

Abstract
Comprehensive two-dimensional gas chromatography with time-of- flight mass spectrometry (GCxGC-TOFMS) coupled with rapid chemometric analysis were used to identify chemical differences in metabolite extracts isolated from yeast cells either metabolizing glucose (repressed (R) cells) via fermentation, or metabolizing ethanol by respiration (derepressed (DR) cells). Principal component analysis (PCA) followed by Parallel Factor Analysis (PARAFAC) in concert with the LECO ChromaTOF software located and identified the differences in composition between the two types of cell extracts and provided a reliable ratio of the metabolite concentrations. In this report, we demonstrate the analytical method developed to provide relatively rapid analysis of three selective mass channels (m/z 73, 205, 387), although in principle all collected mass channels could be analyzed. Twenty-six metabolites that differentiate repressed cells from derepressed cells were identified. The DR/R ratio of metabolite concentrations ranged from 0.02 for glucose to 67 for trehalose. The average biological variation of the sample extracts was 31%. This analysis demonstrates the utility and benefit of using PCA combined with PARAFAC and ChromaTOF software on extremely complex samples to derive useful information from complex three-dimensional chromatographic data objectively and relatively rapidly.
Introduction
Two-dimensional (2D) comprehensive gas chromatography coupled with time-of-flight mass spectrometry (GCxGC-TOFMS) has been used for the analysis of a number of complex mixtures such as jet fuels,1 environmental samples,2 drug screening,3 cigarette smoke condensate4 and pesticides.5 The major advantage of a time-of-flight mass spectrometer (TOFMS) over other GCxGC detectors is its ability to aid in the identification of a large number of compounds in very complex mixtures. Obtaining useful information from the analysis of these mixtures, which may contain literally thousands of different compounds, requires a reliable and relatively rapid procedure to identify compounds of interest and to deconvolute any overlapping mass spectra to yield pure chromatographic peak profiles for accurate quantification.
One way to glean information from GCxGC-TOFMS data is to use chemometric analysis and take advantage of the multivariate selectivity in the third order data.6 An analysis tool that is commonly used to locate distinguishing regions of signal in data sets is principal component analysis (PCA). PCA recognizes patterns in complex data sets based upon the amount of variance in the data sets. The chemical compounds exhibiting the greatest variance are captured on principal component 1, PC1. The next greatest variance in the data set is captured on PC2, and so on. The two outputs from PCA are a scores plot and a loadings plot. The scores plot indicates the degree of similarity between the individual chromatographic profiles and groups them accordingly. The loadings plot identifies the locations in the 2D separation plane of the chemical compounds responsible for these groupings. The loadings values correspond with quantitative differences between classes for each differentiating chromatographic feature. A larger absolute loadings value is associated with a greater class-to-class difference.
Parallel Factor Analysis (PARAFAC) is a quantitative multivariate tool that can be applied to three-dimensional (3D) GCxGC-TOFMS data structures.6 PARAFAC uses the GC column 1, GC column 2 and the mass spectral signal in one sample to obtain pure component profiles (i.e., pure chromatographic and mass spectral peak profiles) by removing the background noise and overlapping peaks. The deconvoluted three-dimensional peak can then be reconstructed by taking the outer product between the first and second dimension chromatographic profiles. This deconvoluted analyte peak can then be integrated to give a total peak volume that is proportional to analyte concentration.
Recently, there has been interest in using GCxGC-TOFMS for metabolite analysis.7–11 Several methods of identifying differences in metabolomic data obtained by gas chromatography have been reported in the literature.8–13 Each of these methods employs different tools to achieve the goal of high- throughput analysis. One method of analysis involves the use of chemometric and multivariate techniques to analyze complex metabolic data.8,9,12,13 Recently, we reported the “DotMap” algorithm, which uses the dot product of the scaled, weighted and normalized library mass spectrum and the observed mass spectrum along each point in the GCxGC-TOFMS data cube to locate analytes of interest.8,9 Using the DotMap algorithm, the analytes of interest must be known prior to analysis. Jonsson and co-workers used multivariate curve resolution (MCR) followed by PCA or PLS-DA on GC-TOFMS data to identify and quantify metabolites of interest.12,13 Using this method all samples were smoothed, baseline subtracted, aligned, resolved using MCR, submitted to a multivariate analysis and exported to a mass spectral search library. The PARAFAC algorithm has also been used to obtain pure mass spectra from GCxGC-TOFMS data derived from a complex derivatized plant metabolite extract.9 This study used the entire mass spectral range collected to obtain pure component peak profiles. Shellie and co-workers used direct chromatogram comparisons and difference plots of the total ion chromatogram (TIC) for GCxGC-TOFMS data to locate differences in mice metabolite data.10–11
Herein, we examined the suitability of PCA for normalized GCxGC-TOFMS chromatographic data followed by the use of ChromaTOF and PARAFAC software for locating, identifying and quantifying metabolites of interest in complex yeast metabolite extracts. Small molecules were extracted from yeast cells either metabolizing glucose via fermentation or metabolizing ethanol by respiration. Growth on the carbon source ethanol (derepressed, DR) should lead to the accumulation within cells of different pools of metabolites than growth on the carbon source glucose (repressed, R). In this study, nine extracts from cells growing on each carbon source were prepared and a total of 70 injections on the GCxGC-TOFMS were processed for comparison. Since the compound classes of interest were known, i.e., derivatized metabolites, three selective mass channels (m/z 73, 205 and 387) were submitted to PCA, thereby utilizing more of the 3D data cube than if only one m/z was examined to locate a significant number of analytes responsible for much of the variability between the sample extracts. The compounds with the highest loadings values were then identified but not quantified using ChromaTOF software. This resulted in the discovery of key analytes that differed in concentration between fermenting (R) and respiring cells (DR). The concentration ratios of these metabolites in the two types of sample extracts were obtained using PARAFAC. Thus, ChromaTOF, PCA and PARAFAC software are used in a complementary way in the reported methodology. The quantitative precision obtained for the metabolites that vary most between growth conditions is also reported along with the mass spectral match values (MV).
Experimental Section
Yeast strain and growth conditions
The yeast strain used in this study was W303-1a (MATa ade2 can1-100 his3-11,15 leu2-13,112 trp1-1 ura3-1). Cells were grown at 30 °C in synthetic complete medium (SC)14 containing either 5% glucose to prepare fermenting cells (R) or 3% ethanol and 0.05% glucose followed by incubation for 6 hours to prepare cells metabolizing ethanol by respiration (DR). Three R and three DR cultures were grown to obtain a measure of biological variability, Figure 1.
Figure 1.

Experimental design and nomenclature for samples. Cultures are labeled as A, B or C. Sample extracts have the aliquot number (1, 2 or 3) in addition to the culture label. An R follows the sample extract number when referring to a fermenting (repressed) sample (A) and DR when referring to a respiring (derepressed) sample (B). The injection number for each extract is written out. For example: repressed sample extract, culture A, extract 2 injection 3 would be written as A2R injection 3.
Extraction and derivatization of metabolites from yeast cells
The metabolic activity of the yeast cells was quenched and small polar metabolites extracted using the method reported by Castrillo et al.15 as a guide. At late log phase a volume of each culture containing 1×107 cells was diluted into 4 volumes of quenching buffer (10 mM tricine, pH 7.4, in 60% methanol) at −40 °C. Three aliquots were taken from each culture for extraction, Figure 1. Each cell suspension was spun at 1000 × g in a Sorvall RC-5B Plus centrifuge at −20 °C for 3 min and the resulting cell pellet was washed once with 1 mL of quenching buffer at −40 °C. Each cell pellet was suspended in 1 mL of extraction buffer (0.5 mM tricine, pH 7.4, in 75% ethanol) at 80 °C and held at this temperature for approximately 3 min. Extracted cell suspensions were cooled on ice for 5 min and then spun twice as described above to pellet large cellular debris. The resulting ethanolic metabolite extracts were dried in a SpeedVac at room temperature and stored at −80 °C under argon prior to GC xGC-TOFMS analysis. The extracted metabolites were methoximated (20 mg/mL methoxyamine in pyridine) and trimethylsilylated (BSTFA: TMCS, 99:1) thus replacing the active hydrogens with the trimethylsilyl (TMS) adducts.16 The samples were warmed at 30 °C for 90 minutes after the addition of 30 μL methoxime solution. Following oximation, 70 μL of TMS reagent was added and the sample was heated at 60 °C for 60 minutes.
Instrumental Parameters
An Agilent 6890N gas chromatograph equipped with an Agilent 7683 auto-injector (Agilent Technologies, Palo Alto, CA, USA) coupled with a LECO Pegasus III time-of- flight mass spectrometer with the commercially available 4D thermal modulator upgrade (LECO, St. Joseph, MI, USA) was used to analyze the yeast extracts. Column 1 was a 20 m × 250 μm i.d. × 0.5 μm RTX-5MS film (Restek, Bellefonte, PA, USA) and column 2 a 2 m × 180 μm i.d. × 0.2 μm RTX-200MS film (Restek, Bellefonte, PA, USA). Injections of 1 μL were made in split- less mode with each extract having 4 replicate injections except A1R and A1DR (nomenclature given in Figure 1), which were injected in triplicate. This resulted in 70 injections (GC x GC-TOFMS chromatograms) for the analysis. The GC inlet and transfer line were set at 280 °C. Column 1 was held at 60 °C for 0.25 minutes and then increased at 8°/min to 280 °C where it was held for 10 minutes. Column 2 was initially set at 70 °C and followed the same temperature program as column 1 giving a total run time of 37.75 minutes. The modulator was kept 40 °C higher than column 1 and the modulation period was 1.5 seconds. A constant flow rate of 1 mL/min was held at the head of column 1. The ion source was set to 200 °C. Mass Channels m/z 40–600 were collected at 100 spectra/second after a 5 minute solvent delay.
Data Analysis
The LECO ChromaTOF software v.2.21 (LECO, St. Joseph, MI, USA) was used to collect the data and obtain raw mass spectral match values (MV). Three mass spectral libraries were searched against: the NIST main library, an in- house metabolite library and the metabolite library obtained from the Max Planck Institute of Molecular and Plant Physiology (http://www.mpimp-golm.mpg.de/mms-library/index-e.html). Data from m/z 73, 205 and 387 were exported as comma separated value (.csv) files to Matlab v.7.0.4 (Mathworks, Natick, MA, USA) where principal component analysis, PCA, was performed, using the PLS toolbox version 3.51 (Eigenvector Research, Manson, WA, USA). All samples were normalized to the summed TIC from regions 430.5 s to 1125 s and 1145 s to the end of the run and mean centered. The regions omitted contained overloaded reagent artifacts that were similar in both the R and DR 2D chromatograms. Only data from m/z 387 required retention time alignment for the greatest amount of class-to-class variance to be captured on PC1. The data was shifted slightly in both separation dimensions, but it was found that only alignment on column 2 was necessary to assist the data analyis, so the peak match one-dimensional retention time alignment algorithm was used.17 The peak match algorithm was sufficient since the magnitude of the shifting on column 2 was less than the typical peak width at the base. The retention time alignment algorithm creates a list of where each peak is located in the specified target chromatogram and all sample chromatograms, where each chromatogram is represented by a single row vector and peaks are defined as local maxima with signal greater than five times the standard deviation of the chromatographic noise. Each peak in a sample chromatogram is then shifted to match the retention time of the nearest peak in the target chromatogram using linear interpolation. The m/z 387 chromatograms were aligned to A1R injection 1 (see Figure 1 for sample extract label nomenclature) and the shifting thresho ld was set to 20. This alignment algorithm did not mismatch peaks due to the scarcity of peaks in the 2D chromatogram at m/z 387. A PARAFAC graphical user interface (GUI) developed in- house and implementing the N-way toolbox was used for the quantification of the metabolites identified by PCA as exhibiting the most variability between R and DR sample extracts. The PARAFAC algorithm in this GUI was implemented from the following website: http://www.models.kvl.dk/courses/.18 The GUI automatically imports raw ChromaTOF data and converts LECO format chromatograms into Matlab variables. The GUI allows the user to quickly specify the sub region of the chromatograms for the analysis as well as the m/z to use for the deconvolution of overlapping mass spectra. The major benefits to using a PARAFAC GUI are the speed and ease of analysis.
Results and Discussion
GCxGC-TOFMS is suitable for the analysis of the very complex methoximated and trimethylsilylated yeast extracts, Figures 2A and B. It is important to no te that even though the modulation period is short, 1.5 s, and some wraparound did occur, sufficient resolution of the eluting components was obtained along with 3 or more modulation periods across each peak to assist the chemometric analysis. Since the trimethylsilyl (TMS) group (-Si(CH3)3) has a m/z of 73, any chemical species in a sample extract that is tagged with one or more TMS group will have a peak at m/z 73. While providing some useful information, the major drawback to using this single m/z for identifying differences in the samples is the complexity of the chromatogram at m/z 73. Of the more than 2500 recognizable peaks present in the 2D chromatogram, many are reagent artifacts along with column bleed. To exclude these artifacts from the analysis and at the same time investigate a certain class of compounds, selected m/z were chosen that would be specific for a known class of compounds of interest, as demonstrated in Figures 2C and D. Trimethylsilyl carbohydrates have a distinguishing mass fragment at m/z 205 (Figure 2C). A majority of TMS sugar phosphates have a distinguishing fragment at m/z 387 (Figure 2D). These m/z showed the metabolites of interest and significantly lowered the complexity of the 2D chromatographic profiles. Only the chromatograms for the R samples are shown for brevity, but the DR samples show a similar number of peaks.
Figure 2.




Two-dimensional plots of a repressed sample extract at selected mass channels. (A) Mass channel (m/z) 73, (B) boxed region of (A) emphasizing the separation resolution achieved. (C) Mass channel 205, which is fairly selective for carbohydrates and (D) mass channel 387, which is selective for sugar phosphates. The selective mass channels significantly reduce the complexity of the 2D chromatograms while providing information of interest.
The complexity of the chromatograms shown in Figure 2 made it difficult to visually observe differences between the R and DR chromatograms. A rapid and objective method for determining the difference between two or more classes of samples is to use PCA on the entire data set of samples. Prior to submission of all 70 chromatograms to PCA, samples were normalized to the sum of the TIC thus correcting for injection volume discrepancies and aligned only if the class-to-class variance was not captured on PC1. The summed TIC signals for the R and DR samples were compared to ensure they fell in the same range and they did. The mean TIC signal for the R samples was 9.0×108 with a standard deviation of 1.1×108 and the mean signal for the DR samples was 9.7×108 with a standard deviation of 1.6×107. Thus, since the standard deviations encompassed the means, the TIC could be objectively used to correct injection volume variation. In the future, it may be prudent to add internal standards from multiple compound classes to correct for variable losses during extract preparation.19 In addition to the major mass channel (m/z 73), PCA was performed individually on m/z 205 and 387 to ascertain the extent of class discrimination followed by the use of the loadings plots to locate differentiating metabolites. Since only two experimental classes, R and DR, were sampled, the class-to-class variance was captured primarily on PC1 as is shown in the scores plots in Figures 3A-C. The 70 injections mentioned previously in the Experimental section are labeled by a chromatogram number. The chromatogram number does not reflect the order in which the samples were derivatized or injected, but rather the order in which the samples were subjected to PCA. At m/z 73 approximately 47% of the variance was captured in PC1 (Figure 3A). This low captured variance is reasonable given that reagent artifacts and column bleed are present at this m/z. The m/z 205 data (Figure 3B) resulted in the greatest amount of variance captured on PC1 (72.1%) and, like m/z 73, was also successful in separating the R and DR samples. The class-to-class variance of the sugar phosphates at m/z 387 was initially obscured by run-to-run retention time variations on column 2, resulting in the R and DR differences being captured on PC2 with 25% of the variance, instead of separation on PC1. Potential reasons for the dependence of the m/z 387 mass channel on alignment are the lower S/N of the eluting compounds as well as smaller differences between the R and DR samples. At m/z 73 and 205 the differentiating peaks were of much higher S/N and with much larger differences between the R and DR samples, so slight retention time shifting was not detrimental to the class-to-class separation. Lower S/N for the sugar phosphate peaks was possibly caused by poor solubility in the derivatization solvent, i.e., pyridine. The scores plot for the aligned m/z 387 data is shown in Figure 3C. Even after alignment and normalization, sample extracts B3R and C3R (chromatograms numbered 31–34 and 55–58) overlapped with sample extracts of the DR cultures and the variance captured by PC1 was only 47%. Although the captured variance was somewhat low, successful location of distinguishing sugar phosphates was also achieved as will be shown.
Figure 3.



Resulting PC1 scores plots after submission of all 70 chromatograms to PCA following normalization to the TIC. PC1 scores plots at (A) m/z 73, (B) m/z 205 and (C) m/z387. Mass channel 387 (C) required alignment in addition to normalization. The circled B3R and C3R sample extracts in (C) overlap with the DR sample extracts.
Following distinction of the R from DR samples on the PC1 scores plot, the PC1 loadings plots were reconfigured into the 2D chromatographic space giving the retention time location of the metabolites showing the biggest difference between the R and DR samples as shown in Figures 4A–C. In order to obtain only the most useful information, a threshold was thoughtfully, yet empirically, set for each of the mass channels to include only metabolites of interest for the proof-of-principle demonstration of this analytical methodology, thus excluding artifacts or peaks of intensity too low to be definitively identified. The loading plot thresholds were set at the absolute values of 0.025, 0.002 and 0.01 for m/z 73, 205 and 387, respectively. Note that m/z 73 had the highest threshold so only the largest intensity differences between the two classes of samples would be discovered. The largest number of compounds that distinguish the DR samples from the R samples were found on the PC1 loadings at m/z 73 as shown in Figure 4A. This was expected due to the large number of peaks at this m/z. The PC1 loadings for m/z 205 revealed an additional three metabolites that were not discovered at m/z 73, one of which (erythrose) was obscured by the larger lysine peak at m/z 73 (Figure 4B). The loadings for m/z 387 revealed nine sugar phosphates that were not identified at either m/z 73 or 205. Examining the selective mass channels proved beneficial as it increased the number of compounds that served to distinguish R and DR sample extracts. Indeed, it is likely that a more exhaustive evaluation of additional m/z would yield a greater amount of information, but further analys is was beyond the scope of the current study.
Figure 4.


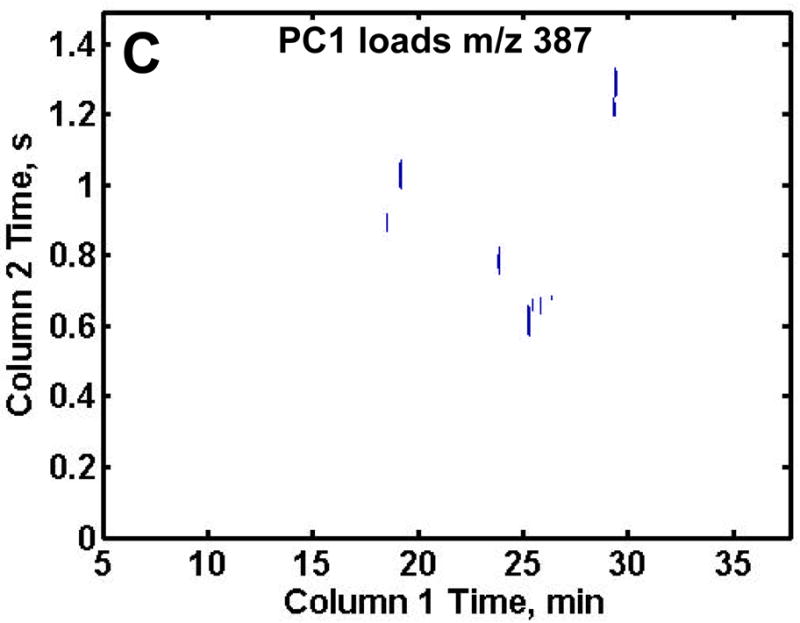
Resulting PC1 loadings plot after submission of all 70 chromatograms to PCA. The thresholds were empirically set to include only metabolites and not reagent artifacts, see text for values. PC1 loadings plot for (A) m/z 73, (B) m/z 205 and (C) m/z 387. Fifteen differentiating compounds were found at m/z 73 (A). Mass channel 205, (B) obtained an additional three metabolites and m/z 387 (C) obtained nine chromatographic peaks not found at m/z 73.
Table 1 lists the differentiating chemical components identified by PC1 in order of increasing column 1 retention time. The mass spectral match value (MV) is a measure of the similarity between the collected mass spectra and the library mass spectra. A perfect MV is 999, but for the purpose of this study, any MV greater than 750 was considered a positive identification. Approximately 77% of the metabolites have spectra that match the library spectra with a MV greater than 750. Some of the lower MV values are a result of the low S/N of the peak or of overlapping chromatographic peaks. Sugar phosphates resulted in two peaks, one each from the α and β isomers, which have been reported previously.20 The loadings values listed in column 6 of Table 1 are the values obtained from PC1 at m/z 73, 205 or 387. As the absolute value of the loadings increases, the difference between the amounts of the metabolite in the two classes of samples also increases. The nega tive loadings values correspond to the metabolites that have higher relative concentration in DR cells than in R cells, and vice versa.
Table 1.
Identification of the most highly loaded metabolites discovered from Figures 2 and 3 along with the column retention times and raw match values (MV) for the repressed (R) and derepressed (DR) samples. The metabolites listed in column 1 were detected as the methoximated and trimethylsilyl derivatives. At least five different classes of compounds are included in this list, including organic acids, amino acids, carbohydrates, sugar phosphates, and a sugar alcohol. The number following the compound name corresponds to anomers or different conformations of the same species. The MV for the C6 sugar phosphates was chosen from the first match to a C6 sugar phosphate in the LECO software. Unless otherwise stated, the loadings values are from figure 3A, m/z 73.
| Metabolites | Col. 1 Time, min | Col. 2 Time, s | MV, R | MV, DR | Load Value |
|---|---|---|---|---|---|
| Glycolic Acid | 7.5 | 0.68 | 893 ± 74 | 951 ± 8 | − 0.0024* |
| Glycerol | 11 | 0.43 | 881 ± 4 | 894 ± 1 | 0.1 |
| Threonine | 12.9 | 0.58 | 945 ± 7 | 939 ± 11 | − 0.062 |
| Arabino hexos-2-ulose | 13.3 | 0.51 | 648 ± 2 | 651 ± 3 | − 0.006* |
| Malic Acid | 14.5 | 0.72 | 896 ± 16 | 925 ± 4 | − 0.061 |
| 5-Oxoproline | 15.1 | 1.36 | 917 ± 2 | 934 ± 2 | − 0.068 |
| Glutaric Acid | 15.9 | 0.59 | 681 ± 10 | --a | 0.059 |
| Glutamic Acid | 16.4 | 0.74 | 861 ± 7 | 853 ± 5 | − 0.035 |
| α-Glycerophosphoric Acid | 18.5 | 0.89 | 782 ± 16 | 869 ± 9 | − 0.013 † |
| 3-Phosphoglycerate | 19.1 | 1.04 | 749 ± 48 | 813 ± 57 | − 0.031 † |
| Ornithine | 19.2 | 0.57 | 863 ± 5 | 844 ± 6 | − 0.075 |
| Citric Acid | 19.3 | 0.66 | 695 ± 117 | 911 ± 8 | − 0.068 |
| Glucopyranose 1 | 20.3 | 0.42 | 899 ± 13 | --a | 0.099 |
| Glucose 1 | 20.4 | 0.42 | 839 ± 8 | 845 ± 13 | 0.15 |
| Erythrose | 20.5 | 0.42 | 733 ± 5 | --a | 0.06* |
| Lysine | 20.5 | 0.59 | 901 ± 13 | 871 ± 27 | − 0.073 |
| Glucose 2 | 20.6 | 0.46 | 837 ± 13 | --a | 0.14 |
| Glucopyranose 2 | 21.3 | 0.41 | 899 ± 21 | --a | 0.11 |
| C6 Sugar Phosphate 1 | 23.9 | 0.76 | 579 ± 99 | --a | 0.014† |
| Glucose -6-Phosphate 1 | 25.3 | 0.58 | 928 ± 19 | 881 ± 29 | 0.29† |
| Glucose -6-Phosphate 2 | 25.5 | 0.63 | 829 ± 39 | 638 ± 64 | 0.066† |
| C6 Sugar Phosphate 2 | 25.9 | 0.62 | 669 ± 117 | 520 ± 108 | 0.087† |
| C6 Sugar Phosphate 3 | 26.4 | 0.645 | 705 ± 66 | --a | 0.051† |
| Fructose Diphosp hate 1 | 29.3 | 1.15 | 810 ± 29 | --a | 0.077† |
| Fructose Diphosphate 2 | 29.4 | 1.22 | 760 ± 37 | --a | 0.12† |
| Trehalose | 29.7 | 0.67 | 920 ± 27 | 953 ± 3 | −0.11 |
The 26 metabolites identified by PCA and the raw mass spectral information (Table 1) was then further analyzed to obtain quantitative information. We sought to obtain the concentration ratio of a given metabolite in the DR samples relative to in the R samples. The concentration ratios for each of the 26 metabolites were obtained by dividing the average amount of each metabolite in the DR sample extracts (NDR), by the average amount of the same metabolite in the R sample extracts (NR). One of the issues with using peak volumes from the raw chromatograms as a measure of the amount of a given metabolite is that co-eluting compounds would bias the quantitative information. Overlapping compounds could significantly contribute to the strength of the chromatographic signal at the selected m/z and interfere with the accurate quantification of the metabolite of interest. Thus, to obtain useful quantitative information, the PARAFAC algorithm was applied since it analyzes the entire 2D peak simultaneously.
Citric acid was chosen to illustrate the use of PARAFAC to obtain accurate quantitative information, in particular in the presence of overlapping compounds. Figure 5A shows the library mass spectrum for citric acid. When the raw mass spectrum, i.e., the unprocessed spectrum output from the ChromaTOF software, is compared to the library mass spectrum, a MV of 752 is obtained (Figure 5B). The raw mass spectrum has significant ion fragments at m/z 157 and 256 that are not present in the library spectrum leading to the low MV that would also result in an inaccurate peak volume. When the m/z 44 and 73–480 are submitted to the PARAFAC GUI, a MV of 858 is obtained and the ion fragments at m/z 157 and 256 are dramatically reduced from the raw spectrum (Figure 5C). This yielded fewer interfering ions and a more accurate concentration ratio. On the other hand, one issue with submitting such a large range of m/z to PARAFAC is the potentially excessive computation time and memory required. This issue can be adequately addressed if only a few selective analyte mass channels are chosen and submitted to the PARAFAC GUI instead of submitting the entire mass range. When submitting only a selected number of mass channels to PARAFAC, the user must realize that some of the signal is being ignored. However, if a concentration ratio is used, as in this report, it is independent of the number of mass channels and can be compared between analytes. Since we seek the concentration ratio of the metabolites in the DR to R sample extracts, a unique set of mass channels can be applied to each metabolite, as will be further described below. Keep in mind that the PARAFAC analysis is complemented by using the commercially available ChromaTOF software to determine the identity of the analyte. The number of mass channels submitted to PARAFAC depends on the mass spectrum of the analyte of interest and the presence of overlapping mass spectra from co-eluting compounds. Generally, the 10 most sensitive mass channels for a given metabolite are sufficient for PARAFAC, however more mass channels can be added if the overlapping components have similar mass spectra or if the analyte of interest has a low S/N. The benefit of using PARAFAC to minimize the impact of overlapping interferences is illustrated in Figure 6. Figures 6A and B show the resulting first and second dimension chromatographic peak profiles for citric acid in a R sample extract after only seven selective m/z were submitted to PARAFAC. Additionally, the resulting deconvoluted mass spectrum is shown in Figure 6C. Fewer than 10 m/z were selected for citric acid to optimize the selectivity and sensitivity of the deconvolution of citric acid from the other interfering species. The m/z 44 data was included to separate the baseline from citric acid. Interfering species overlap the peak profiles of citric acid on both the first and second GC dimensions, i.e. GC column 1 and 2, yet PARAFAC was able to obtain a pure citric acid chromatographic peak profile using the selected m/z. The mass spectral pattern in Figure 6C is in good agreement with the library spectrum in Figure 5A, and the MV increased significantly due to the exclusion of noisy mass channels from the analysis. Using PARAFAC results for citric acid in Figure 6, the reconstructed 2D GC peak profile was obtained from simple linear algebra (outer product of column 1 and 2 peak profiles), and the peak volumes from the three-dimensional peak determined, i.e., the N values. Thus, the peak volume corresponds to the sum of the individual column 2 peaks for a given analyte, obtained from all modulation periods for the elution of the analyte from column 1. For citric acid, on average, NR was 4.9 × 10−4 (13%RSD) and NDR was 6.4 × 10−3 (26%RSD), resulting in a concentration ratio NDR/NR of 13. Even though a subset of m/z was applied with the PARAFAC GUI, the concentration ratio, NDR/NR, is independent of the number of mass channels, since the same number of mass channels is used for both the DR and R sample extracts for a given metabolite. Accordingly, the adjusted MV and the m/z used to obtain the different spectra for all 26 metabolites, which were ultimately used for determining NDR and NR as in the citric acid example, are listed in Table 2. Note that the standard deviation in the R MV for citric acid decreased by a factor of approximately 20 between the raw MV, Table 1, and post PARAFAC value, Table 2. This provides additional evidence that PARAFAC has accurately obtained the citric acid mass spectrum.
Figure 5.

PARAFAC demonstration as a deconvolution and quantification tool for citric acid 4TMS in 2D chromatographic region. Mass spectra from (A) NIST main library, (B) raw and (C) PARAFAC of pure component are shown. The raw spectrum is defined as the spectrum obtained using the ChromaTOF software after baseline correction. The similarity match values (MV) for the raw (B) and PARAFAC (C) spectra are given. The raw spectrum (B) has significant extraneous fragments at 157 and 256.
Figure 6.



Demonstration of using selective mass channels in PARAFAC for quantification. Using only the 7 mass channels labeled, the pure component peak profiles for citric acid 4TMS (solid lines) on column 1 (A), and column 2 (B) was separated from five overlapping interferents (symbolized by the broken and/or dotted lines). (C) The MV and resulting mass spectrum using the selected fragments is shown. The ratio of the fragment intensities is comparable to those in the library spectrum. The protocol for choosing these 7 mass channels is described in the text.
Table 2.
PARAFAC match values (MV) for the derivatized metabolites repressed (R) and derepressed (DR) cells using the selected mass channels. The number of mass channels depended on the selectiveness of the fragments and overlapping components for each metabolite.
| Metabolite | MV, R | MV, DR | Mass Channels |
|---|---|---|---|
| Glycolic Acid | 985 ± 18 | 994 ± 4 | 43,45,66,73,74,75,147,148,177,205 |
| Glycerol | 940 ± 8 | 948 ± 1 | 45,59,73,75,103,117,129,131,133,147,177,193,238,191,205,206,218,263,293,44,219,220,299,314 |
| Threonine | 994 ± 6 | 990 ± 4 | 73,205,147,103,117,218,133,206,75,45,44,159,203,86,291,320,321 |
| Arabino hexos-2-ulose | 869 ± 9 | 894 ± 11 | 45,73,74,75,89,103,117,147,205,234 |
| Malic Acid | 982 ± 1 | 984 ± 4 | 73,147,45,233,75,55,245,133,74,148,44,217,101,307, 335,265,117,175,189,190 |
| 5-Oxoproline | 967 ± 12 | 988 ± 4 | 76,156,45,230,258,147 |
| Glutaric Acid | 928 ± 15 | --a | 73,147,185,259,349,133,44 |
| Glutamic Acid | 899 ± 14 | 884 ± 9 | 44,73,75,84,128,147,156,174,230,246,247,248,348, 363 |
| α-Glycerophosphoric Acid | 638 ± 16 | 643 ± 35 | 73,299,357,147,445,315,44 |
| 3-Phosphoglycerate | 904 ± 25 | 920 ± 26 | 73,357,299,147,103,358,101,75,315,445,44,387 |
| Ornithine | 948 ± 12 | 941 ± 3 | 73,142,174,258,420, 300 |
| Citric Acid | 991 ± 6 | 995 ± 1 | 73,147,273,347,211,465,44 |
| Glucopyranose 1 | 963 ± 15 | --a | 73,74,75,147,191,192,204,205,206,217 |
| Glucose 1 | 948 ± 6 | 930 ± 10 | 45,73,103,129,147,157,160,205,217,319 |
| Erythrose | 975 ± 12 | --a | 45,73,74,75,103,117,147,161,205,206,363 |
| Lysine | 976 ± 9 | 970 ± 9 | 44,73,128,156,174,317,434 |
| Glucose 2 | 877 ± 9 | --a | 45,73,103,129,147,157,160,205,217,319 |
| Glucopyranose 2 | 877 ± 38 | --a | 44,73,74,75,147,191,192,205,206,217,291,319,345,435 |
| C6 Sugar Phosphate 1 | 803 ± 55 | --a | 45,73,147,191,217,299,315,343,357,387 |
| Glucose -6-Phosphate 1 | 895 ± 17 | 880 ± 8 | 73,387,299,147,388,160,315,357,217,471,44,129 |
| Glucose -6-Phosphate 2 | 879 ± 19 | 858 ± 28 | 73,387,299,147,388,160,315,357,217,471,44,129 |
| C6 Sugar Phosphate 2 | 944 ± 21 | 920 ± 10 | 45,73,74,75,129,133,147,160,299,387 |
| C6 Sugar Phosphate 3 | 991 ± 1 | --a | 44,73,74,129,147,204,205,299,357,387,388 |
| Fructose Diphosphate 1 | 997 ± 2 | --a | 44,45,73,74,75,129,147,217,299,315,357,387,459 |
| Fructose Diphosphate 2 | 994 ± 5 | --a | 44,45,73,74,75,129,147,217,299,315,357,387,459 |
| Trehalose | 931 ± 20 | 974 ± 3 | 73,147,191,103,217,129,169,204,361,271,44 |
not detected in sample,
no match value available.
Before going into the details for the quantification of the 26 metabolites, we now turn our attention to a detailed evaluation of the injection-to- injection, extract-to-extract and culture-to-culture precision in the determination of NDR and NR for four metabolites: citric acid, ornithine, malic acid, and glucose 6-phosphate (Figures 7A–D). The injection precision, encompassing instrumental and analytical variation, averaged 8% for the DR sample extracts and 9% for the R sample extracts after normalization. The sample extract variability can be assessed by comparing the values obtained from the three samples of each culture. For future studies, extraction precision could be enhanced by the use of appropriate internal standards. The extraction precision averaged 11% for the DR sample extracts and 31% for the R sample extracts. The culture-to-culture variability or biological precision for these four metabolites averaged 9% for DR sample extracts and 25% for R sample extracts. The R and DR cells were grown, harvested, extracted and derivatized separately. The larger variation in the R samples is likely due to the extraction and derivatization steps and could also arise from the R cells growing faster than the DR cells. Citric acid, ornithine, and malic acid all showed insignificant extraction and culture variation (Figures 7A–C). For these metabolites no concentration overlap was seen between the DR and R sample extracts. Glucose-6-phosphate was more variable between cultures for the reasons described previously (Figure 7D).
Figure 7.




A detailed analysis of the peak volumes N obtained using PARAFAC, illustrating variation in extract-to-extract as well as culture-to-culture comparisons for R and DR samples. The DR is shown in the white bars and the R by the shaded bars. The error bars represent the injection-to- injection standard deviation. In citric acid (A), ornithine (B) and malic acid (C), the R and DR peak volumes from each of the cultures fall within the same range. In glucose-6-phosphate (D) the R and DR are not as clearly separated as in (A), (B) and (C). This is in agreement with what is shown in the scores plot of m/z 387, Figure 2C. If no error bars are shown then only two injections were quantified.
The PARAFAC quantitative results (NDR and NR) and the ratio of NDR divided by NR (labeled NDR/NR) for the 26 most variable metabolites identified by PCA were calculated using PARAFAC on selected mass channels to determine the biological variability due to the cell growth conditions (Table 3). To save analysis time, only one of the replicate sample extracts for each culture was analyzed by PARAFAC. Thus, the average biological relative standard deviation, %RSD, for the R sample extracts was approximately 40% and for DR was 21%. The %RSD values are, on average, almost double those calculated above in the detailed precision study of four of the metabolites (Figure 7), likely due to using fewer of the sample extracts (and their replicates). For these 26 differentiating metabolites the %RSD’s obtained did not hinder the class separation, and the use of fewer replicates was justified. One third of the identified metabolites were detected only in the R sample extracts. These values for biological variability are similar to previous studies of plant metabolites analyzed using GC/MS.21 When numerically defined (neither zero nor infinite), the NDR/NR concentration ratio for the 26 components listed in Table 3 ranged from 0.02 for glucose to 67 for trehalose. All analytes that were located and quantified in both classes of samples were not overloaded, so the quantification was accurate in this regard. Metabolites of lower concentration and exhibiting smaller differences between the R and DR sample extracts were identified in the more selective 205 and 387 mass channels. The results suggest that the use of selective mass channels can facilitate the identification and quantitation of the less abundant metabolites, giving impetus for future study.
Table 3.
Quantification of highest loaded derivatized metabolites discovered in figures 2 and 3. The relative concentrations (NR and NDR) are the average of the peak volumes in one sample extract from cultures A, B and C for both the repressed (R) and (DR) cells. These N values were calculated using the mass channels given in table 2.
| Metabolite | NR | NDR | %RSD, R | %RSD, DR | NDR/NR |
|---|---|---|---|---|---|
| Glycolic Acid | 4.24E-04 | 1.12E-03 | 13.0 | 4.0 | 2.6 |
| Glycerol | 1.99E-02 | 2.58E-03 | 10.7 | 13.5 | 0.1 |
| Threonine | 2.89E-03 | 9.60E-03 | 52.4 | 13.7 | 3.3 |
| Arabino hexos-2-ulose | 5.00E-03 | 6.46E-03 | 27.7 | 11.6 | 1.3 |
| Malic Acid | 4.94E-04 | 3.89E-03 | 34.8 | 15.3 | 7.9 |
| 5-Oxoproline | 6.35E-03 | 1.54E-02 | 27.3 | 16.7 | 2.4 |
| Glutaric Acid | 2.3E-03 | --a | 65.6 | --a | --b |
| Glutamic Acid | 1.41E-03 | 4.40E-03 | 33.7 | 12.7 | 3.1 |
| α-Glycerophosphoric Acid | 1.2E-04 | 3.3E-04 | 18.7 | 18.1 | 2.8 |
| 3-Phosphoglycerate | 1.10E-04 | 2.00E-04 | 22.6 | 33.3 | 1.8 |
| Ornithine | 1.90E-03 | 7.91E-03 | 24.3 | 33.6 | 4.2 |
| Citric Acid | 4.90E-04 | 6.39E-03 | 13.3 | 26.3 | 13.0 |
| Glucopyranose 1 | 5.96E-03 | --a | 58.8 | --a | --b |
| Glucose 1 | 3.28E-02 | 6.36E-04 | 33.7 | 106.2 | 0.02 |
| Erythrose | 1.09E-03 | --a | 59 | --a | --b |
| Lysine | 3.39E-03 | 1.09E-02 | 88.4 | 14.6 | 3.2 |
| Glucose 2 | 1.35E-02 | --a | 38.9 | --a | --b |
| Glucopyranose 2 | 5.88E-03 | --a | 70 | --a | --b |
| C6 Sugar Phosphate 1 | 5.46E-05 | --a | 66.3 | --a | --b |
| Glucose -6-Phosphate 1 | 4.85E-04 | 1.85E-04 | 27.7 | 14.6 | 0.4 |
| Glucose -6-Phosphate 2 | 9.71E-05 | 3.16E-05 | 22 | 8 | 0.3 |
| C6 Sugar Phosphate 2 | 8.42E-05 | 2.97E-05 | 87 | 29 | 0.4 |
| C6 Sugar Phosphate 3 | 6.50E-05 | --a | 13 | --a | --b |
| Fructose Diphosphate 1 | 1.97E-04 | --a | 12.8 | --a | --b |
| Fructose Diphosphate 2 | 3.02E-04 | --a | 12.5 | --a | --b |
| Trehalose | 1.04E-03 | 6.92E-02 | 41.3 | 1.5 | 66.8 |
not detected in sample,
undefined.
Although our main focus in the present study is to demonstrate the feasibility of GCxGC-TOFMS combined with chemometric analysis for the separation, identification and initial quantification of metabolites in yeast extracts, it is important to note that the biological results presented in Table 3 are consistent with the known regulation of the metabolic pathways operative in yeast cells growing in the two conditions tested. When a yeast cell is fermenting glucose, the glycolytic pathway is highly active. Glucose is phosphorylated to glucose-6-phosphate and then converted to fructose 1,6 diphosphate. Glucose-6-phosphate and fructose 1,6 diphosphate are both elevated in the extracts from R cells. Trehalose, a storage carbohydrate, is produced when yeast cells are grown in stressed conditions where growth rate is slow and a carbon source is abundant.22 These conditions are present when cells are metabolizing ethanol by respiration. Trehalose was found to be more abundant in the DR cells metabolizing ethanol. Two intermediates in the tricarboxylic acid (TCA) cycle, malic acid and citric acid were also found to be more abundant in the DR cells. This is consistent with the increased expression of many of the genes encoding the enzymes of the TCA cycle in accord with a more active TCA cycle in these cells.23,24 Thus, the analytical method presented identified and quantified metabolites that differentia te fermenting and respiring cells and which are consistent with the known regulation of the metabolic pathways active in these types of cells.
Conclusions
GCxGC-TOFMS analysis coupled with chemometrics software has the ability to objectively and unambiguously identify and quantitate metabolite differences between yeast cells growing in different media with straightforward and rapid preprocessing of the data. In addition to the major derivatization fragment mass channel at m/z 73, higher mass channels (e.g., 205 and 387) can be used to glean selective information. In the future, it would be highly advantageous to use of all mass channels to determine class differences, hence, a methodology to that uses Fisher ratios to do so is currently under investigation and will be the subject of a forthcoming report. As we show here, multiple mass channels are especially useful in deconvoluting complex mass spectra to resolve individual components and to determine their relative abundance. The use of third order data, two retention times and ion currents at selective masses, is especially important for working with extremely complex samples such as metabolite extracts. Utilizing these methods of analysis to obtain and interpret metabolomic data will narrow the gap between cellular genotype and phenotype.
Acknowledgments
We thank the National Institute of Diabetes and Digestive Diseases of the NIH, grant number DK67276, for support of this work.
References
- 1.Hope J, Sinha A, Prazen B, Synovec R. J Chromatogr A. 2005;1086:185–192. doi: 10.1016/j.chroma.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 2.Sinha AE, Fraga CG, Prazen BJ, Synovec RE. J Chromatogr A. 2004;1027:269–277. doi: 10.1016/j.chroma.2003.08.081. [DOI] [PubMed] [Google Scholar]
- 3.Song SM, Marriott P, Kotsos A, Drummer OH, Wynne P. Forensic Science International. 2004;143:87–101. doi: 10.1016/j.forsciint.2004.02.042. [DOI] [PubMed] [Google Scholar]
- 4.Lu X, Cai J, Kong H, Wu M, Hua R, Zhao M, Liu J, Xu G. Anal Chem. 2003;75:4441–51. doi: 10.1021/ac0264224. [DOI] [PubMed] [Google Scholar]
- 5.Focant J, Sjödin A, Turner WE, Patterson DG., Jr Anal Chem. 2004;76:6313–6320. doi: 10.1021/ac048959i. [DOI] [PubMed] [Google Scholar]
- 6.Sinha AE, Prazen BJ, Synovec RE. Anal Bioanal Chem. 2004;378:1948–1951. doi: 10.1007/s00216-004-2503-7. [DOI] [PubMed] [Google Scholar]
- 7.Hope JL, Prazen BJ, Nilsson EJ, Lidstrom ME, Synovec RE. Talanta. 2005;65:380–388. doi: 10.1016/j.talanta.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 8.Sinha AE, Hope JL, Prazen BJ, Nilsson EJ, Jack RM, Synovec REJ. Chromatogr A. 2004;1058:209–215. [PubMed] [Google Scholar]
- 9.Sinha AE, Hope JL, Prazen BJ, Fraga CG, Nilsson EJ, Synovec RE. J Chromatogr A. 2004;1056:145–154. [PubMed] [Google Scholar]
- 10.Welthagen W, Shellie RA, Spranger J, Ristow M, Zimmerman R, Fiehn O. Metabolomics. 2005;1:65–73. doi: 10.1016/j.chroma.2005.05.088. [DOI] [PubMed] [Google Scholar]
- 11.Shellie RA, Welthagen W, Zrostliková J, Spranger J, Ristow M, Fiehn O, Zimmermann R. J Chromatogr A. 2005;1086:83–90. doi: 10.1016/j.chroma.2005.05.088. [DOI] [PubMed] [Google Scholar]
- 12.Jonsson P, Gullberg J, Nordström A, Kusano M, Kowalczyk M, Sjöström M, Mortiz T. Anal Chem. 2004;76:1738–1745. doi: 10.1021/ac0352427. [DOI] [PubMed] [Google Scholar]
- 13.Jonsson P, Johansson AL, Gullberg J, Trygg JAJ, Grung B, Marklund S, Sjöström M, Antti H, Mortiz T. Anal Chem. 2005;77:5635–5642. doi: 10.1021/ac050601e. [DOI] [PubMed] [Google Scholar]
- 14.Sherman F. Methods Enzymol. 1991;194:3–21. doi: 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- 15.Castrillo JI, Hayes A, Mohammed S, Gaskell SJ, Oliver SG. Phytochemistry. 2003;62:929–937. doi: 10.1016/s0031-9422(02)00713-6. [DOI] [PubMed] [Google Scholar]
- 16.Fiehn O, Kopka J, Trethewey RN, Willmitzer L. Anal Chem. 2000;72:3573–3580. doi: 10.1021/ac991142i. [DOI] [PubMed] [Google Scholar]
- 17.Johnson KJ, Wright BW, Jarman KH, Synovec RE. J Chromatogr A. 2003;996:141–155. doi: 10.1016/s0021-9673(03)00616-2. [DOI] [PubMed] [Google Scholar]
- 18.Andersson CA, Bro R. Chemom Intell Lab Syst. 2000;52:1–4. [Google Scholar]
- 19.Jiye A, Trygg J, Gullberg J, Johansson AI, Jonsson P, Antti H, Marklunk S, Moritz T. Anal Chem. 2005;77:8086–8094. doi: 10.1021/ac051211v. [DOI] [PubMed] [Google Scholar]
- 20.Harvey DJ, Horning MG. J Chromatogr. 1973;76:51–62. doi: 10.1016/s0021-9673(01)97777-5. [DOI] [PubMed] [Google Scholar]
- 21.Fiehn O, Kopka JDörmann P, Altmann T, Trethewey RN, Willmitzer L. Nature Biotechnology. 2000;18:1157–1161. doi: 10.1038/81137. [DOI] [PubMed] [Google Scholar]
- 22.Voit EO. J Theor Biol. 2003;223:55–78. doi: 10.1016/s0022-5193(03)00072-9. [DOI] [PubMed] [Google Scholar]
- 23.Young ET, Dombek KM, Tachibana C, Ideker T. J Biol Chem. 2003;278:26146–26158. doi: 10.1074/jbc.M301981200. [DOI] [PubMed] [Google Scholar]
- 24.Wu J, Zhang N, Hayes A, Panoutsopoulou K, Oliver SG. Proc Natl Acad Sci USA. 2004;101:3148–3153. doi: 10.1073/pnas.0308321100. [DOI] [PMC free article] [PubMed] [Google Scholar]