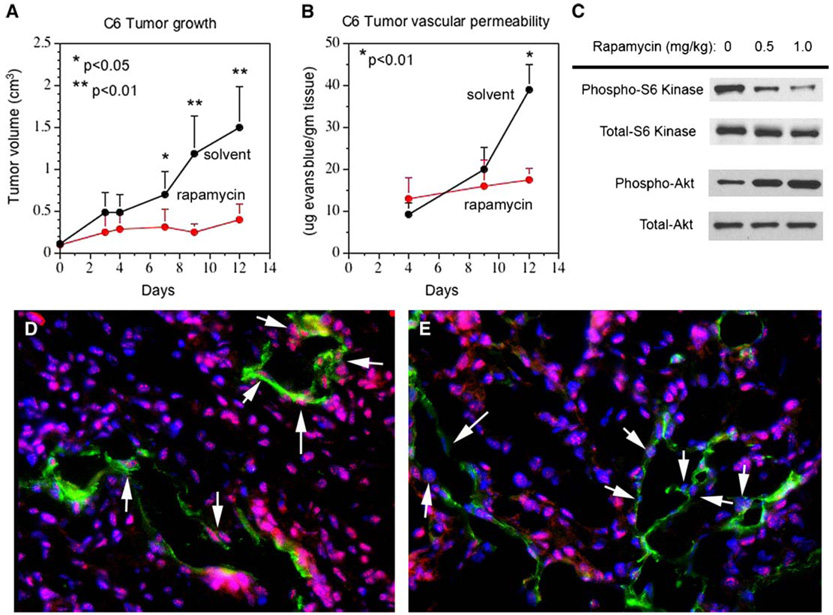
A: Athymic Nu/Nu mice were injected subcutaneously with C6 rat glioma cells expressing VEGF-A. Tumors were allowed to grow to ~0.1 cm3 before randomization into solvent or rapamycin (4 mg/kg/day) treatment group. Tumor size was measured every 2–3 days with a caliper. Tumor growth is presented as the mean ± SD, n = 4 mice per treatment group, two tumor sites per mouse. p values were obtained by comparison with solvent-treated group.
B: The mice in A were injected with Evans blue dye (50 mg/kg, i.v.) and allowed to circulate for 30 min before perfusion with 100 cc of saline to clear intravascular Evans blue. Tumors were resected and Evans blue content was quantified (n = 4 mice per treatment group). p values were obtained by comparison with solvent-treated group.
C: Nu/Nu mice bearing C6 tumors were treated ± rapamycin (0.5 or 1.0 mg/kg/day) for 8 days. Tumors were harvested for Western blot analysis of phosphorylated and total S6 kinase and Akt.
D and E: Frozen sections of C6 tumors from C were immunostained for phosphorylated Akt (red) and CD31 (green) to label blood vessels. Nuclei were counterstained with Hoechst dye (blue). Arrows indicate endothelial cell nuclei positive for phosphorylated Akt in solvent-treated tumors (D) and loss of phosphorylated Akt in endothelial cells in rapamycin-treated tumors (E). Magnification x200.
Discussion
Unlike the normal vasculature, vessels found in solid tumors are irregularly shaped, enlarged, tortuous, leaky, and often deficient in normal pericyte interactions. These features are recapitulated by VEGF-A overexpression in tumors and in nontumor tissues (Dvorak, 2003). We have shown that chronic activation of endothelial Akt signaling alone is sufficient to induce these pathological vessels in a nontumor microenvironment. In our model, we have used a myristoylated Akt1 transgene, the major isoform in endothelial cells. In the recent study by Ackah et al., Akt1 null mice were used in a model of hindlimb ischemia. In response to VEGF-A administration, both angiogenesis and acute permeability were blunted (Ackah et al., 2005). This is consistent with the work of many investigators in vitro and in vivo, with the exception of a recent publication where permeability was assessed in a Matrigel plug assay (Chen et al., 2005). In that model, blood vessel invasion into Matrigel can be accompanied by hemorrhage depending on the experimental conditions. However, since Akt1 null mice have a clotting defect (Chen et al., 2004) and platelet clotting can play an important role in preventing hemorrhage during angiogenesis (Kisucka et al., 2006), it is possible that the resultant bloody plug and the extravasation of Evans blue observed were due to hemorrhage rather than vascular permeability. Our model differs from those of the Akt1 null mice where all cells are deficient for Akt1 and allows us to examine endothelial cell autonomous signaling. Our data are consistent with many studies that report proangiogenic effects of Akt1, and the antiangiogenic effects on vascular function that we observed with rapamycin in mice further support our conclusions. However, we cannot rule out the possibility that myrAkt1 in our model may function to mimic other isoforms or have some other unknown functions not attributable to endogenous Akt1. The hypothesis that Akt1 is more critical for pathological angiogenesis than vascular homeostasis is also similar to what has been observed in the Akt1 null mice with respect to glucose homeostasis (Chen et al., 2001; Cho et al., 2001a).
Our data also shows that the pathological vessels induced by myrAkt1 require sustained Akt signaling for their maintenance even when minimal overexpression in the D4 line is accompanied by gradual angiogenesis over the course of 7 weeks. This is reminiscent of the continued reliance of tumor blood vessels on VEGF-A where removal of VEGF-A in tumor blood vessels leads to regression of newly formed blood vessels (Benjamin et al., 1999; Benjamin and Keshet, 1997). Similarly, “mother vessels” induced by adenoviral vectors expressing VEGF-A, and the dramatic vascular alterations in multiple organs following VEGF-A overexpression, all regress as VEGF-A levels decline (Dor et al., 2002; Nagy et al., 2002; Ozawa et al., 2004; Pettersson et al., 2000). Even some normal vascular beds retain a dependence on VEGF-A (Kamba et al., 2005). These data combined demonstrate the fragility of pathological blood vessels after VEGF-A overexpression. The fragility of Akt-induced blood vessels, even after long periods of time, underscores our hypothesis that Akt signaling downstream of VEGF-A may be mediating many of the VEGF-A-associated phenotypes and strengthens the rationale for Akt inhibition to decrease angiogenesis and excessive vascular permeability in cancer. Although it is beneficial for tumor blood vessels to be permeable to chemotherapy and other drugs, excess vascular leak leads to increased interstitial pressure, which is counterproductive for drug delivery. Thus, the desire for vascular normalization has become an important consideration in antiangiogenic therapy (Jain, 2005).
A major phenotype of endothelial myrAkt1-expressing mice is extensive tissue edema, most notably in the skin, soft tissues, and pleural space accompanied by eventual respiratory failure. Rapamycin was able to prevent VEGF-induced acute permeability and the development of pathological blood vessels and edema in double transgenic mice. However, the eNOS inhibitor AP-Cav only reduced acute vascular permeability in response to VEGF-A. This acute response has been previously thought to be mediated primarily by Akt to eNOS signaling (Bucci et al., 2000; Bucci et al., 2005; Fulton et al., 1999; Six et al., 2002). Whether or not there is a direct role of signaling downstream of mTOR in vascular permeability, or these findings are simply the result of rapamycin-induced Akt loss, remains to be determined. Different molecular pathways for acute and chronic vascular leak may explain why Akt-induced chronic permeability is less responsive to eNOS inhibitors. Similarly, we might have expected that chronic Akt activation in our animals would result in a loss of responsiveness to VEGF-A-induced acute permeability. Not only did we get a normal VEGF-A response in myrAkt1 transgenic animals, but it was notable that we did not get an elevated response. Future studies will be focused on determining whether chronic vascular leak is dependent on downstream pathways in response to Akt activation. Edema is a significant complication in many cancers that is contributed by tumor vascular leak as well as complicated by surgical damage to local lymphatics. The use of MRI for assessing vascular burden and vascular leak could be adapted to investigate the efficacy of antiangiogenic therapy in patients, especially in anti-VEGF-A-based therapy, which should similarly decrease tumor vessel leakage.
Feedback regulation of Akt by mTOR has recently been reported (Sarbassov dos et al., 2005). These studies show that the rictor-mTOR complex can phosphorylate Akt, thus making mTOR both downstream and upstream of Akt. Although this complex is rapamycin insensitive and the preferential reduction in mTORC1 by rapamycin results in increased Akt phosphorylation in short-term treatment, long-term treatment with rapamycin has been shown to partially inhibit Akt activation (Edinger et al., 2003). It has been demonstrated that rapamycin-based pAkt inhibition can be explained by an increasing sequestration of mTOR by rapamycin with secondary consequences on the assembly of mTORC2 (Sarbassov dos et al., 2006). Consistent with the proposed model, at lower rapamycin doses in culture we find elevation of pAkt, which steadily diminishes as the concentration of rapamycin is increased. In a recent study, rapamycin used at 10 mg/kg decreased pAkt levels in xenograft tumors (Sarbassov dos et al., 2006). We found that rapamycin doses between 0.5 and 1 mg/kg provide the recommended clinical blood levels and lead to tumor growth inhibition and stromal suppression of pAkt, but elevation of tumor pAkt. Consistent with our findings, it has been reported that some human cancer cell lines and cancer patients in rapamycin-based clinical trials have elevated tumor levels of pAkt in response to rapamycin (O’Reilly et al., 2006). This may suggest that differences reported in the literature on tumor cell Akt responses can be dose related. If human tumors do indeed depend on sustained endothelial Akt signaling to induce pathological blood vessel formation, our findings suggest that rapamycin treatment may be antiangiogenic via inhibition of endothelial Akt activation. Extrapolation of these findings may suggest that, in addition to reducing tumor vascularization, rapamycin treatment may reduce tumor vascular leak and interstitial pressure, and perhaps ascites accumulation and edema in some cancer patients. There have been concerns over using an immunosuppressant in cancer treatment, with the possibility that debilitating the immune system may work against the host’s ability to fight cancer and infections. Interestingly, one study reported a decrease in Kaposi’s sarcoma in renal transplant patients receiving rapamycin for immunosuppression (Stallone et al., 2005). Whether or not this protection will extend to nonvascular tumors remains to be determined. Clearly, rapamycin is an exciting therapy that should be further explored in cancer treatment as an antiangiogenic agent as well as an effective vascular Akt inhibitor.
Experimental procedures
Mice
The double transgenic mouse model that expresses myrAkt1 in endothelial cells under tetracycline control has been previously described (Sun et al., 2005). The Akt−/− mice were kindly provided by Dr. Morris Birnbaum (Cho et al., 2001b). All studies were conducted in compliance with the Beth Israel Deaconess Medical Center IACUC guidelines.
Inhibitor studies
Mice were given daily i.p. injections of rapamycin (0.1–4 mg/kg) (LLC Laboratories), NG-nitro-L-arginine methyl ester (L-NAME, 25 mg/kg/day), antennapedia-caveolin-1 peptide (AP-Cav, 2.5 mg/kg/day), control antennapedia peptide (AP, 1.2 mg/kg/day), or vehicle controls. Rapamycin was dissolved in solvent solution (0.2% carboxymethylcellulose and 0.25% Tween-80 in sterile H2O). L-NAME was dissolved in PBS. AP-Cav and AP peptides were prepared as stock solutions in DMSO and then dissolved to a final concentration in sterile H2O.
Details of the analysis for tumor growth and vascular function can be found in the Supplemental Data.
MRI analysis
Double transgenic mice and control littermates were taken off tetracycline for 7 days to induce myrAkt1 expression prior to MRI scan. The macromolecular contrast material biotin-BSA-Gadolinium-DTPA (biotin-BSA-Gd-DTPA) was prepared as previously reported (Dafni et al., 2002). Details can be found in the Supplemental Data.
Histology and immunohistochemistry
Frozen and paraffin embedded tissues were processed by standard methods. For evaluation of VVOs, skin tissues were harvested and processed for electron microscopy as previously described (Dvorak et al., 1996). Details for lacZ staining and immunohistochemistry are provided in the Supplemental Data.
Isolation of primary human and mouse endothelial cells
Human dermal microvascular endothelial cells (HDMEC) were isolated from neonatal foreskin as previously described (Richard et al., 1998). Use of human tissue was approved by the Beth Israel Deaconess Medical Center Institutional Review Board. Mouse endothelial cells were isolated from hearts. See Supplemental Data for details.
Statistical analysis
Results were presented as mean ± SD or SEM. Statistical significance of all data was analyzed using InStat 3.0 (GraphPad Software, Inc.). p values ≤ 0.05 in unpaired two-tail Student’s t test were considered to be statistically significant.
Supplementary Material
Acknowledgments
We wish to thank Dr. Morris Birnbaum for the Akt null mice; Dr. Gary Horowitz and Laurie Walsh for performing immunoassays of rapamycin blood levels; and Benjamin Hopkins, Seema Iyer, Shiva Kazerounian, Sharon Shechter, Merav Yoeli-Lerner, and Stuart Robertson for their assistance. This work was supported in part by US Public Health Service NIH grants R01 HL71049-01, CA106263 (L.E.B.), P01 CA09264401 (H.F.D.), and P01 AI50157 (D.M.B.); the Israel Science Foundation (M.N.); and NRSA Training grant T32 HL07893 (T.L.P.).
Footnotes
Supplemental data The Supplemental Data include Supplemental Experimental Procedures and three supplemental figures and can be found with this article online at http://www.cancercell.org/cgi/content/full/10/2/159/DC1/.
References
- Ackah E, Yu J, Zoellner S, Iwakiri Y, Skurk C, Shibata R, Ouchi N, Easton RM, Galasso G, Birnbaum MJ, et al. Akt1/protein kinase Bα is critical for ischemic and VEGF-mediated angiogenesis. J. Clin. Invest. 2005;115:2119–2127. doi: 10.1172/JCI24726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baluk P, McDonald DM. The beta 2-adrenergic receptor agonist formoterol reduces microvascular leakage by inhibiting endothelial gap formation. Am. J. Physiol. 1994;266:L461–L468. doi: 10.1152/ajplung.1994.266.4.L461. [DOI] [PubMed] [Google Scholar]
- Baluk P, Morikawa S, Haskell A, Mancuso M, McDonald DM. Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 2003;163:1801–1815. doi: 10.1016/S0002-9440(10)63540-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin LE, Keshet E. Conditional switching of vascular endothelial growth factor (VEGF) expression in tumors: induction of endothelial cell shedding and regression of hemangioblastoma-like vessels by VEGF withdrawal. Proc. Natl. Acad. Sci. USA. 1997;94:8761–8766. doi: 10.1073/pnas.94.16.8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin LE, Golijanin D, Itin A, Pode D, Keshet E. Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J. Clin. Invest. 1999;103:159–165. doi: 10.1172/JCI5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer. 2003;3:401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat. Rev. Cancer. 2004;4:335–348. doi: 10.1038/nrc1362. [DOI] [PubMed] [Google Scholar]
- Bucci M, Gratton JP, Rudic RD, Acevedo L, Roviezzo F, Cirino G, Sessa WC. In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat. Med. 2000;6:1362–1367. doi: 10.1038/82176. [DOI] [PubMed] [Google Scholar]
- Bucci M, Roviezzo F, Posadas I, Yu J, Parente L, Sessa WC, Ignarro LJ, Cirino G. Endothelial nitric oxide synthase activation is critical for vascular leakage during acute inflammation in vivo. Proc. Natl. Acad. Sci. USA. 2005;102:904–908. doi: 10.1073/pnas.0408906102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- Chen WS, Xu PZ, Gottlob K, Chen ML, Sokol K, Shiyanova T, Roninson I, Weng W, Suzuki R, Tobe K, et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001;15:2203–2208. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, De S, Damron DS, Chen WS, Hay N, Byzova TV. Impaired platelet responses to thrombin and collagen in AKT-1-deficient mice. Blood. 2004;104:1703–1710. doi: 10.1182/blood-2003-10-3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Somanath PR, Razorenova O, Chen WS, Hay N, Bornstein P, Byzova TV. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nat. Med. 2005;11:1188–1196. doi: 10.1038/nm1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, III, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001a;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBα is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J. Biol. Chem. 2001b;276:38349–38352. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- Dafni H, Israely T, Bhujwalla ZM, Benjamin LE, Neeman M. Overexpression of vascular endothelial growth factor 165 drives peritumor interstitial convection and induces lymphatic drain: magnetic resonance imaging, confocal microscopy, and histological tracking of triple-labeled albumin. Cancer Res. 2002;62:6731–6739. [PubMed] [Google Scholar]
- Dimmeler S, Zeiher AM. Akt takes center stage in angiogenesis signaling. Circ. Res. 2000;86:4–5. doi: 10.1161/01.res.86.1.4. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- Dor Y, Djonov V, Abramovitch R, Itin A, Fishman GI, Carmeliet P, Goelman G, Keshet E. Conditional switching of VEGF provides new insights into adult neovascularization and pro-angiogenic therapy. EMBO J. 2002;21:1939–1947. doi: 10.1093/emboj/21.8.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak HF. Leaky tumor vessels: consequences for tumor stroma generation and for solid tumor therapy. Prog. Clin. Biol. Res. 1990;354A:317–330. [PubMed] [Google Scholar]
- Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J. Clin. Oncol. 2002;20:4368–4380. doi: 10.1200/JCO.2002.10.088. [DOI] [PubMed] [Google Scholar]
- Dvorak HF. Rous-Whipple Award Lecture. How tumors make bad blood vessels and stroma. Am. J. Pathol. 2003;162:1747–1757. doi: 10.1016/s0002-9440(10)64309-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak AM, Kohn S, Morgan ES, Fox P, Nagy JA, Dvorak HF. The vesiculo-vacuolar organelle (VVO): a distinct endothelial cell structure that provides a transcellular pathway for macromolecular extravasation. J. Leukoc. Biol. 1996;59:100–115. [PubMed] [Google Scholar]
- Dvorak HF, Nagy JA, Feng D, Brown LF, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Curr. Top. Microbiol. Immunol. 1999;237:97–132. doi: 10.1007/978-3-642-59953-8_6. [DOI] [PubMed] [Google Scholar]
- Edinger AL, Linardic CM, Chiang GG, Thompson CB, Abraham RT. Differential effects of rapamycin on mammalian target of rapamycin signaling functions in mammalian cells. Cancer Res. 2003;63:8451–8460. [PubMed] [Google Scholar]
- Feng D, Nagy JA, Hipp J, Dvorak HF, Dvorak AM. Vesiculo-vacuolar organelles and the regulation of venule permeability to macromolecules by vascular permeability factor, histamine, and serotonin. J. Exp. Med. 1996;183:1981–1986. doi: 10.1084/jem.183.5.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng D, Nagy JA, Pyne K, Hammel I, Dvorak HF, Dvorak AM. Pathways of macromolecular extravasation across micro-vascular endothelium in response to VPF/VEGF and other vasoactive mediators. Microcirculation. 1999;6:23–44. [PubMed] [Google Scholar]
- Feng D, Nagy JA, Brekken RA, Pettersson A, Manseau EJ, Pyne K, Mulligan R, Thorpe PE, Dvorak HF, Dvorak AM. Ultra-structural localization of the vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) receptor-2 (FLK-1, KDR) in normal mouse kidney and in the hyperpermeable vessels induced by VPF/VEGF-expressing tumors and adenoviral vectors. J. Histochem. Cytochem. 2000a;48:545–556. doi: 10.1177/002215540004800412. [DOI] [PubMed] [Google Scholar]
- Feng D, Nagy JA, Dvorak AM, Dvorak HF. Different pathways of macromolecule extravasation from hyperpermeable tumor vessels. Microvasc. Res. 2000b;59:24–37. doi: 10.1006/mvre.1999.2207. [DOI] [PubMed] [Google Scholar]
- Fujio Y, Walsh K. Akt mediates cytoprotection of endothelial cells by vascular endothelial growth factor in an anchorage-dependent manner. J. Biol. Chem. 1999;274:16349–16354. doi: 10.1074/jbc.274.23.16349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun CO, Buerk DG, Huang PL, Jain RK. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc. Natl. Acad. Sci. USA. 2001;98:2604–2609. doi: 10.1073/pnas.041359198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, Ferrara N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J. Biol. Chem. 1998;273:30336–30343. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- Gratton JP, Lin MI, Yu J, Weiss ED, Jiang ZL, Fairchild TA, Iwakiri Y, Groszmann R, Claffey KP, Cheng YC, Sessa WC. Selective inhibition of tumor microvascular permeability by cavtratin blocks tumor progression in mice. Cancer Cell. 2003;4:31–39. doi: 10.1016/s1535-6108(03)00168-5. [DOI] [PubMed] [Google Scholar]
- Guba M, von Breitenbuch P, Steinbauer M, Koehl G, Flegel S, Hornung M, Bruns CJ, Zuelke C, Farkas S, Anthuber M, et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat. Med. 2002;8:128–135. doi: 10.1038/nm0202-128. [DOI] [PubMed] [Google Scholar]
- Harris AL. Hypoxia–a key regulatory factor in tumour growth. Nat. Rev. Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- Kamba T, Tam BY, Hashizume H, Haskell A, Sennino B, Mancuso MR, Norberg SM, O’Brien SM, Davis RB, Gowen LC, et al. VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am. J. Physiol. Heart Circ. Physiol. 2005;290:H560–H576. doi: 10.1152/ajpheart.00133.2005. [DOI] [PubMed] [Google Scholar]
- Kisucka J, Butterfield CE, Duda DG, Eichenberger SC, Saffaripour S, Ware J, Ruggeri ZM, Jain RK, Folkman J, Wagner DD. Platelets and platelet adhesion support angiogenesis while preventing excessive hemorrhage. Proc. Natl. Acad. Sci. USA. 2006;103:855–860. doi: 10.1073/pnas.0510412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Ahmad SA, Reinmuth N, Shaheen RM, Jung YD, Fan F, Ellis LM. Endothelial cell survival and apoptosis in the tumor vasculature. Apoptosis. 2000;5:323–328. doi: 10.1023/a:1009679307513. [DOI] [PubMed] [Google Scholar]
- McDonald DM, Baluk P. Significance of blood vessel leakiness in cancer. Cancer Res. 2002;62:5381–5385. [PubMed] [Google Scholar]
- Meier-Kriesche HU, Kaplan B. Toxicity and efficacy of sirolimus: relationship to whole-blood concentrations. Clin. Ther. 2000;22 SupplB:B93–B100. doi: 10.1016/s0149-2918(00)89026-8. [DOI] [PubMed] [Google Scholar]
- Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, McDonald DM. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 2002;160:985–1000. doi: 10.1016/S0002-9440(10)64920-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy JA, Vasile E, Feng D, Sundberg C, Brown LF, Detmar MJ, Lawitts JA, Benjamin L, Tan X, Manseau EJ, et al. Vascular permeability factor/vascular endothelial growth factor induces lymphangiogenesis as well as angiogenesis. J. Exp. Med. 2002;196:1497–1506. doi: 10.1084/jem.20021244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa CR, Banfi A, Glazer NL, Thurston G, Springer ML, Kraft PE, McDonald DM, Blau HM. Microenvironmental VEGF concentration, not total dose, determines a threshold between normal and aberrant angiogenesis. J. Clin. Invest. 2004;113:516–527. doi: 10.1172/JCI18420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S, Datta K, Mukhopadhyay D. Central role of p53 on regulation of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) expression in mammary carcinoma. Cancer Res. 2001;61:6952–6957. [PubMed] [Google Scholar]
- Pettersson A, Nagy JA, Brown LF, Sundberg C, Morgan E, Jungles S, Carter R, Krieger JE, Manseau EJ, Harvey VS, et al. Heterogeneity of the angiogenic response induced in different normal adult tissues by vascular permeability factor/vascular endothelial growth factor. Lab. Invest. 2000;80:99–115. doi: 10.1038/labinvest.3780013. [DOI] [PubMed] [Google Scholar]
- Rak J, Kerbel RS. Ras regulation of vascular endothelial growth factor and angiogenesis. Methods Enzymol. 2001;333:267–283. doi: 10.1016/s0076-6879(01)33062-8. [DOI] [PubMed] [Google Scholar]
- Rak J, Filmus J, Finkenzeller G, Grugel S, Marme D, Kerbel RS. Oncogenes as inducers of tumor angiogenesis. Cancer Metastasis Rev. 1995;14:263–277. doi: 10.1007/BF00690598. [DOI] [PubMed] [Google Scholar]
- Richard L, Velasco P, Detmar M. A simple immunomagnetic protocol for the selective isolation and long-term culture of human dermal microvascular endothelial cells. Exp. Cell Res. 1998;240:1–6. doi: 10.1006/excr.1998.3936. [DOI] [PubMed] [Google Scholar]
- Sarbassov dos D, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Sarbassov dos D, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- Six I, Kureishi Y, Luo Z, Walsh K. Akt signaling mediates VEGF/VPF vascular permeability in vivo. FEBS Lett. 2002;532:67–69. doi: 10.1016/s0014-5793(02)03630-x. [DOI] [PubMed] [Google Scholar]
- Stallone G, Schena A, Infante B, Di Paolo S, Loverre A, Maggio G, Ranieri E, Gesualdo L, Schena FP, Grandaliano G. Sirolimus for Kaposi’s sarcoma in renal-transplant recipients. N. Engl. J. Med. 2005;352:1317–1323. doi: 10.1056/NEJMoa042831. [DOI] [PubMed] [Google Scholar]
- Sun JF, Phung T, Shiojima I, Felske T, Upalakalin JN, Feng D, Kornaga T, Dor T, Dvorak AM, Walsh K, Benjamin LE. Microvascular patterning is controlled by fine-tuning the Akt signal. Proc. Natl. Acad. Sci. USA. 2005;102:128–133. doi: 10.1073/pnas.0403198102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg C, Nagy JA, Brown LF, Feng D, Eckelhoefer IA, Manseau EJ, Dvorak AM, Dvorak HF. Glomeruloid microvascular proliferation follows adenoviral vascular permeability factor/vascular endothelial growth factor-164 gene delivery. Am. J. Pathol. 2001;158:1145–1160. doi: 10.1016/S0002-9440(10)64062-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.