

Superoxide reductases (SORs) are cysteinate-ligated non–hemeiron enzymes1 that reduce superoxide (O2−) to H2O2 in anaerobicmicrobes.2 The cysteinate of SOR is trans to the O2− binding site, and is proposed to play an important role in promoting the catalytic reaction. Herein, we report a rare example of a functional metalloenzyme active site model, that reduces O2− via atrans thiolate–ligated Fe(III)–peroxo intermediate. The trans thiolate is shown to lower the redox potential, change the spin-state, and dramatically weaken the Fe-O bond, favoring O2− reduction and H2O2 release.
Superoxide is a toxic byproduct of dioxygen chemistry that has been linked to a number of disease states.3 The proposed SOR mechanism involves the oxidative addition of O2− to the open site of the square pyramidal FeIIN4 HisSCys active site2c to afford a trans Scys-ligated FeIII-peroxo intermediate.2d,e This intermediate displays an intense S-to-Fe(III) charge transfer band at ~600(~3500) nm, but has yet to be characterized by vibrational spectroscopy. Iron-peroxo species are extremely difficult to characterize since they are thermally unstable, and photolabile. Vibrational data have been reported for mutant SOR (E47A) peroxos generated via the addition of H2O2.2b,f,g Whether these are identical to the catalytic SOR intermediate remains to be determined. Although a few well-characterized synthetic nitrogen-ligated iron–peroxos have been reported,4a,c there is a paucity of thiolate-ligated analogues.4b Since a thiolate is likely to influence the correlation between peroxide binding mode, vibrational parameters, and spin–state, synthetic thiolate–ligated peroxos are needed to provide benchmark parameters. Prior to the work reported herein, cis thiolate-ligated [FeIII(SMe2N4(tren))(OOH)]+ (1),4b was the only reported example of a synthetic thiolate–ligated FeIII-peroxo.
In situ deprotection and deprotonation of the new macrocyclic ligand cyclam-PrS–Ac·4HCl, afforded [FeII(cyclam–PrS)](BPh4) (2) upon the addition of FeCl2 and NaBPh4. Single crystals were grown from pentane/THF at −30 °C. As shown in the ORTEP (Figure 1), the Fe2+ ion of 2 is ligated by three secondary amines, one tertiary amine, and a tethered apical thiolate in a square pyramidal geometry (τ= 0.13)5 resembling that of SOR. A related tertiary amine cyclam complex [FeII(Me3-cyclam-EtS)]+ (3) was recently reported6 that reacts with H2O2 to afford an Fe(IV)=O.7 Like the SOR active site, 2 is high spin (S= 2; μeff=5.03 BM (MeCN); 4.91 BM (solid)). The Fe–S bond length in 2 (2.286(1) Å) falls in the usual range for synthetic Fe(II)–thiolates,6,8 but is slightly shorter than that of SOR (Fe–S= 2.4 Å), the cysteinate sulfur of which is H-bonded to the protein backbone.2a,c
Figure 1.
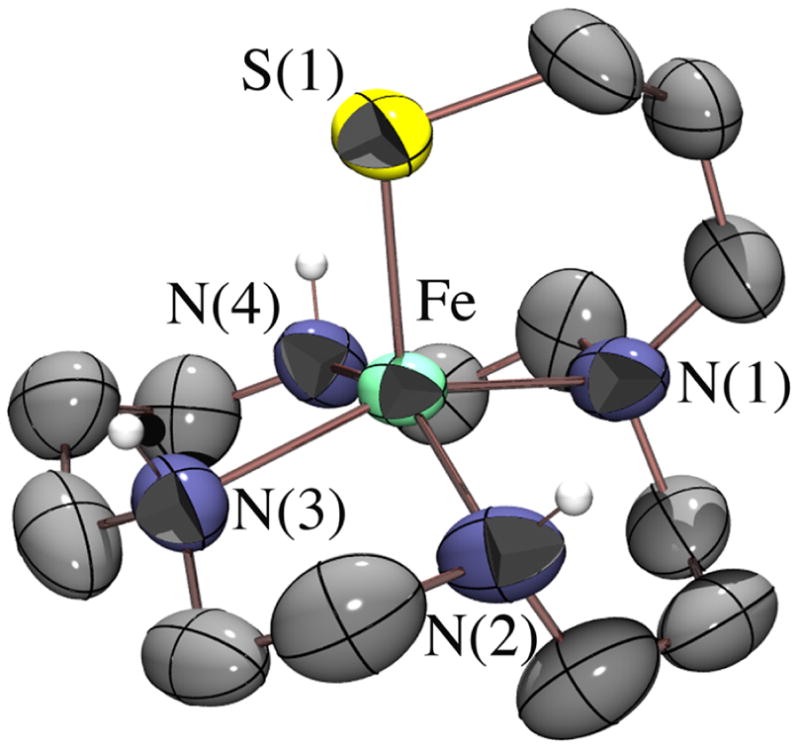
ORTEP of [FeII(cyclam–PrS)]+ (2). Selected bond lengths (Å): Fe–S(1), 2.286(1); Fe–N(1), 2.181(4); Fe–N(2,3,4)avg, 2.16(2).
Thiolate–ligated 2 reacts rapidly with O2 −• (18-crown-6-K+ salt) in CH2Cl2 at −78 °C to afford a metastable burgundy intermediate, as soon as a proton donor (MeOH; 82 equiv) is added. This intermediate is high-spin (g= 7.72, 5.40, 4.15), and displays an absorption band at λmax= 530(1350) nm. Resonance Raman shows νO–O, νFe–O, and νFe–S stretches at 891 cm−1 (Fermi doublet), 419 cm−1, and 352 cm−1, respectively (Figure 2). K18O2 (50% enriched; ICON) causes the νO–O and νFe–O to shift to 856 cm−1 and 400 cm−1, respectively, and addition of D+ (ie, MeOD) causes the Fermi doublet to collapse. These data are consistent with the formation of an Fe–hydroperoxo species, [FeIII(cyclam– PrS)(OOH)]+ (4), via the proton-dependent oxidative addition of superoxide to 2. No reaction occurs in the absence of a proton donor, and O2− does not convert to H2O2 in the absence of 2, under the conditions examined (−78 °C, 82 equiv MeOH). Intermediate 4 represents the first example of a synthetic trans thiolate–ligated FeIII–peroxo SOR intermediate analogue, the characterization of which provides important benchmark parameters.
Figure 2.
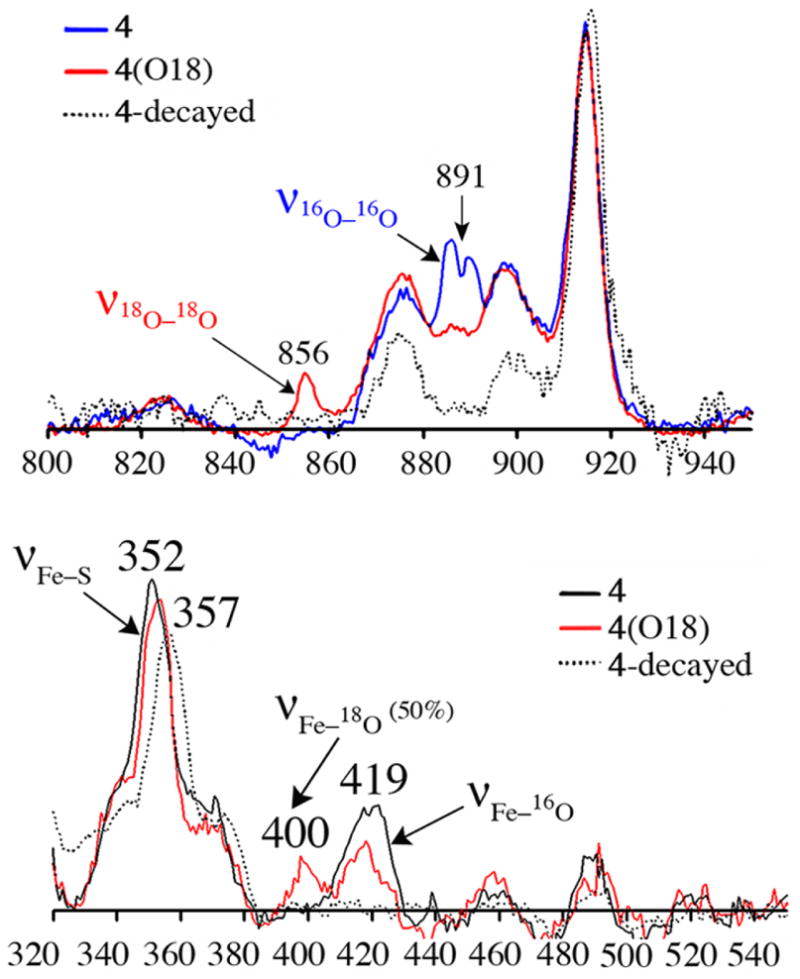
rRaman spectra of 4 generated from 16O2 − (blue), 18O2 − (red), and “decayed” product (dashed black) (571 nm excitation @ 183 K in THF/MeOH (upper panel); @ 77K in CH2Cl2/THF/MeOH (lower panel).
The νFe–O stretch of 4 is significantly lower than all other reported synthetic iron peroxides (range: 450–639 cm−1),4a but compares well with that of the only reported SOR peroxo (438 cm−1).2a,e The νO–O stretch (891 cm−1) is unusually high (reported range: 820–860 cm−1).4a The DFT optimized structure of 4 minimizes with Fe-S and Fe-O distances of 2.36 Å and 1.95 Å, respectively, and a protonated peroxo O-O distance of 1.44 Å. This Fe-O(peroxo) distance is significantly longer than the few reported Fe–(η1-OOH) structures(1.76–1.86 Å)4a reflecting the trans influence of the thiolate sulfur. The calculated νFe–S (345 cm−1), νFe–O (400 cm−1) and νO–O (933 cm−1) stretches are in reasonable agreement with the experimental data. When the thiolate is replaced with an amine, or alkoxide, trans to the peroxo,9 then the calculated νFe–O (495 cm−1 and 420 cm−1, respectively) is considerably higher. These vibrational data, along with the calculated force constant (kFe–O = 1.20 mdynes/cm2 for 4 vs reported range= 2.2-2.1 mdynes/cm2),4a indicate that the Fe– O(peroxide) bond is significantly weakened upon the introduction of a trans thiolate into the coordination sphere.
Addition of HOAc to metastable 4 at −78 °C releases H2O2 (as detected using an amplex red assay), and cleanly affords a new aqua blue species λmax = 604(1350) nm (Figure 3). When this reaction is monitored by EPR, the high–spin signal associated with 4 is replaced with a new low–spin signal at g= 2.37, 2.30, 1.89. The νO–O and νFe–O stretches disappear in the rRaman spectrum, and new stretches are observed at 339, 409, and 421 cm−1. Although this aqua blue species proved too unstable to isolate, it was unambiguously identified by ESI-mass spectrometry as acetate–bound [FeIII(cyclam–PrS)(OAc)]+ (5), a model for Glu–bound SOR.
Figure 3.
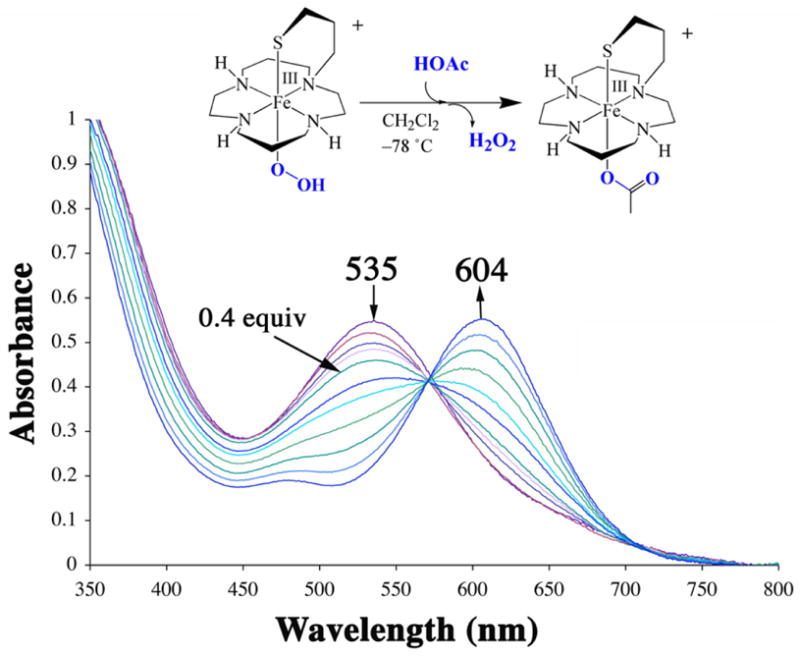
Conversion of peroxo–bound 4 (0.5 mM in CH2Cl2) to acetate–bound [FeIII(cyclam–PrS)(OAc)]+ (5) via the addition of HOAc (0.1 equivaliquots every two minutes) at −78 °C.
Addition of a sacrificial reductant (Cp2Co) to 5 at low temperatures (−78 °C) regenerates 2, which then reacts with a second equiv of O2 −• to re-afford peroxo 4. Addition of a second equiv of HOAc releases H2O2 (Figure 4), thereby mimicking the proposed SOR catalytic cycle involving glutamic acid,2d,e,i and demonstrating that reduction of O2 −• by 2 is catalytic. Thus far, five turnovers have been achieved.
Figure 4.
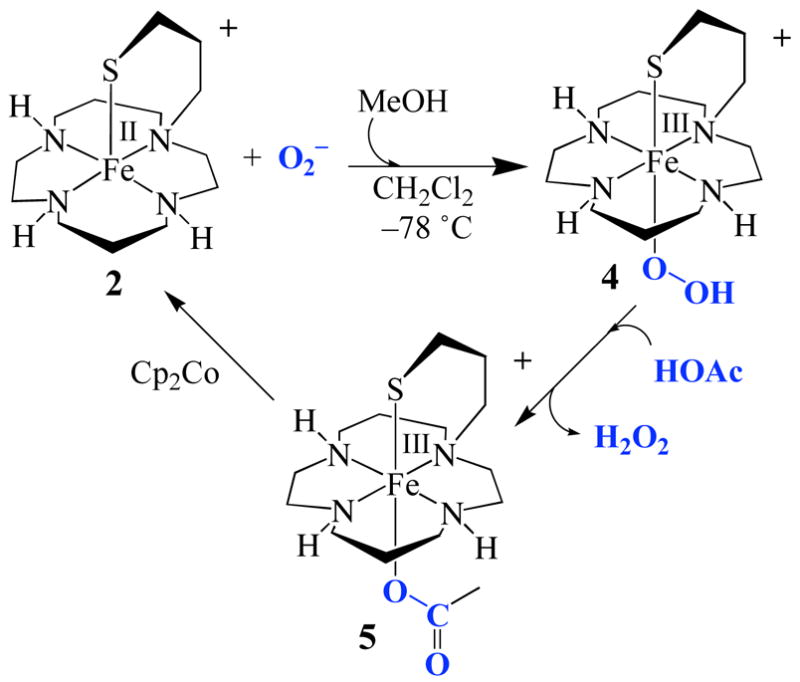
The catalytic cycle involving [FeII(cyclam–PrS)]+ (2) induced superoxide (O2−) reduction
The thiolate ligand and its trans positioning relative to the substrate appear to contribute significantly to the function of our biomimetic catalyst. First, the pendent thiolate arm of 2 causes the redox potential to shift anodically by +480 mV relative to [FeII(cyclam)(MeCN)2] (from +700 mV to +220 mV vs SCE), making it better suited to promote superoxide reduction. Second, the trans thiolate changes the spin–state from S= 1/24b to S= 5/2: the majority of nitrogen-ligated Fe(III)-OOH’s are S= 1/2,4a as is cis-thiolate ligated 1.4b Third, the thiolate dramatically shifts the νFe–O stretch and decreases the kFe–O force constant well-below all other reported iron peroxides.4a Peroxo 4 partially converts to methoxide-bound [FeIII(cyclam–PrS)(OMe)]+ (6; g= 2.34, 2.26, 1.95; νFe–S= 357 cm−1) within minutes at −78 °C, whereas cisligated peroxo 1 takes hours (t1/2= 63.9 hrs) to convert to [FeIII(SMe2N4(tren))(OMe)]+ under the same conditions. Methoxide–bound 6 was identified via its independent synthesis involving Cp2Fe+ oxidation of 2 in MeOH, in the presence of iPr2EtN.
In conclusion, the data described herein indicates that like the enzyme, SOR intermediate–analogue 4 is better suited to promote Fe–O, as opposed to O–O, bond cleavage. This is in contrast to P450 and its analogue 3. Kinetics studies, and studies aimed at determining the pKa of the proximal and distal peroxo oxygens of 4 are currently underway.
Supplementary Material
Detailed ligand syntheses and description of UV/vis monitored catalytic turnover, 1H NMR, ESI mass spec (ligand, 5), EPR, 1/χ vs. T plot and CV of 2, UV/vis of 4, Amplex Red assay, and X-ray tables. This information is available free of charge via the Internet at http://pubs.act.org.
Acknowledgments
This research was supported by NIHGM45881 (J.A.K.), GM40392 (E.I.S.), and F31 GM73583 (P. L.- M.).
References
- 1.Kovacs JA. Chem Rev. 2004;104:825–848. doi: 10.1021/cr020619e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Kurtz DM., Jr Acc Chem Res. 2004;37:902–908. doi: 10.1021/ar0200091. [DOI] [PubMed] [Google Scholar]; (b) Mathé C, Nivire V, Houée-Levin C, Mattioli TA. Biophys Chem. 2006;119:38–48. doi: 10.1016/j.bpc.2005.06.013. [DOI] [PubMed] [Google Scholar]; (c) Yeh AP, Hu Y, Jenney FE, Jr, Adams MWW, Rees DC. Biochemistry. 2000;39:2499–2508. doi: 10.1021/bi992428k. [DOI] [PubMed] [Google Scholar]; (d) Emerson JP, Coulter ED, Cabelli DE, Phillips RS, Kurtz DM., Jr Biochemistry. 2002;41:4348–4357. doi: 10.1021/bi0119159. [DOI] [PubMed] [Google Scholar]; (e) Niviere V, Asso M, Weill CO, Lombard M, et al. Biochemistry. 2004;43:808–818. doi: 10.1021/bi035698i. [DOI] [PubMed] [Google Scholar]; (f) Mathe C, Mattioli TA, Horner O, Lombard M, Latour J-M, Fontecave M, Niviere V. J Am Chem Soc. 2002;124:4966–4967. doi: 10.1021/ja025707v. [DOI] [PubMed] [Google Scholar]; (g) Horner O, Oddou J–L, Niviere V, Fontecave M, Halfen JA, Latour J-M, et al. Biochemistry. 2004;43:8815–8825. doi: 10.1021/bi0498151. [DOI] [PubMed] [Google Scholar]; (h) Mathé C, Nivière V, Mattioli TA. J Am Chem Soc. 2005;127:16436–16441. doi: 10.1021/ja053808y. [DOI] [PubMed] [Google Scholar]; (i) Clay MD, Jenney FE, Jr, Hagedoorn PL, George GN, Adams MWW, Johnson MK. J Am Chem Soc. 2002;124:788–805. doi: 10.1021/ja016889g. [DOI] [PubMed] [Google Scholar]; (j) Jovanovic T, Krebs C, Moura I, Moura JJG, Radolf JD, Huynh BH, Rusnak F, et al. J Biol Chem. 2000;275:28439–28448. doi: 10.1074/jbc.M003314200. [DOI] [PubMed] [Google Scholar]
- 3.(a) Gogun Y, Sakurada S, Kimura Y, Nagumo M. J Clin Biochem Nutr. 1990;8:85–92. [Google Scholar]; (b) Kocaturk PA, Akbostanci MC, Tan F, Kavas GO. Pathophysiology. 2000;7:63–67. doi: 10.1016/s0928-4680(00)00030-4. [DOI] [PubMed] [Google Scholar]; (c) Ihara Y, Chuda M, Kuroda S, Hayabara T. J Neurol Sci. 1999;170:90–95. doi: 10.1016/s0022-510x(99)00192-6. [DOI] [PubMed] [Google Scholar]; (d) De Leo ME, Borrello S, Passantino M, Palazzotti B, Galeotti T, Masullo C, et al. Neurosci Lett. 1998;250:173–176. doi: 10.1016/s0304-3940(98)00469-8. [DOI] [PubMed] [Google Scholar]
- 4.(a) Roelfes G, Ho RYN, Rohde J-U, Feringa BL, Munck E, Que L, Jr, et al. Inorg Chem. 2003;42:2639–2653. doi: 10.1021/ic034065p. [DOI] [PubMed] [Google Scholar]; (b) Shearer J, Scarrow RC, Kovacs JA. J Am Chem Soc. 2002;124:11709–11717. doi: 10.1021/ja012722b. [DOI] [PubMed] [Google Scholar]; (c) Bolland V, Banse F, Mattioli TA, Blondin G, Girerd JJ, et al. Inorg Chem. 2003;42:2470–2477. doi: 10.1021/ic025905n. [DOI] [PubMed] [Google Scholar]
- 5.Addision AW, Rao TN, Reedijk J. J Chem Soc Dalton Trans. 1984:1349. [Google Scholar]
- 6.Fiedler AT, Halfen JA, Brunold TC. J Am Chem Soc. 2005;127:1675–1689. doi: 10.1021/ja046939s. [DOI] [PubMed] [Google Scholar]
- 7.Bukowski MR, Koehntop KD, Stubna A, Bominaar EL, Halfen JA, Munck E, Nam W, Que L., Jr Science. 2005;310:1000–1002. doi: 10.1126/science.1119092. [DOI] [PubMed] [Google Scholar]
- 8.(a) Shearer J, Nehring J, Kaminsky W, Kovacs JA. Irong Chem. 2001;40:5483–5484. doi: 10.1021/ic010221l. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Noveron JC, Olmstead MM, Mascharak PK. J Am Chem. 2001;123:3247–3259. doi: 10.1021/ja001253v. [DOI] [PubMed] [Google Scholar]
- 9.Lehnert N, Ho RYN, Que L, Jr, Solomon EI. J Am Chem Soc. 2001;123:12802–12816. doi: 10.1021/ja011450+. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed ligand syntheses and description of UV/vis monitored catalytic turnover, 1H NMR, ESI mass spec (ligand, 5), EPR, 1/χ vs. T plot and CV of 2, UV/vis of 4, Amplex Red assay, and X-ray tables. This information is available free of charge via the Internet at http://pubs.act.org.