

Abstract
We have added cyanide to oxidized 1Fe and 2Fe superoxide reductase (SOR) as a surrogate for the putative ferric-(hydro)peroxo intermediate in the reaction of the enzymes with superoxide, and have used vibrational and ENDOR spectroscopies to study the properties of the active-site paramagnetic iron center. Addition of cyanide changes the active-site iron center in oxidized SOR from rhombic high-spin ferric (S = 5/2) to axial-like low-spin ferric (S = 1/2). Low-temperature resonance Raman and ENDOR data show that the bound cyanide adopts three distinct conformations in Fe(III)-CN SOR. On the basis of 13CN, C15N, and 13C15N isotope shifts of the Fe–CN stretching/Fe–C–N bending modes, resonance Raman studies of 1Fe-SOR indicate one near-linear conformation (Fe–C–N angle ∼175°) and two distinct bent conformations (Fe–C–N angles < 140°). FTIR studies of 1Fe-SOR at ambient temperatures reveals three bound C–N stretching frequencies in the oxidized (ferric) state and one in the reduced (ferrous) state indicating that the conformational heterogeneity in cyanide binding is a characteristic of the ferric state and is not caused by freezing-in of conformational substates at low temperature. 13C-ENDOR spectra for the 13CN-bound ferric active sites in both 1Fe- and 2Fe-SORs also show three well-resolved Fe–C–N conformations. Analysis of the 13C hyperfine tensors for the three substates of the 2Fe-SOR within a simple heuristic model for the Fe-C bonding gives values for the Fe–C–N angles in the three substates of ca. 123° (C3), 133° (C2), taking a reference value from vibrational studies of 175° (C1 species). Resonance Raman and ENDOR studies of SOR variants, in which the conserved glutamate and lysine residues in a flexible loop above the substrate binding pocket have been individually replaced by alanine, indicate that the side chains of these two residues are not involved in direct interaction with bound cyanide. The implications of these results for understanding the mechanism of SOR are discussed.
INTRODUCTION
Superoxide is well known to be scavenged in aerobic organisms by superoxide dismutase (SOD).1 More recently, an enzyme termed superoxide reductase (SOR), has been found that scavenges superoxide in anaerobic bacteria and archaea (for reviews and perspectives, see (1-5). Instead of the classical disproportionation of superoxide to hydrogen peroxide and dioxygen catalyzed by SODs, SORs catalyze the one-electron reduction of superoxide to hydrogen peroxide, and the reaction does not produce dioxygen. The X-ray crystal structure of SOR from the hyperthermophilic archaeon Pyrococcus furiosus has been determined (6). Each subunit of the homotetramer contains one non-heme iron active site. In the reduced (ferrous) state, the monomeric iron active site is square pyramidal with four imidazole nitrogens from histidines in the equatorial plane and one cysteinyl sulfur in an axial position. In the oxidized (ferric) state, there is an additional axial monodentate glutamate ligand trans to the cysteine. A second class of SOR contains a rubredoxin-like, [Fe(SCys)4] site in addition to the [Fe(NHis)4(SCys)] site. The known 2Fe-SORs are homodimeric, and the crystal structure of Desulfovibrio desulfuricans 2Fe-SOR revealed a [Fe(NHis)4(SCys)] site whose structure is very similar to that described above for the ferrous Pf 1Fe-SOR site (7). Coordination of the conserved glutamate in the ferric form of both 1Fe- and 2Fe-SORs is supported by comparisons of UV-visible absorption and IR spectroscopic properties of wild type and engineered variants in which the glutamate was replaced by alanine (8-14). This common active site is, therefore, hereafter generically referred to as the SOR site. The function of the accessory [Fe(SCys)4] site in 2Fe-SORs is unknown; it is too far away (∼22 Å) from the SOR site for efficient electron transfer, and an engineered variant (C13S) of the D. vulgaris 2Fe-SOR lacking the [Fe(SCys)4] site exhibited a spectroscopically and functionally unperturbed SOR site (15, 16).
The open coordination position trans to the cysteinate sulfur ligand is an obvious candidate for inner-sphere reaction of the ferrous SOR site with superoxide, particularly as exogenous ligands such as azide and cyanide have been shown to bind to reduced SOR at this site, and the site is solvent exposed but is not coordinated by solvent (6, 17). Indeed, as shown in Scheme 1, a transient intermediate observed during the reaction of superoxide with the ferrous SOR site of D. vulgaris 2Fe-SOR has been proposed to be a ferric-(hydro)peroxo, which then decays to the “resting” glutamate-ligated ferric SOR site with loss of hydrogen peroxide (8, 11). A conserved lysine residue (K48 in D. vulgaris 2Fe-SOR) is closest to the putative superoxide binding site in reduced SOR (6), and mutagenesis studies suggest a possible role in protonating or positioning the bound peroxo group (10, 11). The sequentially adjacent lysine and ligating glutamate residues are contained within a flexible loop region above the substrate binding pocket (6, 7).
Scheme 1.
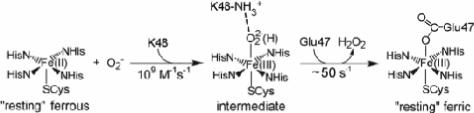
Based on steric and electronic grounds, the (hydro)peroxo of the intermediate was proposed to coordinate to the ferric iron (which could be either high or low spin) with a bent, end-on geometry (5, 18). Others have suggested an adduct resulting from reaction of hydrogen peroxide with the ferrous SOR site, formulated as a seven-coordinate high-spin ferric species with a side-on (η2) coordinated peroxo, as a candidate for the superoxide-derived intermediate (19, 20). However, the formation and decay kinetics of the hydrogen peroxide- and superoxide-derived SOR species differ by orders of magnitude, and, unlike the reaction with superoxide, the hydrogen-peroxide treatment led to irreversible degradation of the SOR site. Thus, the exact nature of the intermediate in the reaction of superoxide with the ferrous SOR site remains to be established.
Cyanide is known to form a low-spin complex with the ferric SOR site (17, 21, 22), in which the cyanide is believed to be C-coordinated trans to the cysteinyl sulfur in place of the glutamate, thus, mimicking the putative ferric-(hydro)peroxo intermediate in the reaction with superoxide. Analysis of the EPR and UV-visible absorption, CD and VTMCD spectra of the cyanide-bound ferric SOR site in P. furiosus 1Fe-SOR has facilitated assignment of the ground-state and charge-transfer/ligand-field excited-state properties and revealed a tetragonally elongated octahedral coordination geometry, with the elongation axis corresponding to an axis with trans-histidyl ligands and the unpaired electron in the Fe dπ orbital available for dπ-pπ interaction with cysteinyl S and the bound CN− (17). Here we report the results of resonance Raman , FTIR and 14,15N- and 13C-ENDOR studies of the cyano-ferric and -ferrous complexes of WT and variant forms of P. furiosus 1Fe-SOR and D. vulgaris 2Fe-SOR. The results reveal that the cyano-ferric enzyme exhibits three distinct conformations that differ in the Fe–C–N angle. Parallel spectroscopic studies of site-directed variants of both 1Fe- and 2Fe-SORs demonstrate that the conserved glutamate and lysine residues, proximal to the substrate binding site, are not involved in specific interactions with the bound cyanide.
MATERIALS AND METHODS
Purification and preparation of SOR samples
P. furiosus 1Fe-SOR
Recombinant wild-type, E14A, and K15A P. furiosus 1Fe-SOR were over-expressed in Escherichia coli and purified to homogeneity using the published protocol (12, 17, 23). Oxidized and reduced forms of P. furisous 1Fe-SOR were prepared by the addition of a 5-fold excess of ammonium hexachloroiridate and ascorbic acid, respectively, using stock solutions in 100 mM TAPS buffer, pH 8.5. Excess oxidant or reductant was removed by Amicon ultrafiltration prior to use. Cyanide-bound samples of WT and variant forms of P. furiosus 1Fe-SOR were prepared by incubating with a 30-fold excess of potassium cyanide for 30 min at room temperature. Both protein and potassium cyanide solutions were in 100 mM TAPS buffer, pH 8.5. The isotopomeric cyanide adducts were prepared in the same way and measured under identical instrumentation conditions. Isotopically labeled potassium cyanide samples (K13CN, KC15N and K13C15N) were 99% enriched and obtained from Isotech Inc. P. furiosus 1Fe-SOR samples were prepared to a final concentration of 4 − 5 mM for all spectroscopic studies. Samples in 2H2O were prepared by Amicon-ultrafiltration buffer exchange using three 10-fold dilution and concentration cycles with the equivalent 2H2O buffer.
D. vulgaris 2Fe-SOR
Recombinant D. vulgaris 2Fe-SOR and its engineered site-directed variants were obtained as described previously (8, 11, 16). 2Fe-SOR ENDOR samples contained ∼1 mM SOR sites and ∼25 mM potassium cyanide in 50 mM TAPS buffer, pH 8.5. The as-isolated 2Fe-SOR contains predominantly ferric [Fe(SCys)4] and ferrous SOR sites. Aerobic incubation of the cyanide-treated samples for 24 hours results in quantitative autoxidation of the SOR site to the ferric state and complexation with cyanide (21, 24).
Spectroscopic Methods
Resonance Raman
Resonance Raman spectra were recorded using an Instruments SA Ramanor U1000 spectrometer fitted with a cooled RCA-31034 photomultiplier tube with 90° scattering geometry. Spectra were recorded digitally using photon counting electronics and improvements in signal-to-noise were achieved by signal averaging multiple scans. Absolute band positions were calibrated using the excitation frequency and CCl4 and are accurate to ±1 cm−1. Lines from a Coherent Sabre 100 10-W Argon Ion Laser or Coherent Innova 200-K2 Krypton Ion Laser were used for excitation, and plasma lines were removed using a Pellin Broca prism premonochromator. Scattering was collected from the surface of a frozen 15-ul droplet of sample using a custom-designed anaerobic sample cell (25), attached to the cold finger of an Air Products Displex Model CSA-202E closed cycle refrigerator. This arrangement enables samples to be cooled down to 17 K, which facilitates improved spectral resolution and prevents laser-induced sample degradation. For assessment of isotope shifts, both the natural abundance and isotopically enriched samples were placed on the sample probe, and frequency calibration was checked before and after measurements to ensure relative accuracy of band positions to at least ±0.1 cm−1.
FTIR
FTIR measurements were recorded using a Bio-Rad FTS 575C spectrometer equipped with an MCT detector. Spectra were collected in the mid-IR region (4000 − 400 cm−1) at room temperature with a resolution of 2 cm−1 and are averages of 256 scans. SOR samples were concentrated to ∼5 mM and placed between Spectra-Tech CaF2 plates with a 50 μm Teflon spacer. A background spectrum corresponding to oxidized SOR at the same concentration as the cyanide-treated sample was subtracted and the baseline was linearized for each of the spectra shown.
EPR and ENDOR Spectroscopy
CW EPR and ENDOR spectra were acquired at 2K on a Q-band CW spectrometer operating in a rapid-passage dispersion mode as described previously (26); 100 KHz bandwidth broadening of radio frequency was introduced to enhance the S/N ratio (27). For a nucleus with spin I = 1/2, the ENDOR pattern to the first order approximation is given by
| (1) |
where νn is the nuclear Larmor frequency, and A is the orientation-dependent hyperfine coupling constant. In Q-band 1H-ENDOR spectra, the two branches, ν±, are a doublet split by |A| and centered at νH (νH > |A/2| ), but in the 13C-ENDOR spectra presented here, the ν± doublet is centered at |A/2| and split by 2νC (νC < |A/2| ) . Full hyperfine tensors were obtained by simulation of 2D field-frequency patterns comprised of multiple ENDOR spectra taken across the EPR envelope, as described (28).
RESULTS
Vibrations of the (Cys)S–Fe–CN unit in cyanide-bound SOR
The visible absorption spectrum of oxidized SOR is dominated by a (Cys)S(pπ)-to-Fe(dπ) CT transition centered at 660 nm which is red-shifted to 690 nm upon the binding of CN− to the mononuclear ferric active site (17;22). Resonance Raman studies of the oxidized SOR site in P. furiosus 1Fe-SOR using 647-nm excitation revealed selective enhancement of modes associated with the Fe–S(Cys) unit and facilitated detailed vibrational assignments (12). As a result of cyanide binding trans to cysteinyl-S, resonance Raman studies of cyanide-bound oxidized SOR in the 200−800 cm−1 region using excitation into the 690 nm absorption band reveal bands associated with the (Cys)S–Fe–CN unit, see Figure 1. Very similar resonance Raman spectra were observed with 647- and 676-nm excitation and assignments to Fe–S(Cys) and Fe–CN modes are readily made based on comparison with oxidized SOR in the absence of cyanide and 13CN, C15N, and 13C15N isotope shifts, see Figure 1. Cyanide binding trans to the coordinated cysteinate induces a high-spin to low-spin transition, but has little effect on the vibrational frequencies of the (Cys)S–Fe unit. The Fe–S(Cys) and cysteine S–Cβ stretching modes, located at 323 and 748 cm−1, respectively, are insensitive to cyanide binding, while the cysteine S–Cβ–Cα bending mode exhibits a +3 cm−1 upshift, from 298 to 301 cm−1 upon the addition of cyanide, see Figure 1.
Figure 1.

Resonance Raman spectra of hexachloroiridate-oxidized P. furiosus 1Fe-SOR: (a) without KCN; (b) with natural abundance KCN; (c) with K13CN; (d) with KC15N; (e) with K13C15N. Spectra were recorded using 647-nm excitation on samples maintained as frozen droplets at 17 K. Each sample was ∼4 mM in SOR and 120 mM in potassium cyanide and was in 100 mM TAPS buffer, pH 8.5. Spectra were recorded using 6 cm−1 resolution by photon counting for 1 sec every 1 cm−1 and each spectrum is the sum of 50 scans. The asterisk indicates a lattice mode of ice and the frequencies of the cyanide isotope-sensitive bands are in bold type.
Resonance Raman spectroscopy has been extensively used to investigate the heterogeneity and geometry of cyanide binding to iron in heme proteins (29-38), and these results provide a firm basis for assigning the cyanide isotope-sensitive bands in cyanide-bound SOR. In principle, the δ( Fe–C–N) bending mode of a linear Fe–C–N complex (180°) should be Raman inactive (30, 36). However, upon steric or electronic distortion of this complex, the bending mode becomes allowed and is observed in the resonance Raman spectrum. Both the ν(Fe–CN) stretching and δ(Fe–C–N) bending modes occur in the low energy region, 300−500 cm−1, and cyanide isotopic labeling (i.e. CN, 13CN, C15N and 13C15N) has been employed to identify these modes and assess the Fe–C–N angle (29-38). In an “essentially” linear Fe–C–N complex (∼175°), where the bending mode is nonetheless allowed, the resonance Raman spectrum typically exhibits a “monotonic-isotope-shifting” ν(Fe–CN) stretching mode with a frequency 30−60 cm−1 higher than the lower energy “zigzag-isotope-shifting” δ(Fe–C–N) bending mode (30, 36, 38). A monotonically-isotope-shifting mode exhibits cyanide isotope shifts that vary as a function of total cyanide mass, with the 13C14N and 12C15N isotopomers having similar frequencies, and is characteristic of the ν(Fe–CN) stretching mode of a near-linear Fe–C–N unit A zigzag-isotope-shifting mode exhibits isotope shifts that primarily vary with isotopic substitution of the carbon atom, and thus move in a “zigzag” fashion with the isotopic sequence CN−, 13CN, C15N and 13C15N; this zigzag isotope shifting is characteristic of a mode primarily composed of δ(Fe–C–N) bending character. However, as the Fe–C–N angle decreases from 180°, the stretching and bending modes mix, the two frequencies move apart with lower frequency mode assuming greater ν(Fe–CN) stretching character, and both bands exhibit partially zigzag-isotope-shifting patterns depending on the extent of the mixing. For example, a bent Fe–C–N unit with an angle of 155° would be expected to display a greater stretching/bending energy separation (70−90 cm−1) with both bands exhibiting a zigzag isotope shift patterns (30, 38).
The cyanide-bound adduct of oxidized P. furiosus 1Fe-SOR has six cyanide isotope-sensitive bands in the low frequency resonance Raman spectrum, see Figure 1, which we assign to a superposition of individual two-line spectra from three distinct Fe–C–N geometries. Each active-site conformer has a higher-frequency band, with frequencies of 525, 497 and 471 cm−1 and a lower energy band, with frequencies of 417, 400 and 383 cm−1. All exhibit zigzag isotope shifting patterns, except for the 471 cm−1 mode which has a monotonic isotope shifting pattern indicative of the ν(Fe–CN) stretching mode of a near-linear (∼175°) Fe–C–N unit, see Figure 1 and Table 1. The lower-frequency bending mode of this conformation is therefore assigned to the band at 417 cm−1 on the basis of the small frequency separation (54 cm−1) and sensitivity to 2H2O/1H2O. Of the six cyanide isotope sensitive bands, only the 471 and 417 cm−1 bands show significant frequency shifts on 2H2O/1H2O buffer exchange, see Figure 2 and Table 1, suggesting that this pair of lines belongs to a near-linear Fe–C–N unit that has a H-bonding interaction with water or an exchangeable protein group. The other four cyanide isotope-sensitive bands are grouped into two sets to avoid unrealistic frequency separations: 497 and 383 cm−1, frequency separation, 114 cm−1; 525 and 400 cm−1, frequency separation, 125 cm−1. These two pairs of lines correspond to Fe–C–N units with progressively smaller angles. Both clearly have angles of less than the 155° associated with an energy separation of 70−90 cm−1 (30, 38), and based on the 13C-ENDOR data presented below, both have angles < 140°, see Table 1. The Raman intensities of bands corresponding to different conformers do not provide a means of quantifying the relative amounts of each conformer, due to differences in resonance enhancement. However, we have noted that the relative intensities of bands originating from the linear and bent conformers vary by as much as a factor of two in different samples at the same pH and in the same buffer solution, whereas the relative intensities of the bent conformers remain essentially constant. The origin of this variability is unclear at present and may relate to sample concentration or rates of freezing.
Table 1.
Stretching (ν) and bending (δ) frequencies and isotope shifts (cm−1) for the Fe—C—N fragment in cyanide-bound oxidized P. furiosus 1Fe-SOR
| ν(Fe—CN) (Δ13C, 15N, 13C15N, 2H2O) | δ(Fe—C—N) (Δ13C, 15N, 13C15N, 2H2O) | ν(C—N) (Δ13C, 2H2O) | <Fe—C—N |
|---|---|---|---|
| 471 (−5, −5, −9, +3) | 417 (−7, −2, −10, +2) | 2111 (−46, +3) | ∼175° |
| 383 (−6, −1, −8, 0)a | 497 (−7, 0, −6, 0)a | 2115 (−46, 0) or 2100 (−45, 0) | ∼133° |
| 400 (−9, −2, −11, 0)a | 525 (−10, −2, −12, 0)a | 2115 (−46,0) or 2100 (−45, 0) | ∼123° |
The ν(Fe—CN) and δ(Fe—C—N) modes are kinematically mixed in bent conformations
Figure 2.

Resonance Raman spectra of hexachloroiridate-oxidized cyanide-bound P. furiosus 1Fe-SOR: (a) WT in 1H2O buffer; (b) WT in 2H2O; (c) E14A variant in 1H2O buffer; (d) K15A variant in 1H2O buffer. Sample and measurements conditions are the same as those described in Figure 1.
The resonance Raman results clearly indicate that cyano-ferric-SOR in frozen solution exists as a mixture of three species, one with a near-linear Fe–C–N unit and two with bent Fe–C–N units. FTIR studies indicate that these conformational substates also exist in fluid solution at ambient temperature, and are not generated during the freezing process. The C–N stretching modes are weak in the resonance Raman spectrum, but are readily observable at room temperature by FTIR. FTIR spectra of the oxidized cyanide-bound adduct of P. furiosus 1Fe-SOR show cyanide isotope-sensitive peaks centered at 2115, 2111, 2100, and 2080 cm−1, see Figure 3, that downshift by 43−46 cm−1 with K13CN. The band at 2080 cm−1 corresponds to the ν(C–N) stretching mode of free cyanide in solution, and studies in 2H2O confirm the absence of a ν(HC–N) stretching mode (expected at 2093 cm−1) (39). Hence the bands at 2115, 2111 and 2100 cm−1 are attributed to the ν(C–N) stretching modes of the three oxidized SOR Fe–C–N conformers. The increase in frequency relative free cyanide is attributed to strong σ-donation from the 5σ* orbital to the ferric iron, resulting in a net increase in the C–N bond order (40). Definitive assignment of each ν(C–N) stretching mode to a specific Fe–C–N conformer is not possible. However, the ν(C–N) stretching mode at 2111 cm−1 is tentatively assigned to the near-linear Fe–C–N conformer as it is the only band that exhibits an isotope shift on exchange into a 2H2O buffer, see Table 1.
Figure 3.
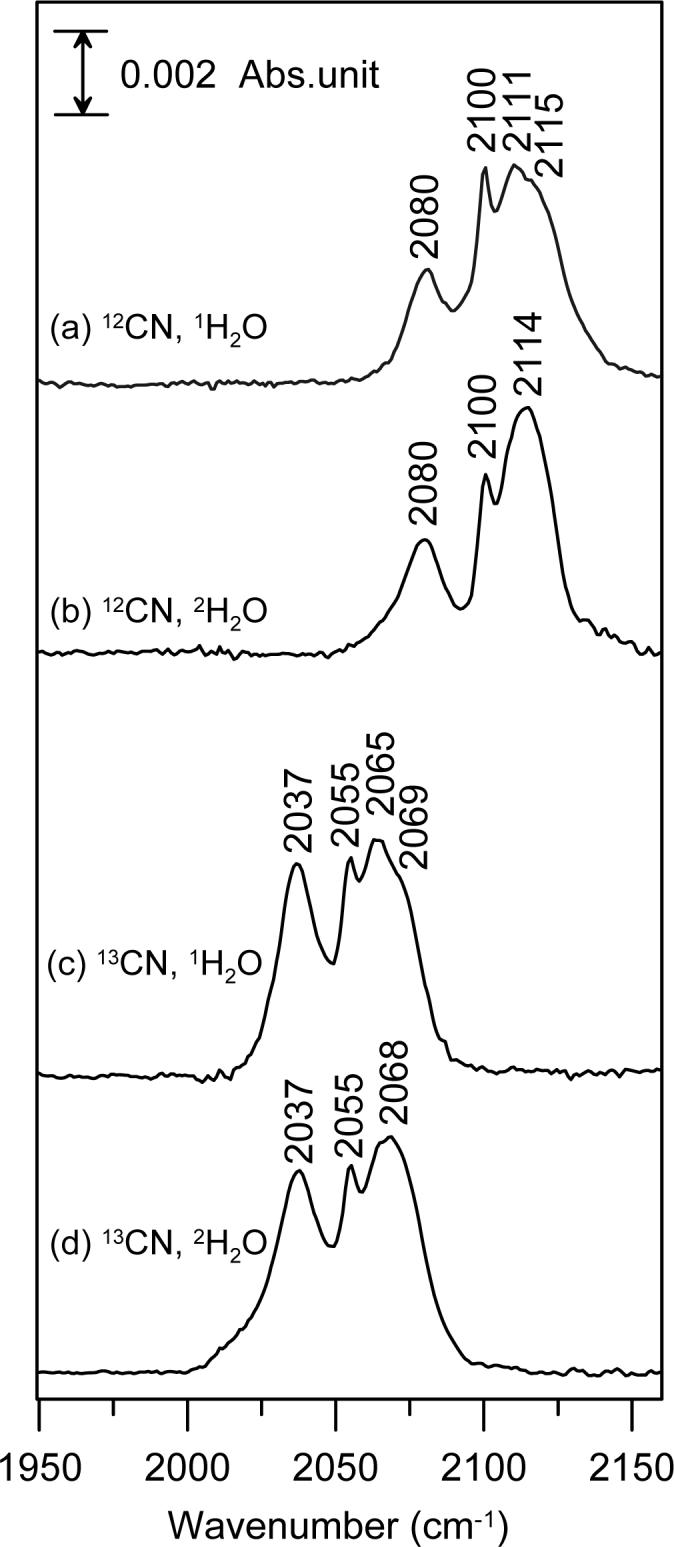
FTIR spectra of cyanide-bound, hexachloroiridate-oxidized P. furiosus 1Fe-SOR in the C–N stretching region: (a) natural abundance KCN in 1H2O buffer; (b) natural abundance KCN in 2H2O buffer; (c) K13CN in 1H2O buffer; (d) K13CN in 2H2O buffer. Each sample was ∼4 mM in SOR and 120 mM potassium cyanide and was in 100 mM 1H2O or 2H2O TAPS buffer, pH 8.5. Spectra were collected in the mid-IR region (400 − 4000 cm−1) at room temperature with a resolution of 2 cm−1 and are represent the average of 256 scans.
FTIR also facilitates characterization of the ν(C–N) stretching mode in ascorbate-reduced P. furiosus 1Fe-SOR, see Figure 4. In contrast to the oxidized cyanide-bound adduct, the reduced cyanide-bound adduct is homogeneous with a single ν(C–N) stretching mode centered at 2037 cm−1 that is insensitive to 2H2O/1H2O exchange. The band at 2080 cm−1 is again attributed to unbound CN−, and both the bound and unbound ν(C–N) stretching modes exhibit the characteristic 42−43 cm−1 downshifts with 13CN. The decrease in the ν(C–N) stretching frequency compared to free CN− is attributed to significant π-back-donation from the ferrous iron into the CN− π* orbitals, suggesting a near-linear Fe–C–N conformation with no solvent-exchangeable H-bonding interactions.
Figure 4.
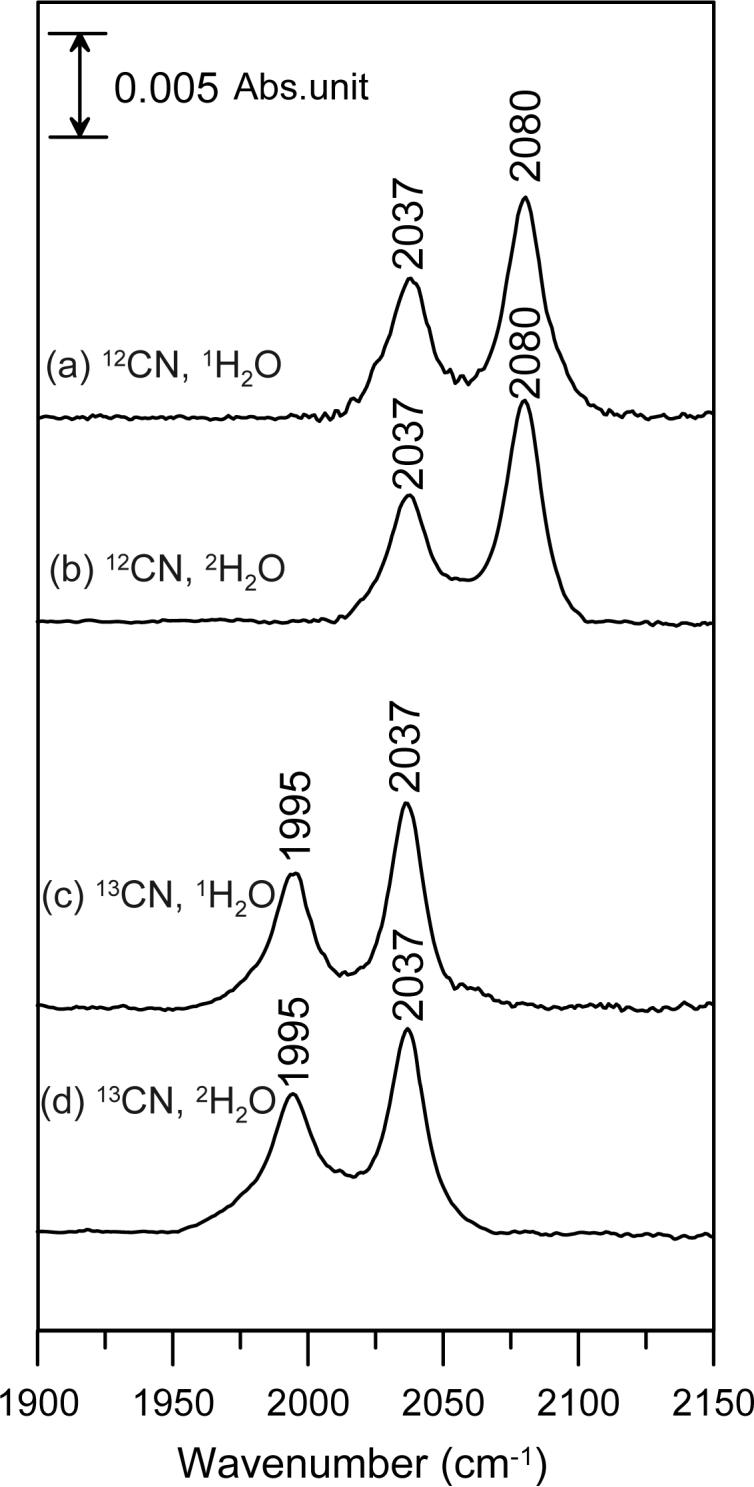
FTIR spectra of cyanide-bound, ascorbate-reduced P. furiosus 1Fe-SOR in the C–N stretching region: (a) natural abundance KCN in 1H2O buffer; (b) natural abundance KCN in 2H2O buffer; (c) K13CN in 1H2O buffer; (d) K13CN in 2H2O buffer. Sample and measurements conditions are the same as those described in Figure 3.
The available crystallographic data (6) suggest an important role for the flexible loop (residues 9−15 in P. furiosus 1Fe-SOR) above the substrate binding pocket, in particular the conserved glutamate and lysine residues (E14 and K15 in P. furiosus 1Fe-SOR), for positioning or protonating bound superoxide and/or mediating peroxide release. Hence the effects of engineered E14A and K15A substitutions on cyanide binding to the oxidized SOR site were assessed using resonance Raman spectroscopy. Except for minor frequency shifts (≤ 3 cm−1), the resonance Raman spectra indicate that E14A and K15A substitutions have little effect on the mode or heterogeneity of cyanide binding at the oxidized SOR site, see Figure 2. We conclude that the heterogeneity in Fe–C–N conformations does not result from specific interactions with either the conserved glutamate or lysine residues.
Q-band EPR of cyanide-bound SOR
Q-band EPR spectra obtained at 2 K for the cyano-ferric adducts of WT P. furiosus 1Fe-SOR and WT, C13S, E47A and K48A D. vulgaris 2Fe-SOR are shown in Figure 5. The WT, E47A and K48A 2Fe-SORs show both the high-spin (S = 5/2) ferric [Fe(SCys4] site, with g1,2,3 = [7.85, 3.91, ∼2] and the low-spin (S = 1/2) SOR site, with g1,2,3 = [2.22, 2.18, 1.96]. For the high-spin [Fe(SCys)4] site, the g values can be interpreted as arising from the transitions within the lowest, MS = ± 1/2 Kramers doublet of the S = 5/2 spin manifold with the following spin Hamiltonian,
| (2) |
where D and E are axial and rhombic zero-field splitting parameters, respectively. From eq. 2 with D >> g0βH > 0, g0 = 2.00 and E/D = 0.09. The g and E/D values are similar to those obtained at X-band at 4.2 K for the protein without addition of cyanide (15). The intense peak at gz = 5.8 seen in the X-band spectrum at 4.2 K, is absent in the Q-band spectrum at 2 K. This feature is attributed to a transition involving the middle doublet, MS = ± 3/2, and its absence is due to the redistribution of Boltzmann spin population between 2 K and 4.2 K. As the temperature is increased to 4.2 K, a very weak gz = 5.7 signal starts to appear (data not shown). This small value for E/D is not common for [Fe(SCys)4] centers, in general, but is observed for the axially-distorted [Fe(SCys)4] centers in desulforedoxins (7, 41) and 2Fe-SORs (42-44) which are ligated by cysteines in CXXC and CC motifs. As the g-values do not change upon cyanide addition, it is clear that cyanide does not bind to the ferric [Fe(SCys)4] site.
Figure 5.

35 GHz Q-band EPR spectra of the cyanide-bound oxidized forms of WT P. furiosus 1Fe-SOR and WT, C13S, E47A and K48A D. vulgaris 2Fe-SOR. Experimental conditions: microwave power = 100 μW; modulation amplitude/frequency = 0.4 mT/100 kHz; time constant = 128 ms; scan speed = 4 min/scan; temperature = 2 K.
Without addition of cyanide, the ferric SOR site in D. vulgaris 2Fe-SOR shows a rhombic, high-spin (S = 5/2) EPR signal with E/D = 0.28 (15). Upon addition of cyanide, this signal converts to a low-spin, S = 1/2, form with a nearly axial g tensor, g1,2,3 = [2.23, 2.19, 1.96], establishing that the SOR site in 2Fe-SOR also binds cyanide, see Figure 5. The cyano-ferric adducts of the C13S, E47A and K48A variants of 2Fe-SOR exhibit low-spin EPR signals identical to that of WT, see Figure 5. Hence cyanide binding at the oxidized SOR site of 2Fe-SORs is not significant perturbed by site-specific changes in the conserved glutamate and lysine residues in the flexible loop region above the substrate binding pocket or elimination of the [Fe(SCys)4] site in the C13S variant. The Q-band EPR spectrum of the cyano-ferric form of P. furiosus 1Fe-SOR also shows a similar low-spin ferric signal, g1,2,3 = [2.27, 2.24, 1.94], with principal g values very similar to those measured previously at X-band and 15 K, g1,2,3 = [2.29, 2.25, 1.94] (17).
Single-crystal-like CW 13C-ENDOR of 13CN-bound SOR
Figure 6 shows the 13C-ENDOR spectra at g1 for the low-spin 13CN− adducts of the SOR site of P. furiosus 1Fe-SOR and WT, C13S, E47A and K48A D. vulgaris 2Fe-SOR. All spectra exhibit the ν+ features from three distinct enzyme substates in which the 13CN− have different 13C hyperfine couplings, and hence different environments. These are labeled as C1, C2, and C3 in the spectrum of the 1Fe-SOR, which shows the best resolution. From equation 1, the ν+ frequencies correspond to 13C hyperfine couplings, A(13C) ∼ 30 MHz, much greater than the Larmor frequency, ν(13C) ∼ 13 MHz; the corresponding lower frequency, ν−, features are not observed, as is often the case in ENDOR measurements at 35GHz and higher microwave frequencies (28). The observation of three distinct environments in the 13C-ENDOR spectra is clearly in accord with the three Fe–C–N conformations that are apparent in IR and Raman studies of P. furiosus 1Fe-SOR. The 13C hyperfine couplings for the WT and variant 2Fe-SORs are essentially the same, but the resolution of the signals from the C2 and C3 substates differs among the variants. The equivalent hyperfine couplings of the WT 2Fe-SORs and its C13S, E47A, and K48A variants provides further confirmation that elimination of the [Fe(SCys)4] site and the absence of the conserved glutamate and lysine residues in the flexible loop above the substrate binding pocket do not significantly perturb cyanide binding at the SOR site. In contrast, the 13C hyperfine couplings of the 1Fe- and 2Fe-SOR differ slightly from each other, see Table 2. Finally, the 13C ENDOR intensities suggest that the C3 substate has a lower occupancy in the 2Fe-SOR than in the 1Fe-SOR (see below), and especially in the E47A and K48A mutants.
Figure 6.

Q-band CW 13C-ENDOR spectra at g1 field for 13CN−-bound oxidized forms of WT P. furiosus 1Fe-SOR and WT, C13S, E47A and K48A D. vulgaris 2Fe-SOR. The EPR conditions are same as in Figure 5 and the rf scanning speed was 1 MHz/sec for ∼ 5 scans.
Table 2.
13C hyperfine coupling constants and Fe—C—N angles in 13CN−-bound SORs.
| 1Fe-SOR WT: | |||
| Aiso (MHz) | Aaniso (MHz) | θ (Fe—C—N) | |
| C1 | −32.8 | [−1.2, −1.2, 2.4] | 175° |
| C2 | −26.7 | [−0.3, −0.3, 0.6] | 133° |
| C3 | −23.2 | [0.2, 3.2, −3.4] | 123° |
| 2Fe-SOR WT: | |||
| Aiso (MHz) | Aaniso (MHz) | θ (Fe—C—N) | |
| C1 | −31.2 | [−0.8, −0.8, 1.6] | 175° |
| C2 | −25.3 | [0.3, 0.3, −0.6] | 133° |
| C3 | −22.3 | [1.3, 1.3, −2.6] | 124° |
| C13S: | |||
| Aiso (MHz) | Aaniso (MHz) | θ (Fe—C—N) | |
| C1 | −31.2 | [−1.3, −0.3, 1.6] | 175° |
| C2 | −25.0 | [0.0, +1.0, −1.0] | 132° |
| C3 | −22.0 | [1.0, 2.0, −3.0] | 123° |
| Transferrin: | |||
| Aiso (MHz) | Aaniso (MHz) | θ (Fe—C—N) | |
| C | −35.5 | [−2.12, −2.35, 4.47] | 0° |
2D CW 13C-ENDOR of 13CN-bound SOR
2D field/frequency 13C-ENDOR patterns comprising numerous spectra taken at fields across the entire EPR envelopes were obtained for the ferric-cyano adducts of all the SOR sites investigated in this work. Portions of the patterns acquired for WT D. vulgaris 2Fe-SOR and P. furiosus 1Fe-SOR are shown in Figure 7 along with simulations of the spectra as the summations of signals from the 13CN of the three substates. The 13C hyperfine tensors obtained from the simulations of WT and C13S 2Fe-SOR and WT 1Fe-SOR are listed in Table 2; the parameters for the E47A and K48A 2Fe-SOR variants are not significantly different from those listed. The three substates in WT 2Fe-SOR and its C13S variant have roughly equal populations (∼ 33%) based on the intensities of 2D ENDOR simulation; the C3 substate is slightly suppressed in the E47A and K48A variants. In contrast, the C1 substate in 1Fe-SOR is in the majority: the intensity ratios are, C1:C2:C3 = 2:1:1.
Figure 7.

Orientation-selective Q-band CW 13C-ENDOR spectra (solid lines) and simulations (dashed lines) for 13CN−-bound oxidized D. vulgaris 2Fe-SOR (left panel) and P. furiosus 1Fe-SOR (right panel). The conditions are same as in Figure 6 except more scans were taken at weak ENDOR signals. Simulation parameters : EPR linewidth = 500 MHz; A and g tensors are coaxial; (1) for 2Fe-SOR: ENDOR linewidth: 1.5 MHz (C1 species) and 2.0 MHz (C2 & C3 species); (2) for 1Fe-SOR: ENDOR linewidth: 1.3 MHz (C1 species) and 2.0 MHz (C2 & C3 species).
The hyperfine interaction for all three 13C resonances in each SOR is dominated by a strong isotropic term attributed to the exchange polarization of the paired Fe-C σ-bonding electrons by the unpaired electron in a dπ orbital of Fe3+ (45, 46), in which case Aiso is negative (46, 47). The magnitude of the C1 coupling, |Aiso (C1)| = 32.8 MHz, is ca. 10% less than that for the cyano-ferric adduct of transferrin, which shows only one Fe–C–N conformation in 13C-ENDOR spectrum (46), see Table 2. 1H ENDOR measurements on the methylene protons of the coordinated cysteine of SOR (data not shown), suggest that this decrease reflects delocalization of spin density from Fe to the cysteinyl sulfur. The isotropic 13C couplings decrease from substates 1 to 3: |Aiso(C1)| = 33 MHz; |Aiso(C2)| = 26 MHz, is ∼ 20% smaller, and is comparable to values found in hemoglobins and myoglobins (48); |Aiso (C3)| ∼ 22 MHz is ∼ 10 % smaller yet.
The anisotropic hyperfine contribution for all three 13C in all three SORs is coaxial with the g tensor. For all three substates of the WT 2Fe-SOR, it is of the dipolar form, T = [-t, -t, 2t], with the unique axis along g3. As the unique dipolar axis must lie along the Fe–C bond (see below), this shows that the direction of g3 corresponds to the normal to the His4 coordination plane. For one or more substates of each of the other SORs, the fit did not yield a dipolar tensor form; we take that to reflect the limits of our ability to fit the overlapping patterns and discuss herein only the 13C tensors for the WT 2Fe-SOR.
As discussed by Snetsinger et al. for the Fe–CN linkage in transferrin (46), the anisotropic term is the sum of a local contribution proportional to the p-orbital spin density induced in the spn orbital involved in the bond to Fe, plus a non-local contribution that is proportional to the spin density in the dπ orbital on Fe and inversely proportional to the cube of r(Fe–C) (49). The two contributions both have the dipolar form with the unique direction along the Fe–C bond; the dipolar parameter for the non-local interaction has a positive sign, 2tnl > 0, whereas that for the polarization-induced local contribution is negative, 2tl < 0, like Aiso. As a result, the observed dipolar coupling can be written, 2t = 2tnl + 2tl ≡ |2tnl| - |2tl|.
As seen in Table 2, the unique value of the anisotropic 13C interaction decreases for the three substates in the order, 2t(C1) > 2t(C2) > 0 > 2t(C3). The Fe–C distance must be essentially the same in all three conformations. 1H ENDOR (not shown) further indicates that the same is true for the S–Fe–C covalency; hence the spin density on Fe is essentially constant. As a result, 2tnl does not vary significantly among substates 1−3, and this sequential decrease in 2t(C) must reflect a progressive increase in the magnitude of the (negative) local dipolar contribution from substates 1 to 3, |2tl(C1)| < |2tl(C2)| < |2tl(C3)|. This progressive increase in |2tl|occurs in synchrony with the corresponding progressive decrease in |Aiso| noted just above, and these correspond to opposing changes in s and p spin density in the spn hybrid orbital on carbon that participates in the Fe–C bond. We now use a simple heuristic model of orbital hybridization to show that these synchronous, but opposing, changes going from substate 1−3 reflect progressive decrease in the Fe–C–N angle with a corresponding increase in n of the spn hybrid, and use this model to attain estimates of the Fe–C–N angles.
For simplicity, we assume that the carbon σ-bonds to Fe and N atoms in Fe–C–N involve equivalent spn hybrids on carbon. These two hybrids can be written, (50)
| (3a) |
| (3b) |
where 2px is a 2p carbon orbital directed along the Fe–C bond, n is the orbital hybridization, and θ is the angle between h1 and h2 hybrids, which in this model is the Fe–C–N angle. Constraining h1 and h2 in equations 3a and 3b to be orthogonal yields,
| (4a) |
which correlates the angle θ and the hybridization. The isotropic coupling is proportional to ρC, the spin density in the spn hybrid: Aiso = a0ρC/(1 + n), where a0 = 3770 MHz (51) is the isotropic coupling for a single 2s electron on carbon. As a result, we can rewrite equation 4a as
| (4b) |
According to equation 4b, the monotonic decrease in Aiso from substates 1−3 reflects a progressive decrease in the Fe–C–N angle. A corresponding equation can be written for 2tl, and the two together could, in principle be solved for a0ρC and thus θ (within the model) for each substate. Unfortunately, the accuracy with which 2tl can be measured is inadequate for this process; indeed, the anisotropic interaction derived from the simulations apparently deviates from the dipolar form in some cases, further complicating such a treatment. Hence this approach is inadvisable. Fortunately, the functional dependence of Aiso(θ) actually constrains the angles for C2 and C3 rather strongly. As discussed above, we may take ρC to be the same in the three substates, unchanged by the bending caused by nonbonding forces in the active site; we further take θ1 = 175° for Fe–C1–N, the most nearly linear linkage, consistent with the resonance Raman data. Equation 4b then yields a0ρC = 66.5(1) MHz, corresponding to ρC ≈ 0.02 spins induced in the carbon spn hybrid. The Aiso for C2 and C3 then give, θ2 = 133(2)° and θ3 = 123(2)° for the C2 and C3 substates. For discussion below, we note that this model involves an axially symmetric Fe-C σ bond, and thus is invariant to possible torsional rotations about that bond that might accompany the Fe–C–N bending.
Mims 15N-ENDOR of C15N-bound 1Fe-SOR
2D orientation-selective Mims 15N-ENDOR spectra have been collected for C15N−-bound P. furiosus 1Fe-SOR, see Figure 8. The 15N is weakly coupled, with A < 1 MHz at all fields. The three different conformations of cyanide could be detected in CW 13C-ENDOR experiments because the coupling is large; small percentage differences in Aiso thus are readily resolved. However, with the small 15N coupling, signals from the individual substate signals are not resolvable; the 2D pattern was simulated in terms of a single (C15N−) adduct of the 1Fe-SOR protein. As with the 13C hyperfine interaction, the 15N isotropic coupling is dominant, Aiso = 0.53 MHz; the anisotropic contribution is dipolar, with t = 0.09 MHz. If treated as arising from a point-dipole interaction with the spin on Fe (ρFe = 0.8), this value is smaller than would be expected; it corresponds to a Fe–N distance of 4.3 Å, which is substantially too long. As with the 13C coupling, this likely reflects a cancellation of local and non-local terms.
Figure 8.
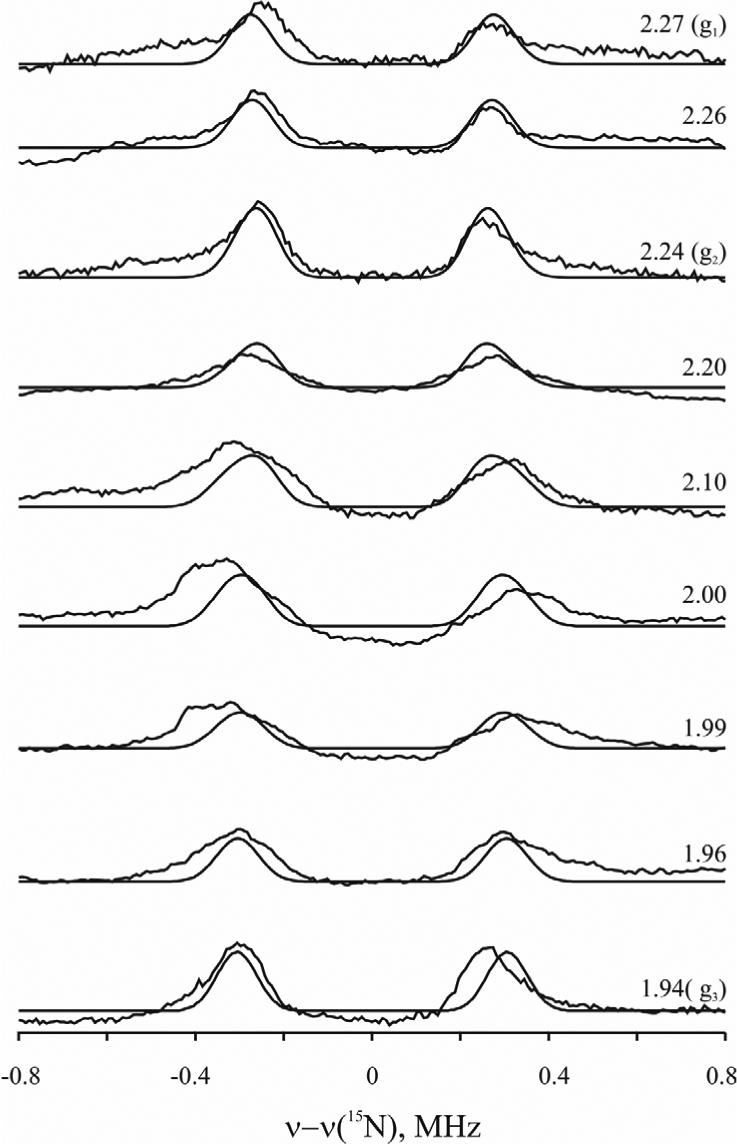
Orientation-selective Q-band Mims 15N-ENDOR spectra (solid lines) and simulations (dashed lines) for C15N−-bound oxidized P. furiosus 1Fe-SOR. Conditions: microwave frequency = 34.78 GHz; microwave pulse length (π/2) = 52 ns; τ =500 ns; rf pulse width = 60μs; spectra resolution = 256 points, 50 samplings of each point, ∼ 10 scans. Simulation parameters: EPR linewidth: 500 MHz; ENDOR linewidth: 0.1 MHz; A = [0.44, 0.44, 0.80] MHz; Euler angles = [45°, 90°] for axial symmetry.
DISCUSSION
We have added CN− to ferric 1Fe-, and 2Fe-SOR and its C13S variant as a surrogate for the putative ferric-(hydro)peroxo intermediate in the reaction of the enzyme with superoxide, and have used vibrational and ENDOR spectroscopies to study the properties of the active-site paramagnetic iron center. Addition of cyanide changes the ferric ion in SOR from rhombic high spin (S = 5/2) to axial-like low spin (S = 1/2). RR, FTIR, and ENDOR data all show that the bound cyanide adopts three distinct conformations in Fe(III)-CN SOR. The majority of the experiments are carried out at cryogenic temperatures, but the central finding of heterogeneity in cyanide binding to the ferric site does not result from freezing-in of conformational substates at low temperature, as the multiplicity of C–N stretching modes persists at ambient temperature in the FTIR spectra. In addition, FTIR studies of the Fe(II)-CN adduct of 1Fe-SOR reveal a homogeneous near-linear Fe–C–N conformation with significant π-back-donation from the ferrous iron into the cyanide π* orbitals, indicating that cyanide bending and structural heterogeneity is confined to the oxidized state.
The FTIR and resonance Raman studies of the SOR active site reported in this work constitute the first characterization of the vibrational modes of a Fe–C–N unit in a non-heme Fe enzyme. Resonance Raman studies of cyanide-bound ferric hemoproteins have been extensively used to identify ν(Fe–CN) stretching and δ(Fe–C–N) bending modes in the low frequency region via cyanide isotopomer difference spectra, due to overlap with intense porphyrin vibrational modes. This approach is not required for cyanide-bound SOR as the ν(Fe–CN) stretching/δ(Fe–C–N) bending modes are strongly enhanced via excitation into the intense (Cys)S(pπ)-to-Fe(dπ) CT transition and occur in the region where there is little overlap with other strongly enhanced vibrational modes. However, the pattern of isotope shifts for the 13C14N, 12C15N and 13C15N isotopomers and the separation between the ν(Fe–CN) stretching and δ(Fe–C–N) bending modes provide a means of assessing the Fe–C–N angle based on the methodology and normal mode calculations developed for cyano-ferric hemoproteins. Multiple conformations involving both near-linear and bent Fe–C–N units are the rule rather than the exception for cyano-ferric hemoproteins, and have been observed in sulfite reductase (37), horseradish peroxidase (29), catalase (33), cytochrome P450 (30;32), myoglobin (36), hemoglobin (36), cytochrome c oxidase (36), and nitrophorins (38). Crystallography has also characterized near-linear and bent forms of Fe–C–N units in cyano-ferric forms of several hemoproteins, e.g. hemoglobins (81−178°) (52), myoglobins (102−177°) (52), myeloperoxiase (157°) (53), heme d1 in cd1 nitrite reductases (146°,164°, and 168°) (54;55). Analysis of the resonance Raman results for the cyano-ferric adduct of SOR in light of these data indicate three well-defined conformations that differ in terms of the angle of the Fe–C–N unit: one near-linear conformer (∼175°) and two distinct bent conformers (< 155°). Moreover, using resonance Raman vibrational data in combination with the 13C-ENDOR data discussed below, provides estimates of ∼175°, ∼133° and ∼123° for the Fe–C–N angles in each of the three conformers, see Table 1.
The paucity of parallel FTIR and resonance Raman studies for cyano-ferric hemoproteins inhibits reliable correlation of ν(C–N) stretching frequencies observed by FTIR with ν(Fe–CN) stretching/ δ(Fe–C–N) bending frequencies observed by resonance Raman for specific conformers. Nevertheless, the FTIR spectrum of cyano-ferric SOR clearly shows three ν(C–N) stretching modes with frequencies ( 2100, 2111, and 2115 cm−1 ) just below those observed for cyano-ferric derivatives of hemoglobin, myoglogin, nitrophorin and peroxidases (2122−2135 cm−1 ) (38;39;56). The primary determinants of the ν(C–N) stretching frequencies are the extent of σ-donation from the filled 5σ* molecular orbital of cyanide, the extent of π-backdonation into the vacant 2π* molecular orbitals of cyanide, and extent of H-bonding interactions involving the bound cyanide (40). Hence, the increased frequencies relative to free cyanide primarily reflect strong σ-donation to ferric iron, and the lower ν(C–N) stretching frequencies in SOR relative to hemoproteins are consistent with increased electron density on the ferric iron as a result of strong dπ-pπ interaction involving the trans cysteinate ligand (17). The ν(C–N) stretching mode at 2111 cm−1 is tentatively assigned to the linear conformation on the basis of sensitivity to 1H2O/2H2O exchange, see Table 1. However, although the sensitivity to 1H2O/2H2O exchange shows that the linear conformation has solvent exchangeable H-bonding interactions, the absence of sensitivity to 1H2O/2H2O exchange does not preclude H-bonding interactions for the bent conformations. Indeed the available mutagenesis and crystallographic evidence for cyano-ferric hemoproteins indicate that solvent or protein-side-chain H-bonding interactions play a major role in stabilizing specific Fe–C–N conformations (32, 52-55).
In all the ferric-cyano SOR derivatives studied here, 13C-ENDOR spectra show three well-resolved Fe–C–N conformations, labeled C1, C2 and C3. On the basis of the ENDOR intensities, the proportion of each substate is approximately equal in the two 2Fe-SORs, while the near-linear conformer (C1) dominates in the 1Fe-SOR. The 13C hyperfine tensors have been determined for the three substates of the 2Fe-SOR. The magnitude of the isotropic coupling, |Aiso|, decreases from substate 1 → 3, while that of the local dipolar coupling, |2tl|, synchronously increases from conformations 1 → 3. The former term is proportional to the 2s contribution to the spn (13C) hybrid that forms a σ bond to Fe, the latter to the 2p contribution. Such opposing changes are shown to be interpretable in terms of the changes in hybridization that accompany a progressive bending of the Fe–C–N linkage in substates 1 → 3, and the heuristic values for the Fe–C–N angles in the three substates derived with a simple model for the bonding ranged from ca. 123° (C3) to the reference 175° (C1 species), see Table 2. Much has been written about the deformation of the Fe–C–O linkage in carboxy-hemoproteins (57), and the degree to which it involves Fe–C–O bending, as opposed to ‘tilting’. It is therefore useful to note that the opposing changes in Aiso and t1 are not interpretable in terms of different spin densities within a rigid spn hybrid on carbon, such as might be induced by different degrees of tilting or of torsional rotation, and can only be interpreted in terms of different degrees of bending.
The structural origin of the heterogeneity in cyanide binding at the ferric site of SOR was addressed by resonance Raman and ENDOR studies of variants involving substitution of conserved glutamate and lysine residues in the flexible loop above the substrate binding pocket (E14 and K15 in 1Fe-SOR and E47 and K48 in 2Fe-SOR). Crystallographic studies indicate that these are the only two charged residues close to the substrate-binding site, with the glutamate bound to iron in the oxidized structure and the lysine closest to the vacant iron coordination site in the reduced structure (6). However, the spectroscopic results for these variants clearly demonstrate that neither residue is responsible for positioning or directly interacting with bound cyanide. Hence it seems unlikely that K15/K48 plays a role in orienting or protonating the putative ferric-peroxo intermediate formed by the interaction of superoxide with reduced SOR (as indicated in Scheme 1). The substantial decrease in the rate of formation of the initial intermediate that has been observed to accompany the lysine to alanine/isoleucine substitutions in 2Fe-SORs (10, 11) is therefore more likely explained in terms of the positively charged lysine helping to direct the negatively charged superoxide to the ferrous active site.
Despite the apparent lack of direct interaction of residues on the flexible loop, the vibrational and ENDOR results for cyano-ferric SOR derivatives clearly indicate a flexible binding pocket for coordination of diatomic molecules. We suggest that this applies to the interaction of superoxide with the active site and the formation of a putative ferric-peroxo intermediate (cf. Scheme 1). A bent Fe–O–O geometry is expected from structural studies of other ferrous-O2 and ferric-hydroperoxo complexes, and would permit facile approach of superoxide ‘below’ the K15/K48 side chain, which overhangs the vacant ferrous SOR coordination site (6, 7, 18). Thus, the bent substates (C1 and C2) are most likely to mimic the binding of a peroxo/hydroperoxo moiety.
It is plausible to further suggest that the two bent Fe–C–N substates might have the CN− ligand interleaved between different pairs of His ring planes. DFT calculations for a bent, end-on peroxo coordination at the ferric SOR site (Fe–O–O ∼120°) (18) indicated that the energetically most favorable range of peroxo torsional angles was centered roughly along the Fe–N bond of what corresponds to His119 in the 2Fe-SOR structure, see Figure 9 (5); the 1Fe-SOR structure is similar and led to the same conclusion, which applies to both high- and low-spin ferric-peroxo moieties. The surrounding side chains of Ala45 and Pro70 in 2Fe-SOR, and homologous residues in 1Fe-SOR, would sterically hinder a bent Fe–C–N pointed between the remaining two His ligand pairs (His49/69 and His69/75, Figure 9). These latter two torsional orientations would also place the cyano nitrogen in relatively unfavorable hydrophobic environments. The two bent Fe–C–N substates observed here are, thus, reasonably assigned to cyano ligands oriented on either side of the Fe-NHis119 bond, i.e., between the rings of His49 and His119 (smallest Fe–C–N bend angle) and His119 and His75 (second smallest Fe–C–N bend angle), thereby providing experimental support for the predicted orientations of the corresponding hydroperoxo/peroxo ligands during SOR turnover.
Figure 9.
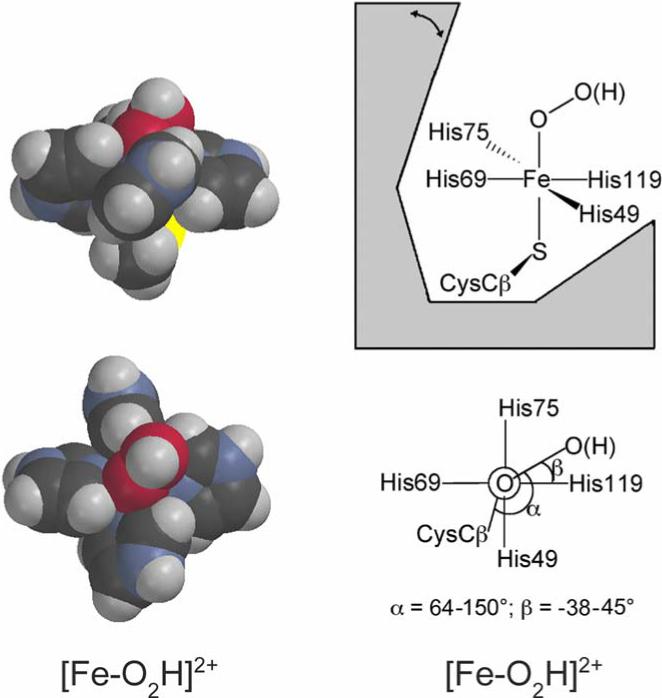
Space-filling depictions of DFT-optimized modeled structures of the putative ferric-(hydro)peroxo intermediate (left), and schematic diagrams showing the geometric restrictions of this intermediate at the SOR site (right), modified with permission from Ref. (5). The modeled structures used imidazoles and methyl thiolate in place of His and Cys ligands, respectively, and the torsion angles of the imidazole rings around the Fe-N bonds were fixed at those in the D. desulfuricans 2Fe-SOR crystal structure (PDB ID 1DFX)(18). Color coding of atoms is carbon, black; hydrogen, gray; oxygen, red; nitrogen, blue; sulfur, yellow, iron, orange. The double-headed arrow indicates movement of the flexible loop region.
Footnotes
This work was supported by grants from the National Institute of Health (GM60329 to M.W.W.A. and M.K.J.; GM40388 to D.M.K. and HL 13531to B.H.M.)
Abbreviations: SOD, superoxide dismutase; SOR, superoxide reductase; ENDOR, Electron Nuclear Double Resonance; VTMCD, variable-temperature magnetic circular dichroism; EPR, Electron Paramagnetic Resonance; CT, charge transfer; WT, wild-type; rf, radiofrequency.
References
- 1.Adams MWW, Jenney FE, Jr., Clay MD, Johnson MK. Superoxide reductase: Fact or fiction. J. Biol. Inorg. Chem. 2002;7:647–652. doi: 10.1007/s00775-002-0359-x. [DOI] [PubMed] [Google Scholar]
- 2.Imlay JA. What biological purpose is served by superoxide reductase? J. Biol. Inorg. Chem. 2002;7:659–663. doi: 10.1007/s00775-002-0361-3. [DOI] [PubMed] [Google Scholar]
- 3.Abreu IA, Xavier AV, LeGall J, Cabelli DE, Teixeira M. Superoxide scavenging by neelaredoxin: dismutation and reduction activities in anaerobes. J. Biol. Inorg. Chem. 2002;7:668–674. doi: 10.1007/s00775-002-0363-1. [DOI] [PubMed] [Google Scholar]
- 4.Nivière V, Fontecave M. Discovery of superoxide reductase: an historical perspective. J. Biol. Inorg. Chem. 2004;9:119–123. doi: 10.1007/s00775-003-0519-7. [DOI] [PubMed] [Google Scholar]
- 5.Kurtz DM., Jr. Microbial detoxification of superoxide: The non-heme iron reductive paradigm for combating oxidative stress. Acc. Chem. Res. 2004;37:902–908. doi: 10.1021/ar0200091. [DOI] [PubMed] [Google Scholar]
- 6.Yeh AP, Hu Y, Jenney FE, Adams MWW, Rees DC. Structures of the superoxide reductase from Pyrococcus furiosus in the oxidized and reduced states. Biochemistry. 2000;39:2499–2508. doi: 10.1021/bi992428k. [DOI] [PubMed] [Google Scholar]
- 7.Coelho AV, Matias P, Fülõp V, Thompson A, Gonzalez A, Carrondo MA. Desulfoferrodoxin structure determined by MAD phasing and refinement to 1.9-Å resolution reveals a unique combination of a tetrahedral FeS4 centre with a square pyramidal FeSN4 centre. J. Biol. Inorg. Chem. 1997;2:680–689. [Google Scholar]
- 8.Coulter ED, Emerson JP, Kurtz DM, Jr., Cabelli DE. Superoxide reactivity of rubredoxin oxidoreductase (desulfoferrodoxin) from Desulfovibrio vulgaris: A pulse radiolysis study. J. Am. Chem. Soc. 2000;122:11555–11556. [Google Scholar]
- 9.Abreu IA, Saraiva LM, Soares CM, Teixeira M, Cabelli DE. The mechanism of superoxide scavenging by Archaeoglobus fulgidus neelaredoxin. J. Biol. Chem. 2001;276:38995–39001. doi: 10.1074/jbc.M103232200. [DOI] [PubMed] [Google Scholar]
- 10.Lombard M, Houée-Levin C, Touati D, Fontecave M, Nivière V. Superoxide reductase from Desulfoarculus baarsii: Mechanism and role of glutamate 47 and lysine 48 in catalysis. Biochemistry. 2001;40:5032–5040. doi: 10.1021/bi0023908. [DOI] [PubMed] [Google Scholar]
- 11.Emerson JP, Coulter ED, Cabelli DE, Phillips RS, Kurtz DM., Jr. Kinetics and mechanism of superoxide reduction by two-iron superoxide reductase from Desulfovibrio vulgaris. Biochemistry. 2002;41:4348–4357. doi: 10.1021/bi0119159. [DOI] [PubMed] [Google Scholar]
- 12.Clay MD, Jenney FE, Jr., Noh HJ, Hagedoorn PL, Adams MWW, Johnson MK. Resonance Raman characterization of the mononuclear iron active-site vibrations and putative electron transport pathways in Pyrococcus furiosus superoxide reductase. Biochemistry. 2002;41:9833–9841. doi: 10.1021/bi025833b. [DOI] [PubMed] [Google Scholar]
- 13.Berthomieu C, Dupeyrat F, Fontecave M, Verméglio A, Nivière V. Redox-dependent structural changes in the superoxide reductase from Desulfoarculus baarsii and Treponema pallidum: An FTIR study. Biochemistry. 2002;41:10360–10368. doi: 10.1021/bi020344x. [DOI] [PubMed] [Google Scholar]
- 14.Nivière V, Asso M, Weill CO, Lombard M, Guigliarelli B, Favaudon V, Houée-Levin C. Superoxide reductase from Desulfoarculus baarsii: Identification of protonation steps in the enzymatic mechanism. Biochemistry. 2004;43:808–818. doi: 10.1021/bi035698i. [DOI] [PubMed] [Google Scholar]
- 15.Clay MD, Emerson JP, Coulter ED, Kurtz DM, Jr., Johnson MK. Spectroscopic characterization of the [Fe(His)4(Cys)] site in 2Fe-superoxide reductase from Desulfovibrio vulgaris. J. Biol. Inorg. Chem. 2003;8:671–682. doi: 10.1007/s00775-003-0465-4. [DOI] [PubMed] [Google Scholar]
- 16.Emerson JP, Cabelli DE, Kurtz DM., Jr. An engineered two-iron superoxide reductase lacking the [Fe(SCys)4] site retains its catalytic properties in vitro and in vivo. Proc. Natl. Acad. Sci. USA. 2003;100:3802–3807. doi: 10.1073/pnas.0537177100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clay MD, Jenney FE, Jr., Hagedoorn PL, George GN, Adams MWW, Johnson MK. Spectroscopic studies of Pyrococcus furiosus superoxide reductase: Implications for active-site structures and the catalytic mechanism. J. Am. Chem. Soc. 2002;124:788–805. doi: 10.1021/ja016889g. [DOI] [PubMed] [Google Scholar]
- 18.Silaghi-Dumitrescu R, Silaghi-Dumitrescu I, Coulter ED, Kurtz DM., Jr. Computational study of the non-heme iron active site in superoxide reductase and its reaction with superoxide. Inorg. Chem. 2003;42:446–456. doi: 10.1021/ic025684l. [DOI] [PubMed] [Google Scholar]
- 19.Mathé C, Mattioli TA, Horner O, Lombard M, Latour J-M, Fontecave M, Nivière V. Identification of iron(III) peroxo species in the active site of the superoxide reductase sor from Desulfoarculus baarsii. J. Am. Chem. Soc. 2002;124:4966–4967. doi: 10.1021/ja025707v. [DOI] [PubMed] [Google Scholar]
- 20.Horner O, Mouesca J-M, Oddou J-L, Jeandey C, Nivière V, Mattioli TA, Mathé C, Fontecave M, Maldivi P, Bonville P, Halfen JA, Latour J-M. Mössbauer characterization of an unusual high-spin side-on peroxo-Fe3+ species in the active site of superoxide reductase from Desulfovibrio baarsii. Density functional calculations on related models. Biochemistry. 2004;43:8815–8825. doi: 10.1021/bi0498151. [DOI] [PubMed] [Google Scholar]
- 21.Romão CV, Liu M-Y, LeGall J, Gomes CM, Braga V, Pacheco I, Xavier AV, Teixeira M. The superoxide dismutase activity of desulfoferrodoxin from Desulfovibrio desulfuricans ATCC 27774. Eur. J. Biochem. 1999;261:438–443. doi: 10.1046/j.1432-1327.1999.00278.x. [DOI] [PubMed] [Google Scholar]
- 22.Silva G, Oliveira S, Gomes CM, Pacheco I, Liu M-Y, Xavier AV, Teixeira M, LeGall J, Rodrigues-Pousada C. Desulfovibrio gigas neelaredoxin. A novel superoxide dismutase integrated in a putative oxygen sensory operon of an anaerobe. Eur. J. Biochem. 1999;259:235–243. doi: 10.1046/j.1432-1327.1999.00025.x. [DOI] [PubMed] [Google Scholar]
- 23.Jenney FE, Verhagen MFJM, Cui X, Adams MWW. Anaerobic microbes: Oxygen detoxification without superoxide dismutase. Science. 1999;286:306–309. doi: 10.1126/science.286.5438.306. [DOI] [PubMed] [Google Scholar]
- 24.Emerson JP, Coulter ED, Phillips RS, Kurtz DM., Jr. Kinetics of the superoxide reductase catalytic cycle. J. Biol. Chem. 2003;278:39662–39668. doi: 10.1074/jbc.M306488200. [DOI] [PubMed] [Google Scholar]
- 25.Drozdzewski PM, Johnson MK. A simple anaerobic cell for low temperature Raman spectroscopy. Appl. Spectrosc. 1988;42:1575–1577. [Google Scholar]
- 26.Werst MM, Davoust CE, Hoffman BM. Ligand spin densities in blue copper proteins by Q-band 1H and 14N ENDOR spectroscopy. J. Am. Chem. Soc. 1991;113:1533–1538. [Google Scholar]
- 27.Hoffman BM, DeRose VJ, Ong J-L, Davoust CE. Sensitivity enhancement in field modulated CW ENDOR via RF bandwidth broadening. J. Magn. Res. 1994;110:52–57. [Google Scholar]
- 28.Hoffman BM, DeRose VJ, Doan PE, Gurbiel RJ, Houseman ALP, Telser J. Metalloenzyme active-site structure and function through multifrequency CW and pulsed ENDOR. In: Berliner LJ, Reuben J, editors. EMR of paramagnetic molecules; biological magnetic resonance. Plenum Press; New York: 1993. pp. 151–218. [Google Scholar]
- 29.Al-Mustafa J, Kincaid JR. Resonance Raman study of cyanide-ligated horseradish peroxidase. Detection of two binding geometries and direct evidence for the ”push-pull” effect. Biochemistry. 1994;33:2191–2197. doi: 10.1021/bi00174a028. [DOI] [PubMed] [Google Scholar]
- 30.Simianu MC, Kincaid JR. Resonance Raman spectroscopic detection of both linear and bent Fe-CN fragments for the cyanide adducts of cytochrome P-450 camphor and its substrate-bound forms. Relevance to the ”charge relay” mechanism. J. Am. Chem. Soc. 1995;117:4628–4636. [Google Scholar]
- 31.Hu S, Treat RW, Kincaid JR. Distinctive heme active-site structure in lactoperoxidase revealed by resonance Raman spectroscopy. Biochemistry. 1993;32:10125–10130. doi: 10.1021/bi00089a031. [DOI] [PubMed] [Google Scholar]
- 32.Deng T-J, Macdonald IDG, Simianu MC, Sykora M, Kincaid JR, Sligar SG. Hydrogen-bonding interactions in the active sites of cytochrome P450cam and its site-directed mutants. J. Am. Chem. Soc. 2001;123:269–278. doi: 10.1021/ja001517d. [DOI] [PubMed] [Google Scholar]
- 33.Al-Mustafa J, Sykora M, Kincaid JR. Resonance Raman investigation of cyanide ligated beef liver and Aspergillus niger catalases. J. Biol. Chem. 1995;270:10449–10460. doi: 10.1074/jbc.270.18.10449. [DOI] [PubMed] [Google Scholar]
- 34.López-Garriga JJ, Oertling WA, Kean RT, Hoogland H, Wever R, Babcock GT. Metal-ligand vibrations of cyanoferric myeloperoxidase and cyanoferric horseradish peroxidase: Evidence for a constrained heme pocket in myeloperoxidase. Biochemistry. 1990;29:9387–9395. doi: 10.1021/bi00492a012. [DOI] [PubMed] [Google Scholar]
- 35.Kim Y, Babcock GT, Surerus KK, Fee JA, Dyer RB, Woodruff WH, Oertling WA. Cyanide binding and active site structure in heme-copper oxidases: Normal coordinate analysis of iron-cyanide vibrations of a32+CN- complexes of cytochromes ba3 and aa3. Biospectroscopy. 1998;4:1–15. doi: 10.1002/(sici)1520-6343(1998)4:1<1::aid-bspy1>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 36.Hirota S, Ogura T, Shinzawa-Itoh K, Yoshikawa S, Kitagawa T. Observation of multiple CN-isotope-sensitive Raman bands for CN− adducts of hemoglobin, myoglobin, and cytochrome c oxidase: Evidence for vibrational coupling between the Fe-C-N bending and porphyrin in-plane modes. J. Phys. Chem. 1996;100:15274–15279. [Google Scholar]
- 37.Han S, Madden JF, Siegel LM, Spiro TG. Resonance Raman studies of Escherichia coli sulfite reductase hemoprotein. 3. Bound ligand vibrational modes. Biochemistry. 1989;28:5477–5485. doi: 10.1021/bi00439a024. [DOI] [PubMed] [Google Scholar]
- 38.Maes EM, Walker FA, Montfort WR, Czernuszewicz RS. Resonance Raman spectroscopic study of nitrophorin 1, a nitric oxide-binding heme protein from Rhodnius prolixus, and its nitrosyl and cyano adducts. J. Am. Chem. Soc. 2001;123:11664–11672. doi: 10.1021/ja0031927. [DOI] [PubMed] [Google Scholar]
- 39.Yoshikawa S, O'Keeffe DH, Caughey WS. Investigations of cyanide as an infrared probe of hemoprotein ligand binding sites. J. Biol. Chem. 1985;260:3518–3528. [PubMed] [Google Scholar]
- 40.Nakamoto K. Infrared and Raman spectra of inorganic and coordination compounds. John Wiley & Sons; New York: 1986. [Google Scholar]
- 41.Moura I, Huynh BH, Hausinger RP, LeGall J, Xavier AV, Münck E. Mössbauer and EPR studies of desulforedoxin from Desulfovibrio gigas. J. Biol. Chem. 1980;255:2493–2498. [PubMed] [Google Scholar]
- 42.Tavares P, Ravi N, Moura JJG, LeGall J, Huang Y-H, Crouse BR, Johnson MK, Huynh BH, Moura I. Spectroscopic properties of desulfoferrodoxin from Desulfovibrio desulfuricans (ATCC 27774) J. Biol. Chem. 1994;269:10504–10510. [PubMed] [Google Scholar]
- 43.Ascenso C, Rusnak FM, Cabrito I, Lima MJ, Naylor S, Moura I, Moura JJG. Desulfoferrodoxin: A modular protein. J. Biol. Inorg. Chem. 2000;5:720–729. doi: 10.1007/s007750000161. [DOI] [PubMed] [Google Scholar]
- 44.Romao MJ, Archer M. Structural versatility of proteins containing rubredoxin-type centers. In: Ferreira GC, Moura JJG, Franco R, editors. Iron Metabolism. Wiley-VCH Verlag; Weinheim: 1999. pp. 341–358. [Google Scholar]
- 45.Kuska HA, Rogers MT. Electron magnetic resonance studies and covalent bonding of cyanide and fluoride complexes of transition metals. J. Chem. Phys. 1964;41:3802–3805. [Google Scholar]
- 46.Snetsinger PA, Chasteen ND, van Willigen H. Structural analysis of a low-spin cyanide adduct of iron(III) transferrin by angle-selected 13C ENDOR spectroscopy. J. Am. Chem. Soc. 1990;112:8155–8160. [Google Scholar]
- 47.Davis DG, Kurland RJ. Carbon-13 NMR of paramagnetic iron-group cyanides. J. Chem. Phys. 1967;46:388–390. [Google Scholar]
- 48.Mulks CF, Scholes CP, Dickinson LC, Lapidot A. Electron nuclear double resonance from high- and low-spin ferric hemoglobins and myoglobins. J. Am. Chem. Soc. 1979;101:1645–1654. [Google Scholar]
- 49.Manikandan P, Choi E-Y, Hille R, Hoffman BM. 35 GHz ENDOR characterization of the ”very rapid” signal of xanthine oxidase reacted with 2-hydroxy-6-methylpurine (13C8): Evidence against direct Mo-C8 interaction. J. Am. Chem. Soc. 2001;123:2658–2663. doi: 10.1021/ja003894w. [DOI] [PubMed] [Google Scholar]
- 50.McWeeny R. Coulson's Valence. Oxford University Press; Oxford: 1979. [Google Scholar]
- 51.Weil JA, Bolton JR, Wertz JE. Electron Paramagnetic Resonance: elementary theory and practical applications. John Wiley & sons; New York: 1995. [Google Scholar]
- 52.Bolognesi M, Rosano C, Losso R, Borassi A, Rizzi M, Wittenberg JB, Boffi A, Ascenzi P. Cyanide binding to Lucina pectinata hemoglobin I and to sperm whale myoglobin: and x-ray crystallographic study. Biophys. J. 1999;77:1093–1099. doi: 10.1016/S0006-3495(99)76959-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blair-Johnson M, Fiedler T, Fenna R. Human myeloperoxidase: Structure of a cyanide complex and its interaction with bromide and thiocyanate substrates at 1.9 Å resolution. Biochemistry. 2001;40:13990–13997. doi: 10.1021/bi0111808. [DOI] [PubMed] [Google Scholar]
- 54.Sun W, Arese M, Brunori M, Nurizzo D, Brown K, Cambillau C, Tegoni M, Cutruzzolà F. Cyanide binding to cd1 nitrite reductase from Pseudomonas aeruginosa: Role of the active site His369 in ligand stabilization. Biochem. Biophys. Res. Commun. 2002;291:1–7. doi: 10.1006/bbrc.2002.6391. [DOI] [PubMed] [Google Scholar]
- 55.Jafferji A, Allen JWA, Ferguson SJ, Fülõp V. X-ray crystallographic study of cyanide binding provides insights into the structure-function relationship for cytochrome cd1 nitrite reducatse from Paracoccus pantotrophus. J. Biol. Chem. 2000;275:25089–25094. doi: 10.1074/jbc.M001377200. [DOI] [PubMed] [Google Scholar]
- 56.Reddy KS, Yonetani T, Tsuneshige A, Chance B, Kushkuley B, Stavrov SS, Vanderkooi JM. Infrared spectroscopy of the cyanide complex of iron(II) myoglobin and comparison with complexes of microperoxidase and hemoglobin. Biochemistry. 1996;35:5562–5570. doi: 10.1021/bi952596m. [DOI] [PubMed] [Google Scholar]
- 57.Spiro TG, Kozlowski PM. Is the CO adduct of myoglobin bent, and does it matter? Acc. Chem. Res. 2001;34:137–144. doi: 10.1021/ar000108j. [DOI] [PubMed] [Google Scholar]