

The full length and the regulatory domain of the LysR-type transcriptional regulator CrgA have been crystallized. Diffraction data were collected from two crystal forms of full-length CrgA to 3.0 and 3.8 Å resolution, respectively. Crystals of the selenomethionine derivative of the C-terminal regulatory domain of CrgA diffracted to 2.3 Å resolution.
Keywords: CrgA, Neisseria meningitidis, LysR-type regulators
Abstract
Although LysR-type regulators (LTTRs) represent the largest family of transcriptional regulators in bacteria, the full-length structure of only one annotated LTTR (CbnR) has been deposited in the PDB. CrgA, a LTTR from pathogenic Neisseria meningitidis MC58, which is up-regulated upon bacterial cell contact with human epithelial cells, has been cloned, purified and crystallized. Crystals of full-length CrgA were obtained after buffer screening with a thermal shift assay and concentration with 0.2 M NDSB-256. Data were collected from two crystal forms of full-length CrgA belonging to space groups P212121 and P21, diffracting to 3.0 and 3.8 Å resolution and consistent with the presence of between six and ten and between ten and 20 copies of CrgA in the asymmetric unit, respectively. In addition, diffraction data were collected to 2.3 Å resolution from the selenomethionine derivative of the regulatory domain of CrgA. The crystals belonged to space group P21 and contained two molecules in the asymmetric unit.
1. Introduction
The LysR family of transcriptional regulators (LTTRs) were first reported by Henikoff et al. (1988 ▶) and are now known to represent the largest family of bacterial transcription factors (Pareja et al., 2006 ▶). LTTRs have molecular weights of between 30 and 36 kDa and comprise an N-terminal DNA-binding domain (∼65 residues) that contains a winged helix–turn–helix motif joined by a linker helix (∼30 residues) to a C-terminal regulatory domain (∼200 residues) that is involved in oligomerization of the protein, typically into tetramers (Schell, 1993 ▶; Lochowska et al., 2001 ▶; Ezezika, Haddad, Neidle et al., 2007 ▶; Smirnova et al., 2004 ▶; Rosario & Bender, 2005 ▶; Muraoka et al., 2003 ▶).
Structural studies of full-length LTTRs have been hampered by their low solubility and tendency to aggregate (Ezezika, Haddad, Clark et al., 2007 ▶; Smirnova et al., 2004 ▶). To date, crystal structures of only one full-length LTTR, CbnR, have been deposited (Muraoka et al., 2003 ▶). Two crystal forms of CbnR were obtained, both comprising protein tetramers but differing in the relative orientation of the subunits. In contrast, several regulatory-domain structures of LTTRs have been described, including CysB, which was derived from a chymotryptic fragment (Tyrrell et al., 1997 ▶), OxyR (Choi et al., 2001 ▶), BenM, CatM (Ezezika, Haddad, Clark et al., 2007 ▶) and Cbl (Stec et al., 2006 ▶), which were produced from C-terminal domain constructs, and DntR, which was crystallized as a full-length protein but only the regulatory domain and a small portion of the linker helix were sufficiently ordered to allow a model to be built (Smirnova et al., 2004 ▶).
CrgA (contact-regulated gene A) is an LTTR from the exclusive human pathogen Neisseria meningitidis (serogroup B) strain MC58, a causative agent of bacterial meningitis and septicaemia (Rosenstein et al., 2001 ▶). N. meningitidis is a common commensal, colonizing the nasopharynx of 8–20% of healthy individuals (Stephens, 2007 ▶) and only rarely causing invasive disease by gaining access to the bloodstream through penetration of the nasopharyngeal mucosa (Rosenstein et al., 2001 ▶). After transmission, in order for successful colonization the meningococci must overcome the host immune system and adhere to the nasopharyngeal epithelial cells (Rosenstein et al., 2001 ▶; Stephens, 2007 ▶). Adhesion can be viewed as a two-step process. During initial adhesion, which is a pilus-mediated event, the meningococci attach as microcolonies to the surface of the host cells. Later in infection, during intimate adhesion, the pili disappear, the microcolonies disband and the meningococci become more closely associated with the host plasma membrane (Merz & So, 2000 ▶). CrgA has been reported to be induced upon contact of N. meningitidis with human epithelial cells and in functional assays meningococci lacking CrgA are unable to undergo intimate adhesion to epithelial cells (Deghmane et al., 2000 ▶, 2002 ▶). Like the majority of LTTRs, CrgA is a dual regulator, activating the expression of the divergently orientated mdaB (modulator of drug activity) gene while repressing its own expression (Deghmane & Taha, 2003 ▶; Ieva et al., 2005 ▶; Schell, 1993 ▶). CrgA has also been shown to bind to the promoter regions of genes encoding surface-associated proteins (PilC, sia and pilE) and is shown to down-regulate these genes. Therefore, it appears to play a role in regulation of the transition between the initial and intimate stages of adhesion (Deghmane et al., 2002 ▶).
CrgA has 299 residues and shares less than 17% (BLASTP and ClustalW) sequence identity to CbnR and other LTTRs currently deposited in the PDB. Here, we report the cloning, purification and crystallization of full-length (FL-CrgA) and regulatory-domain (RD-CrgA) forms of CrgA. The strategy developed to stabilize FL-CrgA prior to crystallization is detailed. Preliminary crystallographic analysis of the data sets collected is presented.
2. Experimental methods and results
2.1. Cloning
Full-length (FL-CrgA) and C-terminal regulatory-domain (RD-CrgA; residues Glu89–Gly299) constructs of CrgA were designed for recombinant expression in Escherichia coli. The crgA gene (NMB1856) was amplified from genomic DNA (N. meningitidis MC58) by PCR using KOD Hot Start DNA Polymerase (Merck) and cloned with In-Fusion technology (Clontech) following the methods reported by Berrow et al. (2007 ▶). The forward primer 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCGAAGGAGATAGAACCATGGCACATCACCACCACCATCACGAAATACCGCAAGGCGTGTTGAGC and the reverse primer 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCTCATTATCCACAGAGATTGTTTCCCAGTTCC were used in the construction of the RD-CrgA expression construct containing a 3C protease-cleavable N-terminal hexahistidine purification tag with sequence MAHHHHHHSSGLEVLFQ↓GP (the 3C protease site is shown in bold, with the cleavage point indicated by an arrow). The forward primer 5′-AGGAGATATACCATGATGAAAACCAATTCAGAAGAACTGAC and the reverse primer 5′-GTGGTGGTGGTGTTTTCCACAGAGATTGTTTCCCAGTTC were used in the construction of the FL-CrgA expression construct containing a C-terminal His tag with sequence KHHHHHH. The sequence-verified expression constructs were tested for soluble expression by semi-automated small-scale screening on a Bio Robot 8000 (Qiagen) using the protocols of Berrow et al. (2007 ▶).
2.2. Purification
Native and selenomethionine-labelled protein was produced from E. coli strain B834 using Power Broth and SelenoMet media (Molecular Dimensions), respectively. Cells were grown in 2 l cultures at 310 K to an OD600 of 0.6, the temperature was reduced to 293 K and recombinant protein expression was induced by addition of IPTG to 0.5 mM. The cultures were grown for a further 20 h and the cells were harvested by centrifugation at 6000g for 15 min. The cells were lysed in binding buffer (50 mM Tris pH 7.5, 500 mM NaCl, 20 mM imidazole) supplemented with 0.2%(v/v) Tween 20 and an EDTA-free protease-inhibitor cocktail tablet (Roche) with a Basic Z Cell Disruptor (Constant Systems). After centrifugation at 24 000g for 30 min, the cleared lysate was applied onto a 1 ml HisTrap FF column attached to an ÄKTA Express protein-purification system (GE Healthcare). The column was washed to a UV (280 nm) baseline with binding buffer and the protein was eluted with 50 mM Tris pH 7.5, 500 mM NaCl, 500 mM imidazole. The eluate was loaded onto a 16/60 HiLoad Superdex 200 column (GE Healthcare) pre-equilibrated in 20 mM Tris pH 7.5, 200 mM NaCl or 20 mM citrate pH 5.0, 200 mM NaCl buffer at 277 K. The N-terminal His tag was removed from RD-CrgA by overnight incubation of the protein with His-tagged 3C protease (the vector was kindly donated by A. Geerlof, EMBL, Hamburg) at 277 K. The 3C protease and any uncleaved protein were removed by passing the sample through a 1 ml HisTrap FF column. The purified proteins were analysed by SDS–PAGE (NuPAGE, Invitrogen) and electrospray mass spectrometry (Nettleship et al., 2008 ▶). RD-CrgA and FL-CrgA had a high level of purity (>98%) and mass spectrometry confirmed full selenomethionine incorporation of the selenomethionine derivative of RD-CrgA.
2.3. Crystallization of RD-CrgA
The selenomethionine derivative of RD-CrgA was concentrated to 3.9 mg ml−1 in 20 mM Tris pH 7.5, 200 mM NaCl, 1 mM TCEP with a Vivaspin 15 concentrator (Vivascience). Crystallization screening experiments were performed at 293 K in nanolitre sitting drops (100 nl protein and 100 nl reservoir) dispensed from a Cartesian robot onto a 96-well Greiner plate as described by Walter et al. (2003 ▶). RD-CrgA crystallized readily in 61 (12.7%) of the 480 screening conditions tested. The crystals used for data collection were optimized from 200 mM ammonium acetate, 25%(w/v) polyethylene glycol 3350, 0.1 M bis-Tris pH 5.5 with protein that had been flash-cooled, stored at 193 K and then thawed, using a drop ratio of 200 nl protein:100 nl reservoir in conjunction with an Additive Screen (Hampton Research) optimization protocol as reported by Walter et al. (2005 ▶).
2.4. Thermal shift assay and crystallization of FL-CrgA
In contrast to RD-CrgA, FL-CrgA concentrated in 20 mM Tris pH 7.5, 200 mM NaCl precipitated out of solution at levels above 0.5 mg ml−1 and consequently significant amounts of protein were lost. Therefore, a thermal shift assay, which monitors the thermal denaturation of a protein, was carried out on FL-CrgA to screen for a buffer that would stabilize FL-CrgA. The relationship between protein thermal stability and solubility is not straightforward, although in general a more stable protein may be less likely to aggregate (Ericsson et al., 2006 ▶). The buffer screen was composed of solutions containing 50 mM buffer ranging from pH 4.0 to 9.0 (sodium acetate pH 4 and pH 4.4, citrate pH 5 and pH 5.4, cacodylate pH 6.0 and pH 6.4, HEPES pH 7 and pH 7.4, Tris pH 8.0 and pH 8.4 and CAPSO pH 9) and 0–500 mM NaCl. 50 µl reactions containing 5 µg FL-CrgA, 1× Sypro Orange dye (Molecular Probes) and the buffer screen were added to white low-profile thin-wall PCR plates (Abgene). After sealing with a Microseal ‘B’ Film (BioRad), the plate was centrifuged at 3000g for 2 min. A BioRad DNA Engine Opticon 2 real-time PCR machine with SYBR Green settings was used to monitor the fluorescence. The fluorescence was read every 0.5 K (after 10 s hold) over a temperature range of 293–368 K. The apparent melting temperature (T m) of the protein was calculated using the Opticon Monitor software from the maximum value of the first derivative curve of the melting curve (Nettleship et al., 2008 ▶).
FL-CrgA was significantly more thermally stable in buffers of mid to lower pH. Varying the concentration of NaCl appeared to be less critical but increasing concentrations were associated with a small stabilization effect. In citrate buffer pH 5.0 containing 200 mM NaCl the apparent T m of CrgA increased by around 8 K compared with the original buffer of pH ∼7.5 (Fig. 1 ▶). On the basis of these results, the protein was concentrated to 2.4 mg ml−1 in 20 mM citrate pH 5.0, 200 mM NaCl and crystallization trials were set up. Although the solubility of CrgA appeared to have improved in this buffer, losses from precipitation still occurred and significantly no diffracting crystals grew from these initial trials.
Figure 1.

Buffer screening of FL-CrgA with a fluorescence-based thermal shift assay. The melting point (T m) of FL-CrgA (y axis) was calculated in buffers of pH 4–9 with 0, 100, 200 or 500 mM NaCl.
Nondetergent sulfobetaines (NDSBs) have been reported to prevent aggregation and to increase the solubility of proteins (Vuillard et al., 1995 ▶). Therefore, 0.2 M NDSB-256 was added to FL-CrgA to help prevent precipitation during concentration. Under these conditions, it proved possible to concentrate the protein to 8.2 mg ml−1 with minimal losses. Crystallization trials were set up and FL-CrgA crystallized in 9 (2.3%) of the 384 screening conditions tested. Crystals from two crystal forms A and B were optimized. Initial crystals of form A, grown from the original condition of 10%(v/v) 2-methyl-2,4-pentanediol in 0.1 M sodium acetate pH 5.0 (Grid Screen MPD, Hampton Research), were small and multiple (Fig. 2 ▶ a). Improved crystals were obtained by streak-seeding; seeds were transferred with a cat’s whisker into 2 µl [1 µl protein + 1 µl reservoir containing 10%(v/v) 2-methyl-2–4-pentanediol (MPD) in 0.1 M sodium acetate pH 5.0] sitting drops pre-equilibrated for 30 min in a Linbro 24-well tissue-culture plate (Fig. 2 ▶ b). Preliminary crystals of form B grown from 0.5 M ammonium sulfate, 1 M lithium sulfate, 0.1 M trisodium citrate pH 5.6 had a cubic morphology but were visibly layered (Fig. 2 ▶ c). After streak-seeding the crystals were single and grew with an elongated pyramid shape (Fig. 2 ▶ d). Seeds were transferred as described above into either 200 nl (100 nl protein + 100 nl reservoir in a Greiner plate) or 2 µl (1 µl protein + 1 µl reservoir in a Linbro plate) sitting drops. The reservoir contained 80% of the original condition above and the drops were pre-equilibrated for 30 min.
Figure 2.

FL-CrgA crystals before and after optimization by streak-seeding. (a) Initial crystals of crystal form A grown in 10%(v/v) 2-methyl-2,4-pentanediol, 0.1 M sodium acetate pH 5 and (b) crystal form A after optimization by streak-seeding. (c) Initial crystals of crystal form B grown in 0.5 M ammonium sulfate, 1 M lithium sulfate, 0.1 M trisodium citrate pH 5.6 and (d) crystal form B after optimization by streak-seeding.
2.5. Data collection and processing
Multiple-wavelength anomalous dispersion data were collected from crystals of the selenomethionine derivative of RD-CrgA on beamline BM14 at the ESRF (Grenoble, France). The crystals had a thin plate-like morphology and despite efforts to optimize them remained comparatively small; the crystals used in the data collection had overall dimensions of approximately 70 × 10 × 5 µm. However, they diffracted strongly and data sets at three different wavelengths were collected from a single crystal to 2.8 Å resolution. A higher resolution data set to 2.3 Å was collected from a second crystal at a single wavelength (Table 1 ▶). The crystals were flash-cooled in a cryosolution consisting of 25%(v/v) glycerol and 75%(v/v) reservoir solution [200 mM ammonium acetate, 25%(w/v) polyethylene glycol 3350, 0.1 M bis-Tris pH 5.5] and maintained at 100 K under a stream of nitrogen gas during data collection.
Table 1. Data-collection and processing statistics for SeMet RD-CrgA.
Values in parentheses are for the last resolution shell.
| Crystal I | Crystal II | |||
|---|---|---|---|---|
| Peak | Remote | Inflection | Peak | |
| X-ray source | BM14 | |||
| Detector | MAR 225 CCD | |||
| Space group | P21 | P21 | ||
| Unit-cell parameters | ||||
| a (Å) | 51.59 | 51.64 | ||
| b (Å) | 65.49 | 65.58 | ||
| c (Å) | 63.31 | 63.50 | ||
| β (°) | 105.27 | 105.02 | ||
| Wavelength (Å) | 0.9780 | 0.9065 | 0.9785 | 0.9795 |
| Resolution range (Å) | 50.0–2.76 (2.86–2.76) | 50.0–2.29 (2.37–2.29) | ||
| Unique reflections | 10553 (1058) | 8620 (235) | 8096 (167) | 15951 (636) |
| Completeness (%) | 99.9 (99.5) | 80.8 (22.1)† | 75.9 (15.7)‡ | 85.8 (34.8)§ |
| Redundancy | 7.4 (6.6) | 4.0 (1.5) | 3.5 (1.3) | 4.1 (1.8) |
| Average I/σ(I) | 20.1 (4.2) | 14.1 (3.3) | 13.2 (3.8) | 18.0 (2.1) |
| Rmerge¶ | 0.108 (0.422) | 0.097 (0.286) | 0.097 (0.214) | 0.073 (0.299) |
| d′′/σ†† | 2.48 (1.27) | 1.38 (0.87) | 1.38 (0.91) | |
Data were processed into the corner of the detector and are essentially complete to 3.5 Å.
Data were processed into the corner of the detector and are essentially complete to 3.8 Å.
Data were processed into the corner of the detector and are essentially complete to 2.7 Å.
R
merge = 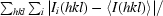
 .
.
As calculated by SHELXC, with data in the resolution ranges 50–8.0 and 5–4.2 Å, respectively.
Diffraction data for native FL-CrgA were collected on beamline ID-14.4, ESRF. Crystals of form A were transferred directly from the drop into a cryosolution consisting of 0.1 M sodium acetate pH 5.0, 25%(v/v) MPD, flash-cooled in liquid nitrogen, transported to the beamline and mounted via the automatic sample changer. A data set was collected from the largest crystal of approximate dimensions 300 × 100 × 30 µm (Fig. 2 ▶ b) to 3.0 Å resolution with 0.5° oscillations. Although smaller crystals froze well, the freezing of this crystal was unsatisfactory. Therefore, the data were only 83.1% complete owing to a lack of usable reflections in the resolution range 3.56–3.78 Å from the ice ring. For crystal form B, prior to the drop being fully opened to the air, perfluoropolyether PFO-X125/03 oil (Lancaster Synthesis) was added directly to the top of the drop through a small hole in the film covering the Greiner plate. A crystal was then transferred out of the drop through the layer of oil into a cold stream (100 K) of nitrogen gas. A data set was collected to 3.8 Å resolution. Data were processed with DENZO and SCALEPACK from the HKL-2000 suite (Otwinowski & Minor, 1997 ▶). The data-collection and processing statistics are presented in Table 2 ▶.
Table 2. Data-collection and processing statistics for native FL-CrgA.
Values in parentheses are for the last resolution shell.
| Crystal form A | Crystal form B | |
|---|---|---|
| X-ray source | ID14-4 | |
| Space group | P212121 | P21 |
| Detector | Q315r ADSC CCD | |
| Unit-cell parameters | ||
| a (Å) | 104.64 | 123.17 |
| b (Å) | 119.09 | 204.99 |
| c (Å) | 250.20 | 124.80 |
| β (°) | 93.10 | |
| Wavelength (Å) | 0.9765 | 0.9765 |
| Resolution range (Å) | 30.0–3.00 (3.11–3.00) | 30.0–3.80 (3.94–3.80) |
| Unique reflections | 52736 (5140) | 60401 (5853) |
| Completeness (%) | 83.1† (82.3) | 99.6 (97.2) |
| Redundancy | 3.5 (2.7) | 3.7 (3.3) |
| Average I/σ(I) | 14.1 (1.11) | 10.4 (4.10) |
| Rmerge‡ | 0.066 (0.774) | 0.138 (0.365) |
The 3.56–3.78 Å resolution shell is 23.7% complete.
R
merge =
.
2.6. Preliminary crystallographic analysis of the CrgA data sets
The RD-CrgA crystals belong to space group P21. Based on Matthews coefficient calculations, it is most likely that there are two molecules in the asymmetric unit, with a Matthews coefficient of 2.25 Å3 Da−1 and a solvent content of 45%. The anomalous signal from the selenomethionines was monitored using the SHELX suite and although relatively weak, possibly as a result of the presence of only two methionines in the sequence separated by a single amino acid, extended to around 4 Å (Table 1 ▶). FL-CrgA crystal form A belongs to the primitive orthorhombic space group P212121. The asymmetric unit is likely to contain between six and ten copies of FL-CrgA, resulting in a solvent content of between 68 and 47%. A native Patterson suggests the presence of a pseudo-translation with a peak at fractional coordinates (0.5, 0.5, 0.1). Crystals of form B were monoclinic (P21). In this case between ten and 20 molecules are expected to be present in the asymmetric unit, corresponding to a solvent content of 73–47%; no pseudo-translation was detected.
3. Summary
Obtaining crystal structures of full-length LTTRs has proved more challenging than for their regulatory domains and our experience with CrgA was consistent with this. Thus, purified full-length CrgA was less soluble and more difficult to crystallize than the C-terminal regulatory domain, which accounts for two-thirds of the molecule. The reasons for the poor solubility of full-length LTTRs is not clear but may relate to the extended linker helix region which joins the DNA-binding and regulatory domains. Diffracting crystals of the full-length protein were produced after buffer optimization using a thermal shift assay and concentration in 0.2 M NDSB-256, emphasizing the potential benefits of stabilizing ‘problematic’ proteins prior to crystallization. The protocol employed in this project may be generally applicable to other full-length LTTRs, aiding the concentration and crystallization of this class of proteins. Structure determination of RD-CrgA using MAD techniques is ongoing and if successful would provide a suitable molecular-replacement model for the subsequent determination of the full-length structure from the native data that have been collected.
Acknowledgments
The Oxford Protein Production Facility is funded by the Medical Research Council with additional finance from the Biotechnology and Biological Science Research Council. We thank the staff at BM14 and ID14-4 at the ESRF for help with data collection. SS was supported by an MRC studentship.
References
- Berrow, N. S., Alderton, D., Sainsbury, S., Nettleship, J., Assenberg, R., Rahman, N., Stuart, D. I. & Owens, R. J. (2007). Nucleic Acids Res.35, e45. [DOI] [PMC free article] [PubMed]
- Choi, H., Kim, S., Mukhopadhyay, P., Cho, S., Woo, J., Storz, G. & Ryu, S. (2001). Cell, 105, 103–113. [DOI] [PubMed]
- Deghmane, A. E., Giorgini, D., Larribe, M., Alonso, J. M. & Taha, M. K. (2002). Mol. Microbiol.43, 1555–1564. [DOI] [PubMed]
- Deghmane, A. E., Petit, S., Topilko, A., Pereira, Y., Giorgini, D., Larribe, M. & Taha, M. K. (2000). EMBO J.19, 1068–1078. [DOI] [PMC free article] [PubMed]
- Deghmane, A. E. & Taha, M. K. (2003). Mol. Microbiol.47, 135–143. [DOI] [PubMed]
- Ericsson, U. B., Hallberg, B. M., DeTitta, G. T., Dekker, N. & Nordlund, P. (2006). Anal. Biochem.357, 289–298. [DOI] [PubMed]
- Ezezika, O. C., Haddad, S., Clark, T. J., Neidle, E. L. & Momany, C. (2007). J. Mol. Biol.367, 616–629. [DOI] [PubMed]
- Ezezika, O. C., Haddad, S., Neidle, E. L. & Momany, C. (2007). Acta Cryst. F63, 361–368. [DOI] [PMC free article] [PubMed]
- Henikoff, S., Haughn, G. W., Calvo, J. M. & Wallace, J. C. (1988). Proc. Natl Acad. Sci. USA, 85, 6602–6606. [DOI] [PMC free article] [PubMed]
- Ieva, R., Alaimo, C., Delany, I., Spohn, G., Rappuoli, R. & Scarlato, V. (2005). J. Bacteriol.187, 3421–3430. [DOI] [PMC free article] [PubMed]
- Lochowska, A., Iwanicka-Nowicka, R., Plochocka, D. & Hryniewicz, M. M. (2001). J. Biol. Chem.276, 2098–2107. [DOI] [PubMed]
- Merz, A. J. & So, M. (2000). Annu. Rev. Cell Dev. Biol.16, 423–457. [DOI] [PubMed]
- Muraoka, S., Okumura, R., Ogawa, N., Nonaka, T., Miyashita, K. & Senda, T. (2003). J. Mol. Biol.328, 555–566. [DOI] [PubMed]
- Nettleship, J. E., Brown, J., Groves, M. R. & Geerlof, A. (2008). Methods Mol. Biol.426, 299–318. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pareja, E., Pareja-Tobes, P., Manrique, M., Pareja-Tobes, E., Bonal, J. & Tobes, R. (2006). BMC Microbiol.6, 29. [DOI] [PMC free article] [PubMed]
- Rosario, C. J. & Bender, R. A. (2005). J. Bacteriol.187, 8291–8299. [DOI] [PMC free article] [PubMed]
- Rosenstein, N. E., Perkins, B. A., Stephens, D. S., Popovic, T. & Hughes, J. M. (2001). N. Engl. J. Med.344, 1378–1388. [DOI] [PubMed]
- Schell, M. A. (1993). Annu. Rev. Microbiol.47, 597–626. [DOI] [PubMed]
- Smirnova, I. A., Dian, C., Leonard, G. A., McSweeney, S., Birse, D. & Brzezinski, P. (2004). J. Mol. Biol.340, 405–418. [DOI] [PubMed]
- Stec, E., Witkowska-Zimny, M., Hryniewicz, M. M., Neumann, P., Wilkinson, A. J., Brzozowski, A. M., Verma, C. S., Zaim, J., Wysocki, S. & Bujacz, G. D. (2006). J. Mol. Biol.364, 309–322. [DOI] [PubMed]
- Stephens, D. S. (2007). FEMS Microbiol. Rev.31, 3–14. [DOI] [PubMed]
- Tyrrell, R., Verschueren, K. H., Dodson, E. J., Murshudov, G. N., Addy, C. & Wilkinson, A. J. (1997). Structure, 5, 1017–1032. [DOI] [PubMed]
- Vuillard, L., Braun-Breton, C. & Rabilloud, T. (1995). Biochem. J.305, 337–343. [DOI] [PMC free article] [PubMed]
- Walter, T. S., Diprose, J., Brown, J., Pickford, M., Owens, R. J., Stuart, D. I. & Harlos, K. (2003). J. Appl. Cryst.36, 308–314.
- Walter, T. S. et al. (2005). Acta Cryst. D61, 651–657. [DOI] [PMC free article] [PubMed]