

Abstract
Kynureninase is a member of a large family of catalytically diverse but structurally homologous pyridoxal-5′-phosphate dependent enzymes known as the aspartate aminotransferase superfamily or α-family. The Homo sapiens and other eukaryotic constitutive kynureninases preferentially catalyze the hydrolytic cleavage of 3-hydroxy-L-kynurenine to produce 3-hydroxyanthranilate and L-alanine, while L-kynurenine is the substrate of many prokaryotic inducible kynureninases. The human enzyme was cloned with an N-terminal hexahistidine tag, expressed, and purified from a bacterial expression system using Ni-metal ion affinity chromatography. Kinetic characterization of the recombinant enzyme reveals classic Michaelis-Menten behavior, with a Km= 28.3 ± 1.9 μM, and a specific activity of 1.75 μmol min-1 mg-1 for 3-hydroxy-DL-kynurenine. Crystals of recombinant kynureninase were obtained that diffracted to 2.0 Å, and the atomic structure of the PLP-bound holoenzyme was solved by molecular replacement using the Pseudomonas fluorescens kynureninase structure (PDB accession 1qz9) as the phasing model. A structural superposition with the P. fluorescens kynureninase revealed that these two structures resemble the “open” and “closed” conformations of aspartate aminotransferase. The comparison illustrates the dynamic nature of these proteins’ small domains and reveals a role for Arg-434 similar to that in other AAT α-family members. Docking of 3-hydroxy-L-kynurenine into the human kynureninase active site suggests that Asn-333 and His-102 are involved in substrate binding and molecular discrimination between inducible and constitutive kynureninase substrates.
Mammalian kynureninase [E.C. 3.7.1.3] is a pyridoxal-5′-phosphate (PLP) dependent constitutive (1) enzyme that catalyzes the hydrolytic cleavage of 3-hydroxy-L-kynurenine to 3-hydroxyanthranilic acid and L-alanine (Eq. 1).
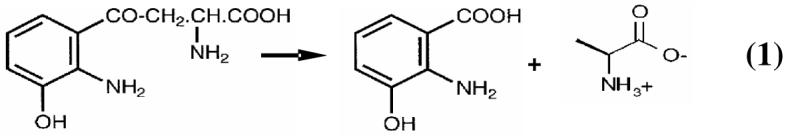
In contrast, prokaryotic inducible kynureninases preferentially hydrolyze L-kynurenine to yield anthranilic acid and L-alanine. Mammalian kynureninase is a member of the catabolic cascade known as the “kynurenine pathway” (2) through which the majority of dietary tryptophan is hepatically degraded (3), and provides for the de novo biosynthesis of NAD+ in the absence of niacin (2, 4). In extrahepatic cells, kynurenine pathway genes are expressed in immune system cells such as macrophages and microglia, although the primary metabolite produced in these cells appears to be quinolinic acid and not NAD+ (5, 6). Quinolinic acid (QA) is a known agonist of NMDA sensitive glutaminergic ionotropic receptors (7) and is thus an excitotoxin. QA is a known necrotic agent of cultured neuron cells (8), and prolonged exposure causes excitotoxic damage at concentrations ten times below physiological levels (9). Excessive QA in CNS tissues resulting from the overstimulation of the kynurenine pathway is thought to contribute to the etiology of many neurodegenerative diseases including AIDS-related dementia complex, Alzheimer’s, stroke, epilepsy, and Huntington’s disease (10-18). Since kynureninase acts at a metabolic branch point that directs metabolites to either the production of quinolinate via kynureninase, or xanthurenic acid via 3-hydroxykynurenine transaminase (3), inhibitors of this enzyme could prevent the overproduction of quinolinate. Thus, inhibitors that specifically target constitutive kynureninases have potential therapeutic value. To this end, the availability of the three dimensional structure of human kynureninase is an important step for rational drug design.
Kynureninase belongs to the PLP-dependent aspartate aminotransferase superfamily fold (19, 20) (or α-family), subgroup IVa (21). This catalytically diverse group shares very low sequence identity but retains significant structural homology (22). Only a few amino acid residues are conserved among all members: 1) the PLP-enzyme Schiff base forming lysine; 2) an active site aspartate; and 3) a substrate binding arginine invariantly located on a small domain β-hairpin loop (19). In aspartate aminotransferase, this arginine plays an important role in substrate binding and catalysis by stabilizing and orienting the substrate within the active site. Also, the small domains of aspartate aminotransferases are known to undergo conformational changes upon ligand binding (23).
In this work, we report the first cloning, expression, purification, characterization, and crystallization of Homo sapiens kynureninase (Hkyn) using a bacterial expression system and the atomic structure to 2.0 Å resolution.
Experimental Procedures
Protein Expression and Purification
The Homo sapiens kynureninase was cloned into a pET100 plasmid using a TopoTA-Kit (Invitrogen) after PCR amplification from a placental cDNA clone obtained from the American Type Culture Collection (ATCC MGC-5080; Genbank accession number U57721.gb-pr3) to generate an N-terminal hexahistidine recombinant protein fusion. The resulting plasmid was sequenced to confirm identity with the Genbank cDNA sequence. The PCR oligonucleotide primers used for amplification were: 5′-GAGCCTTCATCTCTTGAGCTGCC-3′ (forward), and 5′-GTTGCCCTAGAAAACACTGCTAA-3′ (reverse). A single colony of Escherichia coli BL21 (DE3) cells transformed with the recombinant plasmid was inoculated into 1 L of ZYP-5052 medium (24) containing 100 mg L-1 ampicillin and grown at 37 °C for 20 hours with shaking at 300-325 rpm. Prior to harvesting, the cultures were pre-chilled for 1 hour at 4 °C and then collected by centrifugation at 2500 × g for 15 minutes at 4 °C. Cell pellets were resuspended in 30 mL of ice-cold buffer (0.3 M NaCl, 0.05 M KPi, pH 7.0, and 0.1 mM PLP) and sonicated for four cycles of 1 minute sonication intervals followed by 2 minutes on ice. Cell debris was removed by centrifugation for 20 minutes at 25,000 × g and 4 °C. The supernatant was allowed to stand at room temperature for 20 minutes followed by the drop-wise addition of four mL of a 2% protamine sulfate solution (in resuspension buffer), and the resultant cloudy solution was centrifuged for 40 minutes at 25,000 × g at 4 °C. The supernatant was applied to a Ni-CAM (Sigma) resin column pre-equilibrated with buffer A (0.3 M NaCl, 0.05 M KPi, pH 7.0, 0.1 mM PLP, 4 °C) at a rate of 0.5 mL min-1 at 4 °C. After a 1 hour wash with buffer A (1.5 mL min-1), kynureninase was eluted from the column with a 360 minute 0% to 100% buffer B (0.3 M NaCl, 0.05 M KPi, pH 7.0, 0.1 mM PLP, 150 mM imidazole, 4 °C) linear gradient (0.5 mL min-1). SDS-PAGE analysis of fractions eluting from the column was used to determine appropriate samples to be pooled and concentrated. Pooled fractions were dialyzed for 6 hours at 4 °C in a 3.5 L solution containing 0.1 M KPi, pH 7.0, 0.1 mM PLP, and then exchanged, via two tenfold dilutions, into 0.05 M HEPES, pH 5.2, and 0.2 mM PLP using Amicon Centriprep centrifugal filter units (YM-30 membrane). Protein concentration was determined using a Bradford (Bio-Rad) assay (25).
Kinetic Assay
Kynureninase activity was measured at 37 °C by following the absorbance change at λmax = 370 nm as 3-hydroxykynurenine (Δε = 4500 M-1 cm-1) is converted to 3-hydroxyanthranilate and L-alanine in 30 mM KPi, pH 7.0, and 40 μM PLP. Specific activity assays contained 70 μM 3-hydroxy-DL-kynurenine, 40 μM PLP, and 30 mM KPi, pH 7.0. Km measurements were performed in 5 μM steps from 0 μM to 100 μM 3-hydroxy-DL-kynurenine in quadruplicate assays.
Crystallization, Structure Determination, and Refinement
High throughput crystallization trials were conducted at the Hauptman-Woodward Institute (26). Screenings were performed with a 11 mg mL-1 kynureninase solution in several buffers: 1) 30 mM MOPS, pH 7, 0.2 mM PLP, 2) 30 mM KPi, pH 7.5, 0.2 mM PLP, 3) 50 mM HEPES, pH 5.2, 0.2 mM PLP, and 4) 30 mM MOPS, pH 7, 0.2 mM PLP, 10% Glycerol.
Kynureninase crystals were grown in-house at 23.5 °C using the microbatch under-oil method (27) by mixing 2 μL of a 9.2 mg mL-1 kynureninase solution in 50 mM HEPES, pH 5.2, 0.2 mM PLP with 2 μL of 0.05 M MgCl2, 0.1 M Tris-Cl, pH 8.0, 25% PEG 3000. Crystals were flash frozen in liquid nitrogen with cryoprotectant containing 0.055 M MgCl2, 0.11 M Tris-Cl, pH 8.0, 33% PEG 3000. X-ray synchrotron data were collected (detector distance 200 mm, 200 frames, 1° oscillations, 5 second exposure, λ = 0.97934 Å) at the Advanced Photon Source SBC-CAT beamline 19-BM in Argonne, IL (28). Data were processed, scaled, and merged using HKL2000 (29). The merged SCALEPACK (30) intensities were used as input for the MolRep (31) utility in the CCP4 crystallographic suite (32), and a suitable molecular replacement solution was identified using the Pseudomonas fluorescens (Pkyn) (PDB (33) accession code 1qz9 (21)) as a model (initial Rvalue = 55.7). All water molecules, hetero atoms, and pyridoxal-5′-phosphate coordinates were deleted from the molecular replacement model. The electron density for the PLP cofactor was used to evaluate the success of phasing methods. Phases of the highest scoring MolRep solution and the Hkyn sequence were used as input for the automated building package RESOLVE (34) with default set parameters and no input model. The highest scoring RESOLVE solution was refined with Refmac5 (35) in the CCP4 suite with ARP/wARP (36, 37) automated water addition (3 cutoff). Refined phases were subsequently used as input into the ARP/wARP automated free atom model building utility in CCP4. Two tandem RESOLVE-ARP/wARP cycles produced a kynureninase model covering approximately 83% of the predicted amino acid sequence. Manual building of the remainder of the model and PLP docking were carried out with the XtalView/Xfit (38) CCP4 utility, followed by restrained refinement in Refmac5. A final TLS refinement was used with the following residue segments: 6-52, 53-65, 66-101, 102-111, 112-134, 135-180, 181-200, 201-252, 253-267, 268-277, 278-285, 286-291, 292-306, 307-329, 330-375, 376-391, 392-413, 414-460. TLS groups were defined based on secondary structure, and were chosen to maximize the improvement in Rfree. MolProbity (39) and Procheck (40) were used to identify errors during the building/refinement process and evaluate the quality of the final structure.
Structural Superpositions and Ligand Docking
Structural superpositions were done using the Hkyn monomer as a reference model with STRAP (41) and the Iterative and Magic Fit utilities in the Swiss PDB viewer (SPDBV) (42). 3-Hydroxy-L-kynurenine was docked into the Hkyn dimer using the molecular docking algorithm Moldock (43) in the Molegro Virtual Docker. Active site cavities were identified using the Molegro Van der Waals molecular surface prediction algorithm with a grid resolution of 0.5 Å. Kynureninase and 3-hydroxy-L-kynurenine atoms were defined using the Molegro atom definition tool. The pyridoxal-5′-phosphate was defined as a cofactor, and the protonation and hybridization state of individual PLP atoms were defined to correspond to the internal aldimine dipolar ionic tautomer. All water molecules were deleted from the model and hydrogens were added using the Molegro protonation wizard. Arg-434 was modeled to resemble the position of Arg-375 in 1qz9 with XtalView/Xfit. Side chain atoms in Arg-434, Asn-333* (residues having * are associated with a second subunit of the dimeric biological unit), Ser-75, and the 3-hydroxy-L-kynurenine 2-amino and 3-hydroxyl group were defined as hydrogen bond donors. The 3-hydroxy-L-kynurenine α-carboxylate and γ-carbonyl groups were defined as hydrogen bond acceptors. A total of 50 docking runs with a population size of 200 were calculated over a 15 Å radius surrounding the predicted active site cavity, with a grid resolution of 0.2 Å, and a maximum of 5000 iterations per pose. Pre-positioned ligands were randomized in the predicted active site cavity prior to each docking run, and docking was constrained to the predicted active site cavity.
Results
Protein Purification and Kinetic Analysis
Although Homo sapiens kynureninase was expressed in E. coli cells from a pET vector under lac control, a pilot expression study revealed that the addition of IPTG or the use of Rosetta cells containing rare tRNAs did not increase the final yield. Also, a large pellet of insoluble protein, which has the same SDS-PAGE molecular weight as the soluble kynureninase fractions, results from each culture. No attempt was made to resolubilize the insoluble protein pellet. The final yield was only increased (by 2 mg per liter) when ZYP-5052 (24) was used as the growth media (as opposed to LB broth). The expression and purification from E. coli yields ∼ 8.4 mg of crystallization purity grade protein from a 1 L culture. The recombinant enzyme has a specific activity of 1.75 μmol min-1 mg-1, a kcat of 3.5 s-1, a Km for 3-hydroxy-DL-kynurenine of 28.3 ± 1.9 μM, and a kcat/ Km of 2.5 × 105 s-1 μM-1 for 3-hydroxy-L-kynurenine.
Crystallization
High-throughput screens of kynureninase in 50 mM HEPES, pH 5.2, and 0.2 mM PLP yielded two crystallization conditions that produced two distinct morphologies. Crystals grow under a range of pH values in HEPES buffers (5.2-7.5) but the best crystals grow with HEPES, pH 5.2. Exchanging the protein buffer from KPi, pH 7.0, to HEPES, pH 5.2, results in a 20%-30% loss of protein mass. Kynureninase is subsequently stable for several weeks at 4 °C in 50 mM HEPES, pH 5.2, 0.1 mM PLP. Crystallization solutions contained: A) 0.1 M tribasic KPi, 0.1 M Tris-Cl, pH 8.0, 40% PEG 4000, and B) 0.05 M MgCl2, 0.1 M Tris-Cl, pH 8.0, 25% PEG 3000. Crystals grown in solution A appeared after two days and were small (0.5 × 0.2 × 0.2 mm) pale yellow rhombohedra with Laue symmetry 4/m and cell dimensions a = b= 98.8, c = 231.1 Å, α=β=γ=90°. Crystals grown in solution B appeared after three weeks and were dark yellow and block shaped (0.17 × 0.1 × 0.17 mm) with Laue symmetry 2/m, had cell constants a = 74.1, b = 76.8, c = 93.2, β = 108.7 Å, and belonged to space group C2. Only data collected with a single crystal grown with crystallization solution B is presented henceforth.
Model Geometry
The final kynureninase model was refined to 2.0 Å with an RFree and R value of 0.195 and 0.150, respectively, (see Table 1 for model statistics) with 98% of the cDNA predicted amino acid residues (residues 6-460) fitted to the electron density. An eight residue segment between helix H16 and β-strand S17 (residues 379-386) could not be modeled due to poor electron density and is not included in the PDB file. Cysteine-45 had additional electron density protruding from the γ sulfur atom which was modeled as a cysteine sulfenic acid. The structure had good geometry with r.m.s. delta deviations from ideal bond lengths of 0.008 Å, and bond angles 1.135° with 97.3%, 2.5%, and 0.2% residues in the favorable, generously allowed, and disallowed regions of the Ramachandran plot, respectively. (Table 1). The conformation of the single residue in the disallowed region, Thr-353, appears to be forced into an unusual geometry by a strong hydrogen bond (2.50 Å) between its side chain γ oxygen and the Ala-352 carbonyl oxygen. Electron density is clearly visible between these two atoms. This residue is located on a three residue turn that connects the first (helix H15) and last (helix H16) helices of the large and small domains, respectively, and has excellent electron density at contour levels as high as 1.8 σ. The C2 crystallographic unit cell contained one monomer per asymmetric unit, with approximately 53% bulk solvent, and there are 535 water molecules in the final model.
Table 1.
Summary of Crystallographic analysis
| Space Group | C2 |
|---|---|
| Resolution range (outer shell), Å | 88.39-2.00 (2.08-2.01) |
| No of reflections (outer shell) | 31583 (2269) |
| Redundancy (outer shell) | 3.7 (3.3) |
| % of Reflections with I > 3σ (outer shell) | 82 (53.9) |
| Rsym (outer shell) | 0.055 (0.264) |
| Rfactor (outer shell) | 0.150 (0.186) |
| Rfree (outer shell) | 0.195 (0.255) |
| Mean B value | 21.97 Å2 |
| r.m.s delta. from ideal geometry: | |
| Bond angles | 1.135° |
| Bond distances | 0.008 Å |
| Ramachandran plot residues in favored/allowed/disallowed (%) regions | 97.3/2.5/0.2 |
Rfree calculated with 5.1% of total data that was excluded from refinement.
Overall Fold
The biological dimer (Figure 1) can be generated by applying the crystallographic symmetry operator 1-x, y,-z to the monomer. Monomers are related by a two-fold rotation axis located at the interface of the large domains. Evaluation of the crystal contacts between molecules revealed only a dimeric kynureninase form; no higher polymerization forms were identified. Each monomer (Figure 2) consists of a small domain (residues 9-74 and 354-462) and a large domain (residues 75-353), which have αβ-sandwich and Greek key αβα architecture, respectively. The large domain is composed of a seven stranded mixed β-pleated sheet of topology AGFEDBC (β-strands S5-S8, S10, and S11) surrounded by α-helices H8-H14. The small domain is composed of a four stranded anti-parallel β-pleated sheet (β-strands S17, S18, S20, and S21) of topology ABED surrounded by α-helices H17-H20. The organization of the secondary structure elements in the small and large domains is consistent with other PLP dependent α-family members.
Figure 1.
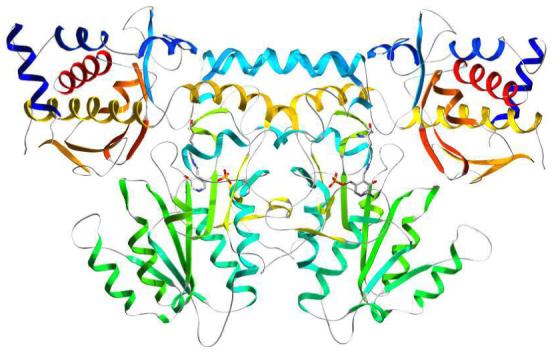
Ribbon drawing of the kynureninase biologically active unit with secondary structure elements colored in a blue to red progressing scheme. The dimer can be generated by applying the crystallographic symmetry operator 1-x, y,-z to the monomer.
Figure 2.

Stereo ribbon drawing of the kynureninase monomer with secondary structure elements colored in a blue to red progressing scheme. β-Strands are labeled with the prefix S, α-helices with the prefix H. The pyridoxal-5′-phosphate cofactor is drawn in CPK scheme and can be seen in the middle portion of the monomer between β-strand S8 and β-strand S11. The large domain is colored green, aquamarine, and yellow (H15, S15, and S16). Small domain elements are colored red, dark blue, orange, and yellow (H16).
Important dimerization contacts are provided by: 1) a two helix bundle formed between residues on α-helix H4 and its two-fold related counterpart, 2) interactions between residues on β-strand S2 with residues on α-helix H4, and 3) hydrogen bonding between Ala-164/Asn-318*, Ser-167/Trp-305*, His-142/Val-303*, Ser-174/Val-303*. The active site is formed at the bottom of a large cavity found at the interface of the large and small domains; it is amphiphilic in nature and has dimensions of 10 Å × 23 Å wide and 16 Å deep. The large domain face of the active site is lined with the hydrophobic side chains of Tyr-226, Phe-225, Leu-310*, Phe-314*, Met-316*, Trp-305*, and Phe-165, which form a large hydrophobic patch directly above the PLP cofactor. On the opposite side of the active site cleft, the small domain face contains the charged side chains of Gln-402, Asp-426, Arg-434, Arg-428, and Asn-429. The bottom of the active site is lined with the side chains of Ser-75 and His-102*.
PLP-Binding Site
The electron density returned from the molecular replacement solution showed continuous density between the ε nitrogen of Lys-276 and the 4′ carbon atom of the PLP cofactor (Figure 3). This observation is consistent with the presence of an enzyme-PLP internal aldimine complex and follows the observed spectroscopic characteristics of the active enzyme which has a 422 nm absorption maximum. The pyridoxal-5′phosphate molecule is stabilized and properly oriented at the monomer-monomer interface via extensive hydrogen bonding with residues contributed by two-fold related monomer chains (Figure 4). Hydrogen bonding between PLP phosphate oxygens and the side chains of residues involved in monomer-monomer contacts (Trp-305*/Ser-167, Asn-333*/Gly-278, Asn-333*/Tyr-275) coordinate residues from both monomer chains within the active site. PLP-phosphate oxygens are within hydrogen bonding distance of atoms on Trp-305*, Tyr-275, Asn-333*, Ser-332*, and Thr-138. Also, a water bridge appears to coordinate one of the PLP-oxygens with the main chain amide nitrogen of Leu-137. The coenzyme pyridine ring is stabilized via π-π stacking interactions with the phenyl ring of Phe-165. Phe-165 is conserved in all kynureninases and it is likely that these enzymes stabilize the PLP pyridine ring through π stacking with the side chain of this phenylalanine residue. The PLP pyridine nitrogen is hydrogen bonded with the side chains of Asp-168 and Asp-250, which are strictly conserved among kynureninases. Density can be observed between the δ oxygen of Asp-168 and the PLP pyridine nitrogen. The δ oxygens of Asp-168 and Asp-250 share a strong hydrogen bond (2.59 Å) and electron density can be seen extending between these atoms. Aspartate-250 is the expected conserved aspartate involved in PLP binding and maintaining the PLP pyridinium nitrogen protonated (44) among α-family members. The PLP phenolic 3′ oxygen is stabilized by hydrogen bonds with the His-253 δ nitrogen (2.95 Å) and the Lys-276-PLP aldimine NH (ζ nitrogen -3′oxygen distance 2.58 Å). The interactions of Asp-250 and His-253 with PLP atoms fix the pyridine ring as the dipolar ionic tautomer, resulting in the 422 nm absorption peak. The active site is highly solvated with two well ordered water molecules interacting with one of the PLP-phosphate oxygens, and another with the PLP 3′ oxygen. Also, water molecules connect active site residues of the large and small domains through a continuous string of seven waters between the δ oxygen and nitrogen atoms of Asn-333*, the His-102* ε nitrogen, the main chain carbonyl oxygen of Ser-75, across the active site cavity and passing directly over the aldimine NH towards the His-253 ε nitrogen, and ending at the main chain carbonyl oxygen of Phe-225 in the small domain. Four of the water molecules closest to the side chains of Asn-333* and His-102* occupy a large space in the active site that is devoid of any amino acid side chains.
Figure 3.
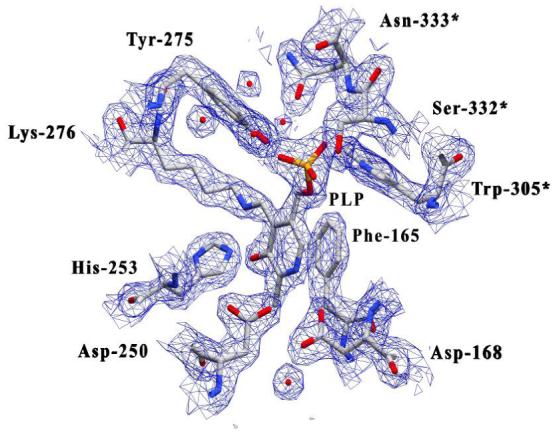
Map calculated with likelihood-weighted 2Fo-Fc coefficients for the area surrounding the pyridoxal-5′-phosphate-Lys-276 internal aldimine complex (contoured at 1.5σ). The active site is well solvated and several water molecules can be observed near the side chains of Asn-333*, Asp-168, and Tyr-275. Other active site residues have been omitted for clarity. Residues contributed from the symmetry related monomer are labeled with an asterisk (*).
Figure 4.

Stereo image of the kynureninase active site PLP hydrogen bonding interactions. Hydrogen bonds were drawn using a 3.55 Å cutoff. Residues contributed from the symmetry related monomer are labeled with an asterisk (*).
Comparison with the Pseudomonas fluorescens Kynureninase
Homo sapiens kynureninase shares 26% sequence identity with the Pseudomonas fluorescens ortholog, and a structural superposition reveals a high level of homology with a 1.2 Å2 R.M.S.D. over 317 amino acid residues. Human kynureninase has an additional α-helix (helix H1) that is part of a 25 amino acid extension at the N-terminus. There are three additional loops in Hkyn: 1) a fourteen amino acid loop connecting β-strand S2 and β-strand S3 (residues 52-65), 2) a ten amino acid loop between helix H11 and β-strand S7 (residues 191-200), and 3) a nine amino acid loop between helix H16 and β-strand S17 (378-387). For the most part, these extensions are found exclusively in constitutive kynureninase orthologs. Residues that play a role in stabilizing the PLP cofactor are well conserved. However, residues surrounding the PLP-phosphate moiety are much less conserved and include the following variations: ThrPkyn96 to LeuHkyn137, SerPkyn97 to ThrHkyn138, ThrPkyn131 to SerHkyn167, GlyPkyn281* to SerHkyn332*, and ThrPkyn282* to AsnHkyn333*. Absolutely conserved active site residues include Phe-165, Asp-168, Asp-250, His-253, Cys-273, Tyr-275, Lys-276, Arg-434, and Trp-305*.
The structural superposition of the Hkyn with Pkyn reveals differences in the quaternary structures not related to the primary structures. These high-order variations lie within elements of the small domain and in a large domain loop between β-strands S15 and S16 (residues 310-320), henceforth referred to as β-strand S15-S16loop, that extends above the active site cavity (Figure 5). Conformational differences in the small domain are observed in the β-hairpin formed by β-strands S20 and S21. This hairpin, the S20-S21 β-hairpin, contains two arginines conserved among kynureninases and other α-family members (ArgHkyn434 and ArgHkyn428). Of these, ArgHkyn434, is known to be dynamically and catalytically important in substrate binding and catalysis (45). The side chain ζ carbons of these arginines are separated in the superpositioned structures by 5.9 Å (between ArgHkyn434 and ArgPkyn375) and 11.18 Å (between ArgHkyn428 and ArgPkyn369), respectively. In Hkyn, the side chain of Arg-434 is thoroughly stabilized at the small/large domain interface with hydrogen bonds to the backbone carbonyls of Lys-427 and Tyr-226, through a water bridge to the conserved Asp-426, and by a hydrogen bond with the side chain of Arg-428. In Pkyn, the Arg-375 side chain is also extensively hydrogen bonded, but is instead stabilized within the active site cavity with hydrogen bonds to Asp-367 (equivalent to AspHkyn426), Tyr-176, with the PLP-aldimine through a chloride ion, and with atoms on a PEG molecule. The Pkyn Arg-375 guanidino nitrogens are thus much closer (by 6.5 Å) to the PLP-Lys-276 imine bond than in the Hkyn structure. The Hkyn and Pkyn S20-S21 β-hairpins are approximately 11.2° apart if measured from the Pkyn PLP-Lys aldimine. At their maximum separation point, α-carbons within this β-hairpin (of the conserved ProHkyn430 and ProPkyn371) are 5.9 Å away (Figure 5). Behind the S20-S21 β-hairpin, residues on helix H17 and β-strand S18 are 5.8 Å away (between α carbons of ProHkyn410 and ProPkyn350) from those of their prokaryotic counterparts (Figure 5). Also, the small domain helices H16 and helix H18 are each separated from their prokaryotic counterparts by 2.7 Å (between α carbons of LeuHkyn371 and LeuPkyn320) and 3.7 Å (between α carbons of IleHkyn457 and IlePkyn398), respectively. These conformational changes bring the entire small domain of the bacterial kynureninase closer to the Lys-276-PLP atoms and large domain active site residues.
Figure 5.
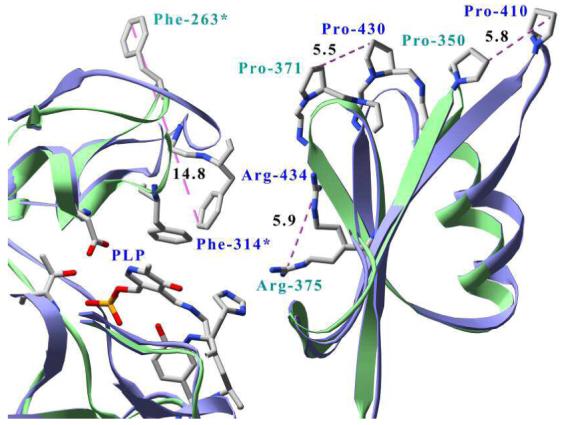
Structural superposition between dimers of human (ribbons and labels blue, atoms CPK) and Pseudomonas fluorescens (ribbons and labels green, atoms CPK) kynureninases. For clarity, only P. fluorescens residues Phe-263*, Arg-375, Pro-371, Pro-350 and their human equivalents Phe-314*, Arg-434, Pro-430, and Pro-410 are labeled. Only distances between conserved residues are shown and are in angstroms. Residues contributed from the symmetry related monomer are labeled with an asterisk (*). Unlabeled residues surrounding the pyridoxal-5′-phosphate cofactor (PLP) are all contributed from the human chain and are displayed for perspective only.
Large domain residues that line the active site cavity also exhibit considerable differences between kynureninase crystal structures. The PheHkyn314 side chain (Figure 5) is found within the active site cavity, 14.8 Å from its prokaryotic counterpart (PhePkyn263), although PhePkyn263 was observed to have poor electron density in the bacterial structure (21). Here, the side chain of PheHkyn314 forms part of a large hydrophobic patch directly above the PLP-phosphate moiety with the side chains of Ile-110*, Phe-165, Trp-305*, Phe-306*, and Met-316*. The side chains of Arg-313* and Met-316* are 7.5 Å and 5.5 Å away from their prokaryotic counterparts, respectively (not shown).
Docking of 3-Hydroxykynurenine into the Hkyn Active Site
Each Molegro output pose was evaluated based on MolDock score, protein interaction, hydrogen bonding, and affinity interaction energies. The pose with the best overall energy values with these criteria had values (in kJ/mol) of -114.5, -124.7, -18.9, and -25.8, respectively. This pose is positioned within the active site cavity with the α-amino moiety within hydrogen bonding distance of the side chains of small domain residues Arg-434, and Ser-75. The aromatic ring moiety is centered among the side chains of His-102*, Ile-110*, Trp-305*, Phe-306*, Phe-314*, Asn-333*, and Tyr-275 (Figure 6). The main chain carbonyl oxygen of Ser-75 is also within hydrogen bonding distance of the aromatic ring 2-amino group, whereas the 3-hydroxy group is within 2.9 Å of the Asn-333* δ nitrogen.
Figure 6.

Stereo image of 3-hydroxy-L-kynurenine docked with human kynureninase. Human kynureninase bonds and labels are colored white with CPK colored atoms. 3-hydroxy-L-kynurenine (3-HK) bonds and labels are colored blue. Residues contributed from the symmetry related monomer are labeled with an asterisk (*).
Discussion
Previously, the human enzyme was expressed in sf9 insect cells using a baculovirus expression vector (46), and in COS-1 cells transfected with human kynureninase cDNA (47). Until now, its expression has not been reported using a bacterial expression system. The Km values, and specific activity for 3-hydroxykynurenine that have been reported for the constitutive forms of kynureninase range from 3 μM (46) to 49 μM (47), and 0.16 μmol min-1 mg-1 from sf9 cells (46) to 5.5 μmol min-1 mg-1 for the enzyme expressed in COS-1 cells (46), respectively. The Michaelis constant (Km = 28.3 μM), and specific activity (1.75 μmol min-1 mg-1) of our enzyme for 3-hydroxykynurenine agree best with that for enzyme expressed in COS-1 cells (46). We find this to be a good indicator that the expression of the human enzyme in a bacterial system is functionally comparable to that from a eukaryotic expression system. We also determined the Km of the human enzyme for the bacterial substrate, L-kynurenine, to be 493 ± 46 μM. This agrees well with that reported for the human enzyme expressed in COS-1 (46) cells. All of our data were fit with HYPERO (48), Prism (GraphPad), and EnzFitter (Biosoft), and found to exhibit classical Michaelis-Menten behavior, which also contrasts with reports of sigmoid kinetic behavior from the enzyme expressed in sf9 cells (47). Attempts to fit our data to a Hill equation were unsuccessful. We measured the kinetic properties of a pseudo-native enzyme (with the hexa-histidine tag proteolitically removed) and found no change in Km and only a slight loss of activity (less than 3%). This enzyme had classic Michaelis-Menten behavior as well.
Like other members of the PLP-dependent α-family, kynureninase appears to undergo conformational changes during catalysis. In this work we identify two regions in the small and large domains that could play a role in opening and closing the active site cavity: the small domain S20-S21 β-hairpin, and the large domain S15-S16loop. The S20-S21 β-hairpin in particular emerges as a dynamic region since a structural superposition between Hkyn and the structures of Pkyn (PDB ID 1qz9), 2-aminoethylphosphonate transaminase (PDB ID 1m32) (49), NifS (PDB ID 1jf9) (50), cystathionine beta-lyase (PDB ID 1ibj) (51), CsdB (PDB ID 1i29) (52), alanine:glyoxylate aminotransferase (PDB ID 1h0c) (53), histidinol phosphate aminotransferase (PDB ID 1fg3) (54), malY (PDB ID 1d2f) (55), serine hydroxymethyltransferase (PDB ID 1bj4) (56), and 3-hydroxykynurenine transaminase (PDB ID 2ch2) (57) reveals that the difference in the S20-S21 β-hairpin conformational states observed between kynureninases is also clearly visible with other members of the α-family (Figure 7). Based on the structural superposition, it appears that the conformation of the conserved Arg-434 in Hkyn is a function of the positioning of the small domain, in particular that of the S20-S21 β-hairpin. Furthermore, the conformational changes required to position the Hkyn S20-S21 β-hairpin, Arg-434, and small domain elements closer to active site residues (thus resembling the structures of 1qz9) would be very similar to the movements observed between aspartate aminotransferase open (PDB ID 1ama) (58) and closed (PDB ID 9aat) (59) states. Although the small domains of kynureninases and aspartate aminotransferases do not superimpose well, in part due to a difference in length between the small-large domain linker α-helix (32 residues in aspartate aminotransferase versus 21 residues in kynureninase), the relative motion that must be undertaken by the entire small domain is very similar between the AAT open/closed and Pkyn/Hkyn pairs (Figure 8). Even though the small domain movements in aspartate aminotransferase are much more pronounced, it is possible that neither the Pkyn or Hkyn structures demonstrate a fully closed/open state, but rather an intermediate between them. Thus, the investigation of new kynureninase conformational states presents an interesting avenue of research. It is also possible that the second crystal morphology of kynureninase crystals grown in solution A might contain this different conformational state. It is important to note that these conditions would require some optimization in order to produce crystals with desirable diffraction properties.
Figure 7.
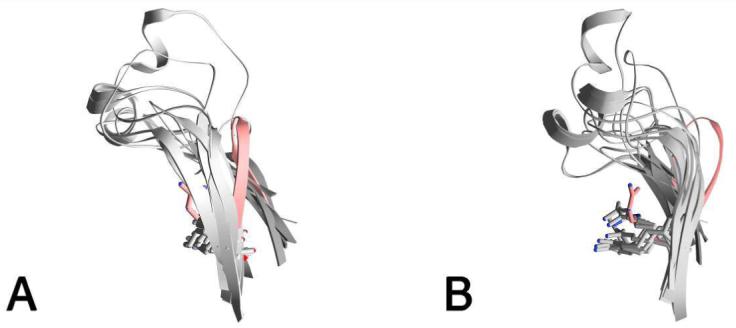
A) Ribbon drawing of the Hkyn small domain S20-S21 β-hairpin (red) and equivalent hairpins (gray) from the structures with PDB IDs 1qz9, 1m32, 1jf9, 1ibj, 1i29, 1h0c, 1fg3, 1d2f, 1bj4, and 2ch2. All structures were superimposed using STRAP with the Hkyn monomer as the reference model. B) view as in (A) rotated 90 degrees. The Hkyn Arg-434 is colored red and all other equivalent arginine residues are colored gray.
Figure 8.
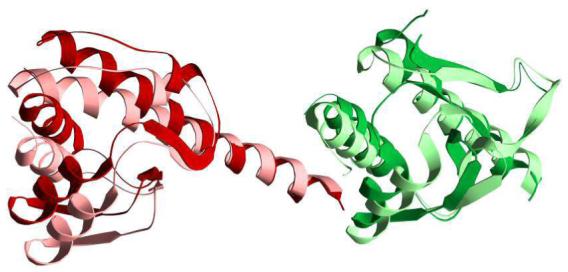
Comparison of the superimposed small domains of aspartate aminotransferase in open (PDB ID 1ama, colored dark red) and closed (PDB ID 9aat, colored light red) with the Homo sapiens kynureninase (colored dark green) and the Pseudomonas fluorescens kynureninase (PDB ID 1qz9, colored light green).
The two kynureninase structures taken together suggest that the ArgHkyn434/ArgPkyn375 side chain is dynamic near the vicinity of the PLP aldimine bond. This conserved residue plays an important role in the substrate binding and catalysis of other PLP β-family members. Site directed mutagenesis studies with aspartate aminotransferase and serine hydroxymethyltransferase have shown that this residue is important for both substrate specificity and regulating the conformational changes that accompany substrate binding in these enzymes. Arginine to lysine mutants of serine hydroxymethyltransferase have a 15-fold reduction in affinity and 0.03% of the wild type catalytic activity (60). Arginine to alanine mutants do not bind substrate or have catalytic activity (61). Also, aspartate aminotransferase arginine to aspartate mutants have catalytic properties that are several orders of magnitude slower than wild type (23). The structural superposition between these kynureninase orthologs also suggests that it is reasonable to presume that Arg-434 is involved in substrate binding as well. Human kynureninase crystals shatter within seconds of exposure to a mother solution containing 100 μM 3-hydroxy-DL-kynurenine. Thus, we speculate that substrate binding causes a conformational change that breaks crystal contacts and causes these crystals to shatter. This conformational change is likely to be initiated by an interaction between the substrate’s α carboxyl group and the Arg-434 guanidino nitrogens, as is seen in aspartate aminotransferase (62). This is an important step in the closure mechanism since aspartate aminotransferase arginine to alanine mutants cannot adopt fully closed conformational states (62). Subsequently, the aromatic ring of the substrate can occupy an amphiphilic pocket near the PLP-phosphate moiety between the charged side chains of Ser-75, His-102*, Tyr-275, Asn-333* and the hydrophobic side chains of Ile-110*, Phe-306*, Phe-314*, and Trp-305* (Figure 6). Substrate binding within the active site would tether the S20-21 β-hairpin, and consequently the rest of the small domain, to the large domain through the side chain of Arg-434 and stabilize the closed state until product release. The binding of substrate to both small and large domain residues would place a certain amount of strain on substrate bonds which would be released as the substrate is cleaved. In this way the large and small domains act as a spring loaded catalytic unit that puts strain on the substrate bonds and facilitates hydrolysis. The docking of 3-hydroxy-L-kynurenine in the kynureninase active site reveals that substrate α-carboxylate oxygens are likely to form hydrogen bonds with the side chain of Arg-434. The docking also suggests that the 2-amino and 3-hydroxyl moieties of the substrate are likely to be within hydrogen bonding distance to atoms on Asn-333*, Ser-75, and His-102*, and in a favorable distal quadrupole π-π stacking interaction with the side chain of the conserved Trp-305*. Several atoms in the kynureninase-3-hydroxy-L-kynurenine docking solution occupy the positions of atoms on a PEG molecule found in the active site of 1qz9 (not shown). The Moldock docking algorithm positions the α nitrogen of 3-hydroxy-L-kynurenine in close proximity to the PLP-Lys-276 Schiff base aldimine as required to form the 3-hydroxy-L-kynurenine external aldimine, which is the first intermediate in the kynureninase catalytic mechanism (63). 3-Hydroxy-L-kynurenine is also properly oriented such that the α carbon can be properly protonated by Lys-276 (21) in subsequent catalytic steps to form the appropriate product enantiomer (L-alanine). Also, the position of the top scoring docking pose from our calculations is oriented within the active site in a similar fashion to that of 4-(2-aminophenyl)-4-oxobutanoic acid in the crystal structure of A. gambiae 3-hydroxykynurenine transaminase (PDB ID 2ch2) (57) (superimposed figure in supplementary material). In both cases, α carboxylate atoms are near small domain residues, whereas the hydrophobic ring moiety is anchored on the large domain side of the active site cavity.
Inducible kynureninases preferentially catalyze the hydrolytic cleavage of kynurenine, which is not hydroxylated at the 3′ position. On the other hand, constitutive orthologs preferentially utilize 3-hydroxykynurenine as a substrate. Currently, there is a poor understanding of the molecular basis of the discrimination process between these two very similar molecules. The problem is further complicated by the fact that these enzymes have active sites that are highly conserved. Many of these conserved residues are found near the pyridoxal-5′-phosphate cofactor and there are currently no substrate or inhibitor kynureninase complex crystal structures to help identify residues involved in substrate differentiation. It is thus difficult to predict with certainty which residues convey molecular specificity. However, a structural superposition of the docked Hkyn-3-hydroxy-L-kynurenine structure with Pkyn, reveals contacts that can aid in directing experiments to identify these residues (Figure 9). Specifically, the TrpPkyn69* side chain phenyl group and the γ carbon of ThrPkyn282* form a hydrophobic patch unfavorable for hydrogen bonding with the 3-hydroxyl moiety of 3-hydroxykynurenine. These groups are more suitable to accommodate the kynurenine 3′ methyne carbon. On the other hand, the δ nitrogen of AsnHkyn333* and the smaller HisHkyn102* side chains form an environment in human kynureninase that would allow for this group (3-hydroxyl) to interact favorably with their side chains and not be sterically hindered (by Trp-69* in Pkyn). Interestingly, these two point mutations are conserved among constitutive and inducible kynureninases, with inducible kynureninases having a Thr-Trp pair whereas constitutive orthologs contain the Asn-His pair. Based on these deductions, we have designed site-directed mutagenesis experiments to determine whether these residues play a role in substrate binding and specificity.
Figure 9.
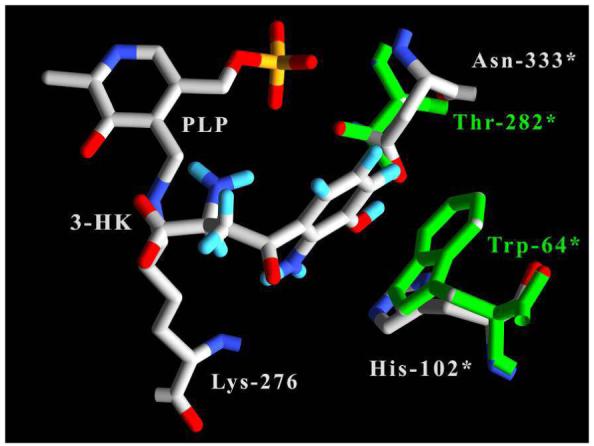
Active site amino acid differences between human and P. fluorescens kynureninases in the vicinity of the 3-hydroxyl moiety of docked 3-hydroxy-L-kynurenine (3-HK). 3-hydroxy-L-kynurenine and Hkyn labels and bonds are colored white with CPK-colored atoms. Pkyn residues and labels are colored green. Residues contributed from the symmetry related monomer are labeled with an asterisk (*).
Supplementary Material
Acknowledgements
We would like to thank the Hauptman-Woodward Institute for their instrumental role in identifying crystallization conditions. We would also like to thank the staff at the SBC-CAT beamline 19-BM at the Advanced Photon Source, Argonne, IL for their assistance during data collection.
Abbreviations
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- Hkyn
Homo sapiens kynureninase
- KPi
potassium phosphate
- LB
Luria Bertrani Broth
- MME
monomethylether
- PEG
polyethylene glycol
- Pkyn
Pseudomonas fluorescens kynureninase
- PLP
pyridoxal-5′-phosphate
- KPi
potassium phosphate
Footnotes
This work was partially supported by a grant from the National Institutes of Health (GM42588) to RSP.
References
- 1.Shetty AS, Gaertner FH. Kynureninase-Type enzymes of Penicillum roqueforti, Aspergillus niger, Rhizopus stolonifer, and Pseudomonas fluorescens: further evidence for distinct kynureninase and hydroxykynureninase activities. J Bacteriol. 1975;122:235–244. doi: 10.1128/jb.122.1.235-244.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Soda K, Tanizawa K. Kynureninases: enzymological properties and regulation mechanism. Adv Enzymol Relat Areas Mol Biol. 1979;49:1–40. doi: 10.1002/9780470122945.ch1. [DOI] [PubMed] [Google Scholar]
- 3.Schwarcz R, Pellicciari R. Manipulation of brain kynurenines: glial targets, neuronal effects, and clinical opportunities. J Pharmacol Exp Ther. 2002;303:1–10. doi: 10.1124/jpet.102.034439. [DOI] [PubMed] [Google Scholar]
- 4.Magni G, Amici A, Emanuelli M, Orsomando G, Raffaelli N, Ruggieri S. Enzymology of NAD+ homeostasis in man. Cell Mol Life Sci. 2004;61:19–34. doi: 10.1007/s00018-003-3161-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Espey MG, Chernyshev ON, Reinhard JF, Jr., Namboodiri MA, Colton CA. Activated human microglia produce the excitotoxin quinolinic acid. Neuroreport. 1997;8:431–434. doi: 10.1097/00001756-199701200-00011. [DOI] [PubMed] [Google Scholar]
- 6.Moffett JR, Els T, Espey MG, Walter SA, Streit WJ, Namboodiri MA. Quinolinate immunoreactivity in experimental rat brain tumors is present in macrophages but not in astrocytes. Exp Neurol. 1997;144:287–301. doi: 10.1006/exnr.1996.6365. [DOI] [PubMed] [Google Scholar]
- 7.Stone TW, Perkins MN. Quinolinic acid: a potent endogenous excitant at amino acid receptors in CNS. Eur J Pharmacol. 1981;72:411–412. doi: 10.1016/0014-2999(81)90587-2. [DOI] [PubMed] [Google Scholar]
- 8.Chiarugi A, Meli E, Moroni F. Similarities and differences in the neuronal death processes activated by 3OH-kynurenine and quinolinic acid. J Neurochem. 2001;77:1310–1318. doi: 10.1046/j.1471-4159.2001.00335.x. [DOI] [PubMed] [Google Scholar]
- 9.Giulian D, Vaca K, Noonan CA. Secretion of neurotoxins by mononuclear phagocytes infected with HIV-1. Science. 1990;250:1593–1596. doi: 10.1126/science.2148832. [DOI] [PubMed] [Google Scholar]
- 10.Guillemin GJ, Brew BJ, Noonan CE, Takikawa O, Cullen KM. Indoleamine 2,3 dioxygenase and quinolinic acid immunoreactivity in Alzheimer’s disease hippocampus. Neuropathol Appl Neurobiol. 2005;31:395–404. doi: 10.1111/j.1365-2990.2005.00655.x. [DOI] [PubMed] [Google Scholar]
- 11.Achim CL, Heyes MP, Wiley CA. Quantitation of human immunodeficiency virus, immune activation factors, and quinolinic acid in AIDS brains. J Clin Invest. 1993;91:2769–2775. doi: 10.1172/JCI116518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heyes MP, Brew B, Martin A, Markey SP, Price RW, Bhalla RB, Salazar A. Cerebrospinal fluid quinolinic acid concentrations are increased in acquired immune deficiency syndrome. Adv Exp Med Biol. 1991;294:687–690. doi: 10.1007/978-1-4684-5952-4_94. [DOI] [PubMed] [Google Scholar]
- 13.Heyes MP, Swartz KJ, Markey SP, Beal MF. Regional brain and cerebrospinal fluid quinolinic acid concentrations in Huntington’s disease. Neurosci Lett. 1991;122:265–269. doi: 10.1016/0304-3940(91)90874-s. [DOI] [PubMed] [Google Scholar]
- 14.Saito K, Crowley JS, Markey SP, Heyes MP. A mechanism for increased quinolinic acid formation following acute systemic immune stimulation. J Biol Chem. 1993;268:15496–15503. [PubMed] [Google Scholar]
- 15.Saito K, Nowak TS, Jr., Markey SP, Heyes MP. Mechanism of delayed increases in kynurenine pathway metabolism in damaged brain regions following transient cerebral ischemia. J Neurochem. 1993;60:180–192. doi: 10.1111/j.1471-4159.1993.tb05836.x. [DOI] [PubMed] [Google Scholar]
- 16.Saito K, Nowak TS, Jr., Suyama K, Quearry BJ, Saito M, Crowley JS, Markey SP, Heyes MP. Kynurenine pathway enzymes in brain: responses to ischemic brain injury versus systemic immune activation. J Neurochem. 1993;61:2061–2070. doi: 10.1111/j.1471-4159.1993.tb07443.x. [DOI] [PubMed] [Google Scholar]
- 17.Sei S, Saito K, Stewart SK, Crowley JS, Brouwers P, Kleiner DE, Katz DA, Pizzo PA, Heyes MP. Increased human immunodeficiency virus (HIV) type 1 DNA content and quinolinic acid concentration in brain tissues from patients with HIV encephalopathy. J Infect Dis. 1995;172:638–647. doi: 10.1093/infdis/172.3.638. [DOI] [PubMed] [Google Scholar]
- 18.Sei Y, Paul IA, Saito K, Layar R, Hartley JW, Morse HC, 3rd, Skolnick P, Heyes MP. Quinolinic acid levels in a murine retrovirus-induced immunodeficiency syndrome. J Neurochem. 1996;66:296–302. doi: 10.1046/j.1471-4159.1996.66010296.x. [DOI] [PubMed] [Google Scholar]
- 19.Mehta PK, Hale TI, Christen P. Aminotransferases: demonstration of homology and division into evolutionary subgroups. Eur J Biochem. 1993;214:549–561. doi: 10.1111/j.1432-1033.1993.tb17953.x. [DOI] [PubMed] [Google Scholar]
- 20.Jansonius JN. Structure, evolution and action of vitamin B6-dependent enzymes. Curr Opin Struct Biol. 1998;8:759–769. doi: 10.1016/s0959-440x(98)80096-1. [DOI] [PubMed] [Google Scholar]
- 21.Momany C, Levdikov V, Blagova L, Lima S, Phillips RS. Three-dimensional structure of kynureninase from Pseudomonas fluorescens. Biochemistry. 2004;43:1193–1203. doi: 10.1021/bi035744e. [DOI] [PubMed] [Google Scholar]
- 22.Paiardini A, Bossa F, Pascarella S. Evolutionarily conserved regions and hydrophobic contacts at the superfamily level: The case of the fold-type I, pyridoxal-5′-phosphate-dependent enzymes. Protein Sci. 2004;13:2992–3005. doi: 10.1110/ps.04938104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jager J, Moser M, Sauder U, Jansonius JN. Crystal structures of Escherichia coli aspartate aminotransferase in two conformations. Comparison of an unliganded open and two liganded closed forms. J Mol Biol. 1994;239:285–305. doi: 10.1006/jmbi.1994.1368. [DOI] [PubMed] [Google Scholar]
- 24.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 25.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 26.Luft JR, Collins RJ, Fehrman NA, Lauricella AM, Veatch CK, DeTitta GT. A deliberate approach to screening for initial crystallization conditions of biological macromolecules. J Struct Biol. 2003;142:170–179. doi: 10.1016/s1047-8477(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 27.Chayen NE, Shaw Stewart PD, Blow DM. Microbatch crystallization under oil-a new technique allowing many small-volume crystallization trials. J. Crystal Growth. 1992;122:176–180. [Google Scholar]
- 28.Use of the Advanced Photon Source was supported by the U. S. Department of Energy. O. o. S., Office of Basic Energy Sciences; under Contract No. W-31-109-Eng-38. [Google Scholar]
- 29.Otwinowski Z, W. M. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 30.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 31.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J. Appl. Cryst. 1997;30:1022–1025. [Google Scholar]
- 32.Collaborative Computational Project, N. The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. 1994;D50 doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 33.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Terwilliger TC. Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr D Biol Crystallogr. 2003;59:38–44. doi: 10.1107/S0907444902018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 36.Lamzin VS, Perrakis A, Wilson KS. The ARP/WARP suite for automated construction and refinement of protein models. In Int. Tables for Crystallography. F. Dordrecht, Kluwer Academic Publishers; The Netherlands: 2001. [Google Scholar]
- 37.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 38.McRee DE. XtalView/Xfit--A versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 39.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 40.Morris AL, MacArthur MW, Hutchinson EG, Thornton JM. Stereochemical quality of protein structure coordinates. Proteins. 1992;12:345–364. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- 41.Gille C, Frommel C. STRAP: editor for STRuctural Alignments of Proteins. Bioinformatics. 2001;17:377–378. doi: 10.1093/bioinformatics/17.4.377. [DOI] [PubMed] [Google Scholar]
- 42.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 43.Thomsen R, Christensen MH. MolDock: a new technique for high-accuracy molecular docking. J Med Chem. 2006;49:3315–3321. doi: 10.1021/jm051197e. [DOI] [PubMed] [Google Scholar]
- 44.Alexander FW, Sandmeier E, Mehta PK, Christen P. Evolutionary relationships among pyridoxal-5′-phosphate-dependent enzymes. Regio-specific alpha, beta and gamma families. Eur J Biochem. 1994;219:953–960. doi: 10.1111/j.1432-1033.1994.tb18577.x. [DOI] [PubMed] [Google Scholar]
- 45.Han GW, Schwarzenbacher R, Page R, Jaroszewski L, Abdubek P, Ambing E, Biorac T, Canaves JM, Chiu HJ, Dai X, Deacon AM, DiDonato M, Elsliger MA, Godzik A, Grittini C, Grzechnik SK, Hale J, Hampton E, Haugen J, Hornsby M, Klock HE, Koesema E, Kreusch A, Kuhn P, Lesley SA, Levin I, McMullan D, McPhillips TM, Miller MD, Morse A, Moy K, Nigoghossian E, Ouyang J, Paulsen J, Quijano K, Reyes R, Sims E, Spraggon G, Stevens RC, van den Bedem H, Velasquez J, Vincent J, von Delft F, Wang X, West B, White A, Wolf G, Xu Q, Zagnitko O, Hodgson KO, Wooley J, Wilson IA. Crystal structure of an alanine-glyoxylate aminotransferase from Anabaena sp. at 1.70 A resolution reveals a noncovalently linked PLP cofactor. Proteins. 2005;58:971–975. doi: 10.1002/prot.20360. [DOI] [PubMed] [Google Scholar]
- 46.Toma S, Nakamura M, Tone S, Okuno E, Kido R, Breton J, Avanzi N, Cozzi L, Speciale C, Mostardini M, Gatti S, Benatti L. Cloning and recombinant expression of rat and human kynureninase. FEBS Lett. 1997;408:5–10. doi: 10.1016/s0014-5793(97)00374-8. [DOI] [PubMed] [Google Scholar]
- 47.Walsh HA, Botting NP. Purification and biochemical characterization of some of the properties of recombinant human kynureninase. Eur J Biochem. 2002;269:2069–2074. doi: 10.1046/j.1432-1033.2002.02854.x. [DOI] [PubMed] [Google Scholar]
- 48.Cleland WW. Statistical analysis of enzyme kinetic data. Methods Enzymol. 1979;63:103–138. doi: 10.1016/0076-6879(79)63008-2. [DOI] [PubMed] [Google Scholar]
- 49.Chen CC, Zhang H, Kim AD, Howard A, Sheldrick GM, Mariano-Dunaway D, Herzberg O. Degradation pathway of the phosphonate ciliatine: crystal structure of 2-aminoethylphosphonate transaminase. Biochemistry. 2002;41:13162–13169. doi: 10.1021/bi026231v. [DOI] [PubMed] [Google Scholar]
- 50.Lima CD. Analysis of the E. coli NifS CsdB protein at 2.0 A reveals the structural basis for perselenide and persulfide intermediate formation. J Mol Biol. 2002;315:1199–1208. doi: 10.1006/jmbi.2001.5308. [DOI] [PubMed] [Google Scholar]
- 51.Breitinger U, Clausen T, Ehlert S, Huber R, Laber B, Schmidt F, Pohl E, Messerschmidt A. The three-dimensional structure of cystathionine beta-lyase from Arabidopsis and its substrate specificity. Plant physiology. 2001;126:631–642. doi: 10.1104/pp.126.2.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mihara H, Fujii T, Kato S, Kurihara T, Hata Y, Esaki N. Structure of external aldimine of Escherichia coli CsdB, an IscS/NifS homolog: implications for its specificity toward selenocysteine. J Biochem (Tokyo) 2002;131:679–685. doi: 10.1093/oxfordjournals.jbchem.a003151. [DOI] [PubMed] [Google Scholar]
- 53.Zhang X, Roe SM, Hou Y, Bartlam M, Rao Z, Pearl LH, Danpure CJ. Crystal structure of alanine:glyoxylate aminotransferase and the relationship between genotype and enzymatic phenotype in primary hyperoxaluria type 1. J Mol Biol. 2003;331:643–652. doi: 10.1016/s0022-2836(03)00791-5. [DOI] [PubMed] [Google Scholar]
- 54.Sivaraman J, Li Y, Larocque R, Schrag JD, Cygler M, Matte A. Crystal structure of histidinol phosphate aminotransferase (HisC) from Escherichia coli, and its covalent complex with pyridoxal-5′-phosphate and l-histidinol phosphate. J Mol Biol. 2001;311:761–776. doi: 10.1006/jmbi.2001.4882. [DOI] [PubMed] [Google Scholar]
- 55.Clausen T, Schlegel A, Peist R, Schneider E, Steegborn C, Chang YS, Haase A, Bourenkov GP, Bartunik HD, Boos W. X-ray structure of MalY from Escherichia coli: a pyridoxal 5′-phosphate-dependent enzyme acting as a modulator in mal gene expression. The EMBO journal. 2000;19:831–842. doi: 10.1093/emboj/19.5.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Renwick SB, Snell K, Baumann U. The crystal structure of human cytosolic serine hydroxymethyltransferase: a target for cancer chemotherapy. Structure. 1998;6:1105–1116. doi: 10.1016/s0969-2126(98)00112-9. [DOI] [PubMed] [Google Scholar]
- 57.Rossi F, Garavaglia S, Giovenzana GB, Arca B, Li J, Rizzi M. Crystal structure of the Anopheles gambiae 3-hydroxykynurenine transaminase. Proc Natl Acad Sci U S A. 2006;103:5711–5716. doi: 10.1073/pnas.0510233103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McPhalen CA, Vincent MG, Picot D, Jansonius JN, Lesk AM, Chothia C. Domain closure in mitochondrial aspartate aminotransferase. J Mol Biol. 1992;227:197–213. doi: 10.1016/0022-2836(92)90691-c. [DOI] [PubMed] [Google Scholar]
- 59.McPhalen CA, Vincent MG, Jansonius JN. X-ray structure refinement and comparison of three forms of mitochondrial aspartate aminotransferase. J Mol Biol. 1992;225:495–517. doi: 10.1016/0022-2836(92)90935-d. [DOI] [PubMed] [Google Scholar]
- 60.Delle Fratte S, Iurescia S, Angelaccio S, Bossa F, Schirch V. The function of arginine 363 as the substrate carboxyl-binding site in Escherichia coli serine hydroxymethyltransferase. Eur J Biochem. 1994;225:395–401. doi: 10.1111/j.1432-1033.1994.00395.x. [DOI] [PubMed] [Google Scholar]
- 61.Almo SC, Smith DL, Danishefsky AT, Ringe D. The structural basis for the altered substrate specificity of the R292D active site mutant of aspartate aminotransferase from E. coli. Protein Eng. 1994;7:405–412. doi: 10.1093/protein/7.3.405. [DOI] [PubMed] [Google Scholar]
- 62.Graber R, Kasper P, Malashkevich VN, Sandmeier E, Berger P, Gehring H, Jansonius JN, Christen P. Changing the reaction specificity of a pyridoxal-5′-phosphate-dependent enzyme. Eur J Biochem. 1995;232:686–690. doi: 10.1111/j.1432-1033.1995.tb20861.x. [DOI] [PubMed] [Google Scholar]
- 63.Koushik SV, Moore JA, 3rd, Sundararaju B, Phillips RS. The catalytic mechanism of kynureninase from Pseudomonas fluorescens: insights from the effects of pH and isotopic substitution on steady-state and pre-steady-state kinetics. Biochemistry. 1998;37:1376–1382. doi: 10.1021/bi971130w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.