

The transition-metal mediated intramolecular carboamination of alkenes is a direct method for complex nitrogen heterocycle synthesis. A number of research groups have investigated this transformation in recent years.1 The catalytic asymmetric carboamination of alkenes is an obvious challenge for these reactions, and has been realized rarely.1d Herein is described a novel copper(II)-catalyzed asymmetric carboamination reaction that involves intramolecular addition of arylsulfonamides across terminal alkenes to provide chiral sultams. Sultams and sulfonamides are common components of biologically active small molecules.2
The copper-facilitated synthesis of heterocycles via addition of heteroatoms to alkenes and alkynes is an important area in organic synthesis.3 We have recently reported the first copper(II)-facilitated intramolecular carboamination of alkenes, a net oxidative cyclization process (Scheme 1).1c,f In depth mechanistic studies led us to conclude that the stereochemistry determining C-N bond forming step occurred via syn aminocupration of the alkene (cf. 3 → 5 via 4, Scheme 1).1f If the copper salt is involved in the stereochemistry-determining step, chiral ligands on copper could allow for a stereocontrolled synthesis of the product. In the proposed C-N bond-forming transition state 4, the substrate occupies two coordination sites on a tetracoordinate copper(II),4 leaving two coordination sites available for a bidentate ligand (Scheme 1). Inspired by this analysis, we initiated a search for an appropriate oxidant for copper turnover and ligand for asymmetric induction. Herein is reported the first catalytic as well as catalytic asymmetric variant of this copper(II)-facilitated carboamination reaction.
Scheme 1.
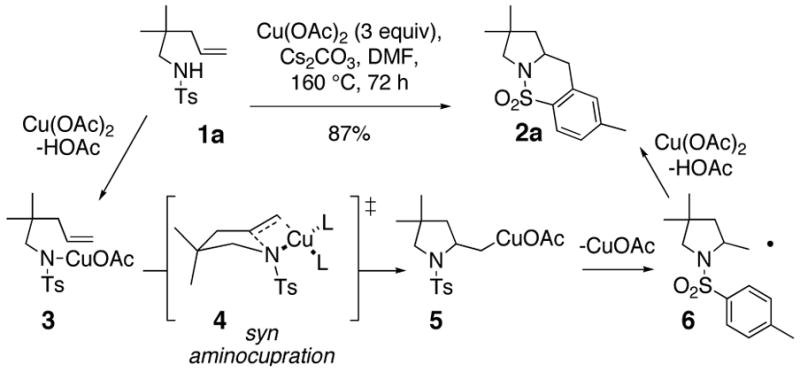
Copper(II)-Promoted Carboamination and Mechanism
In the search for conditions catalytic in copper, we screened a number of oxidants [O2, PhI(OAc)2, oxone, Me3NO, MnO2],with and without ligands in different solvents for the conversion of 1a to 2a with a catalytic amount (0.2 equiv) of copper(II) ethylhexanoate. The highest conversions were obtained with MnO2 (3 equiv) as oxidant in trifluorotoluene in the presence of ligands (Table 1). Under the optimized conditions but in the absence of copper(II), no reaction occurs. In toluene, a mixture of carboamination and hydroamination products 2a and 7 was observed (Table 1, entries 3 and 4). We hypothesized the hydroamination product 7 is formed via carbon radical (e.g. 6) capture of a hydrogen atom from solvent. Gratifyingly, changing the solvent to trifluorotoluene substantially decreased formation of the hydroamination adduct (Table 1, entries 5, 6 and 8). The carboamination reactions stoichiometric in copper(II) (cf. Scheme 1) start out blue [due to copper(II)] and terminate as orange-brown heterogeneous mixtures, indicating formation of copper(0), possibly from disproportionation of copper(I) to copper(II) and copper(0). Reoxidation of copper(0) under the mildly basic reaction conditions used in these reactions is challenging. We hypothesized that ligands would stabilize copper(I) and copper(II) in preference to copper(0).3 Addition of diethylsalicylamide 8 (0.2 and 0.8 equiv) increased the reaction yields, likely due to improved copper turnover (Table 1, compare entries 1 and 2 with 3–6).5 We changed the copper(II) source to Cu(OTf)2 to achieve better ligand chelation (entries 6–8). The bipyridine ligand 9 (0.2 equiv) gave more efficient conversion when Cu(OTf)2 was used (compare entry 6 to 8).
Table 1.
Dependence on Solvent and Liganda
 | |||||
|---|---|---|---|---|---|
| entry | R | solvent | ligand (equiv) | yield (%) 2 | yield (%) 7 |
| 1 | EH | PhCH3 | -- | <5 | trace |
| 2 | EH | PhCF3 | -- | 7 | trace |
| 3 | EH | PhCH3 | 8 (0.2) | 41 | 7 |
| 4 | EH | PhCH3 | 8 (0.8) | 63 | 17 |
| 5 | EH | PhCF3 | 8 (0.8) | 75 | <5 |
| 6 | OTf | PhCF3 | 8 (0.8) | 29 | <5 |
| 7 | OTf | PhCH3 | 9 (0.2) | 59 | 12 |
| 8 | OTf | PhCF3 | 9 (0.2) | 60 | <5 |
Reaction conditions: Substrate 1a was dissolved in solvent (0.1 M) and treated with K2CO3 (1 equiv), MnO2 (3 equiv), CuR2 (0.2 equiv) and the specified amount of ligand and stirred in a sealed tube at 120 °C for 24 h.
Yields refer to amount of compound isolated after chromatography on SiO2. Remainder of material was always unreacted starting 1a. EH = 2-ethylhexanoate.
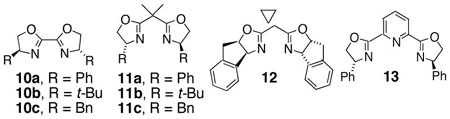
We screened chiral ligands 10–13 by precomplexing Cu(OTf)2 (0.2 equiv) with ligand (0.2 equiv) followed by addition of substrate 1a, K2CO3 (1 equiv) and MnO2 (3 equiv) and heating in a sealed tube for 24 h in PhCF3 (Table 2).
Table 2.
Chiral Ligand Screeninga
 | |||||
|---|---|---|---|---|---|
| entry | equiv Cu(OTf)2 | ligand (equiv) | yield (%) | %eec | ERc |
| 1 | 0.2 | (R,R)-10a (0.2) | 54 | 24 | 62:38 |
| 2 | 0.2 | (S,S)-10b (0.2) | 53 | 14 | 43:57 |
| 3 | 0.2 | (S,S)-10c (0.2) | 73 | 28 | 36:64 |
| 4 | 0.2 | (S,S)-11a (0.2) | 75 | 86 | 7:93 |
| 5 | 0.2 | (R,R)-11a (0.2) | 85 | 92 | 96:4 |
| 6 | 0.2 | (S,S)-11b (0.2) | 50 | 24 | 38:62 |
| 7 | 0.2 | (R,R)-11c (0.2) | 55 | 82 | 91:9 |
| 8 | 0.2 | (R,S)-12 (0.2) | <5 | -- | -- |
| 9 | 0.2 | (R,R)-13 (0.2) | 18 | 4 | 48:52 |
| 10 | 0.15 | (R,R)-11a (0.15) | 64 | 92 | 96:4 |
| 11 | 0.05 | (R,R)-11a (0.05) | 34 | -- | -- |
| 12d | 0.2 | (R,R)-11a (0.2) | 72 | 94 | 97:3 |
Reaction conditions: Cu(OTf)2 and ligand were combined, dissolved in PhCF3 (0.1 M w/r to 1a) and heated at 50 °C for 1 h. Substrate 1a, MnO2 (3 equiv) and K2CO3 (1 equiv) were added and the reaction tube sealed and heated at 120 °C for 24 h unless otherwise noted.
Yield refers to amount of isolated 2a after purification by flash chromatography on SiO2.
Enantiomeric excess and ratios were determined by chiral HPLC analysis (chiralcel OD-H).
Reaction was run at 110 °C.
We quickly found that 2,2-bis[(4R)-4-phenyl-2-oxazolin-2-yl]propane (11a) gave the highest asymmetric induction, providing carboamination adduct 2a in 85% isolated yield, 92% enantiomeric excess (Table 2, entry 5). We were unable to reduce the catalyst loading below 0.2 equiv without adversely affecting the product yield (Table 2, entries 10 and 11). Lowering the reaction temperature to 110 °C provided the product in slightly lower yield (72%) and 94% enantiomeric excess (Table 2, entry 12). Ligands 12 and 13 rendered the copper(II) complex less reactive (entries 8 and 9).
The generality of the reaction was examined as shown in Table 3. γ-Alkenyl arylsulfonamides 1 cyclized in 45–85% yield and 80–94% ee.6 The 2-sulfamido thiophene substrate 1 4 reacted very sluggishly but with good enantioselectivity. N-Tosyl-2-allylaniline 16 reacted efficiently but with low (46%) enantioselectivity and N-tosyl-2-allylbenzylamine 18 reacted sluggishly and in moderate yield and enantioselectivity to provide tetrahydroisoquinoline 19 (entry 12).
Table 3.
Scope of Enantioselective Carboamination with (R,R)-11aa
| entry | substrate | product | yield (%)b | ee (%)c | ERc |
|---|---|---|---|---|---|
|
|
||||
| 1 | 1a, R1 = Me, R2 = Me | 2a | 85 | 92 | 96:4 |
| 2 | 1b, R1= Me, R2 = H | 2b | 73 | 92 | 96:4 |
| 3 | 1c, R1= Me, R2 = Cl | 2c | 45 | 92 | 96:4 |
| 4 | 1d, R1= Me, R2 = OMe | 2d | 75 | 94 | 97:3 |
| 5 | 1e, R1= Ph, R2 = Me | 2e | 78 | 94 | 97:3 |
| 6 | 1f, R1 = -CH2(CH2)2CH2-, R2= Me | 2f | 83 | 92 | 96:4 |
| 7 | 1g, R1= -CH2(CH2)3CH2-, R2 = Me | 2g | 68 | 92 | 96:4 |
| 8 | 1h, R1= H, R2 = Me | 2h | 68 | 80 | 90:10 |
| 9 | 1i, R1 = H, R2= H | 2i | 77 | 82 | 91:9 |
| 10d |
14 |
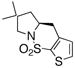
15 |
30 | 86 | 93:7 |
| 11 |
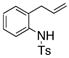
16 |
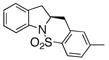
17 |
75 | 46 | 73:27 |
| 12d |
18 |
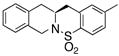
19 |
63 | 72 | 86:14 |
Reaction conditions: Cu(OTf)2 (0.2 equiv) and (R,R)-11a (0.2 equiv) were combined and treated with PhCF3 (0.1 M w/r to substrate) and heated at 50 °C for 1h. Substrate (1 equiv), MnO2 (3 equiv) and K2CO3 (1 equiv) were added and the reaction tube was sealed and heated at 120 °C for 24 h unless otherwise noted. All reactions were run at least two times to ensure reproducibility.
Yields refer to amount isolated after purification by flash chromatography on SiO2.
Enantiomeric excess and ratios were determined by chiral HPLC anlaysis (chiralcel OD-H or AD-RH).
Reactions were run for 96 h.
X-Ray crystal structures of sultams 2g and 15 indicate the (S)-configuration. The other carboamination adducts in Table 3 are assigned the S stereochemistry by analogy. Sultam 2i was converted to the known 2(S)-benzylpyrrolidine 20,7 an intermediate used in the synthesis of a potent calcium-sensing receptor antagonist,8 by reductive removal of SO2 (Scheme 2). Pyrrolidine (S)-20 is thus available by this method in three steps from commercially available starting materials (see Supporting Information for complete details).
Scheme 2.
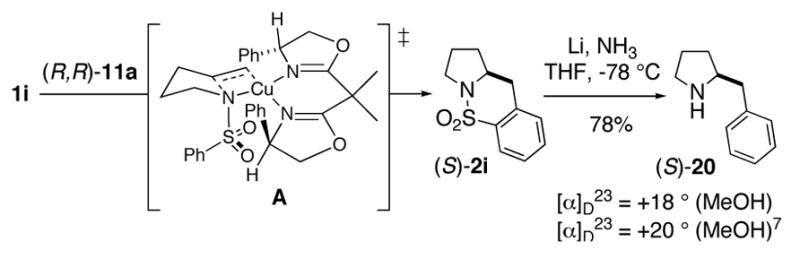
Transition State Model and Removal of SO2
The observed stereochemistry is consistant with transition state A (Scheme 2), where the substrate’s N-substituent is on the face opposite of the oxazoline phenyl substituent it is cis to about the distorted square planar copper center.4
The method reported herein provides access to enantiomerically enriched nitrogen heterocycles. Applications of this reaction toward the synthesis of bioactive compounds are underway. This method will also inspire the development of other copper(II)-catalyzed enantioselective amination reactions.
Supplementary Material
Experimental procedures, compound characterization data, X-ray structures with determination of absolute configuration and HPLC traces with determination of enantiomeric excess. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The financial support of the NIH/NIGMS RO1 GM078383 is gratefully acknowledged. Dr. Shao-Liang Zheng (SUNY, Buffalo X-ray crystallography facility) is gratefully acknowledged for obtaining crystal structures of 2g and 15.
References
- 1.(a) Larock RC, Yang H, Weinreb SM, Herr RJ. J Org Chem. 1994;59:4172. [Google Scholar]; (b) Harayama H, Abe A, Sakado T, Kimura M, Fugami K, Tanaka S, Tamaru Y. J Org Chem. 1997;62:2113. doi: 10.1021/jo961988b. [DOI] [PubMed] [Google Scholar]; (c) Sherman ES, Chemler SR, Tan TB, Gerlits O. Org Lett. 2004;6:1573. doi: 10.1021/ol049702+. [DOI] [PubMed] [Google Scholar]; (d) Yip KT, Yang M, Law KL, Zhu NY, Yang D. J Am Chem Soc. 2006;128:3130. doi: 10.1021/ja060291x. [DOI] [PubMed] [Google Scholar]; (e) Scarborough CC, Stahl SS. Org Lett. 2006;8:3251. doi: 10.1021/ol061057e. [DOI] [PubMed] [Google Scholar]; (f) Sherman ES, Fuller PH, Kasi D, Chemler SR. J Org Chem. 2007;72:3896. doi: 10.1021/jo070321u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Peng J, Lin W, Yuan S, Chen Y. J Org Chem. 2007;72:3145. doi: 10.1021/jo0625958. [DOI] [PubMed] [Google Scholar]; (h) Bertrand MB, Leathen ML, Wolfe JP. Org Lett. 2007;9:457. doi: 10.1021/ol062808f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Nakhla JS, Wolfe JP. Org Lett. 2007;9:3279. doi: 10.1021/ol071241f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Wolfe JP. Eur J Org Chem. 2007:571. and references therein. [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Sammes PG. Chap. 7.1. In: Hansch C, Sammes PG, Taylor JB, editors. Comprehensive Medicinal Chemistry. Vol. 2 Pergamon Press; Oxford: 1990. [Google Scholar]; (b) Wilkening RR, Ratcliffe RW, Wildonger KJ, Cama LD, Dykstra KD, DiNinno FP, Blizzard TA, Hammond ML, Heck JV, Dorso KL, Rose ESt, Kohler J, Hammond GG. Bioorg Med Chem Lett. 1999;9:673. doi: 10.1016/s0960-894x(99)00070-0. [DOI] [PubMed] [Google Scholar]
- 3.Chemler SR, Fuller PH. Chem Soc Rev. 2007;36:1153. doi: 10.1039/b607819m. and references therein. [DOI] [PubMed] [Google Scholar]
- 4.Johnson JS, Evans DA. Acc Chem Res. 2000;33:325. doi: 10.1021/ar960062n. [DOI] [PubMed] [Google Scholar]
- 5.Kwong FY, Buchwald SL. Org Lett. 2003;5:793. doi: 10.1021/ol0273396. [DOI] [PubMed] [Google Scholar]
- 6.A homologue of 1a, one carbon longer in the chain, did not react.
- 7.Burgess LE, Meyers AI. J Org Chem. 1992;57:1656. [Google Scholar]
- 8.Yang W, Wang Y, Roberge JY, Ma Z, Liu Y, Lawrence RM, Rotella DP, Seethala R, Feyen JH, Dickson JK. Bioorg Med Chem Lett. 2005;15:1225. doi: 10.1016/j.bmcl.2004.11.071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures, compound characterization data, X-ray structures with determination of absolute configuration and HPLC traces with determination of enantiomeric excess. This material is available free of charge via the Internet at http://pubs.acs.org.