

Abstract
A series of five-coordinate thiolate-ligated complexes [MII(tren)N4SMe2]+ (M = Mn, Fe, Co, Ni, Cu, Zn; tren = tris(2-aminoethyl)amine) are reported, and their structural, electronic, and magnetic properties are compared. Isolation of dimeric [NiII(SN4(tren)–RSdang)]2 (“dang”= dangling, uncoordinated thiolate supported by H–bonds) using the less bulky [(tren)N4S]1− ligand, pointed to the need for gem-dimethyls adjacent to the sulfur in order to sterically prevent dimerization. All of the gem-dimethyl derivatized complexes are monomeric, and with the exception of [NiII(SMe2N4(tren)]+, are isostructural and adopt a tetragonally distorted trigonal bipyramidal geometry favored by ligand constraints. The nickel complex uniquely adopts an approximately ideal square pyramidal geometry, and resembles the active site of Ni-superoxide dismutase (Ni-SOD). Even in coordinating solvents such as MeCN, only five-coordinate structures are observed. The MII–S thiolate bonds systematically decrease in length across the series (Mn–S > Fe–S > Co–S > Ni–S ~ Cu–S < Zn–S) with exceptions occurring upon the occupation of σ* orbitals. The copper complex, [CuII(SMe2N4(tren)]+, represents a rare example of a stable CuII–thiolate, and models the perturbed “green” copper site of nitrite reductase. In contrast to the intensely colored, low–spin Fe(III)-thiolates, the M(II)-thiolates described herein are colorless to moderately colored, and high–spin (in cases where more than one spin-state is possible), reflecting the poorer energy match between the metal d– and sulfur–orbitals upon reduction of the metal ion. As the d–orbitals drop in energy proceeding across the across the series M2+ (M= Mn, Fe, Co, Ni, Cu), the sulfur-to-metal charge transfer transition moves into the visible region, and the redox potentials cathodically shift. The reduced M+1 oxidation state is only accessible with copper, and the more oxidized M+4 oxidation state is only accessible for manganese.
Introduction
A comprehensive understanding of the influence of thiolate ligands on the electronic, magnetic, and reactivity properties of first-row transition metal ions is essential if we are to fully understand why nature utilizes cysteinate residues to promote specific biological functions in metalloenzymes.2–4 Cysteinate-ligated metalloenzymes and metalloproteins promote a number of critical biological processes including electron transfer,5–7 toxic radical scavenging,8–13 the degradation and excretion of toxic substances,14 strong bond activation,2,14,15 enzyme activation,16,17 and the formation of key metabolic intermediates.18,19 Metal ions are essential components of biological systems since they readily promote reactions (such as electron transfer, strong bond activation, and controlled radical reactions) that would otherwise be impossible.2–4 Mechanistic pathways in metalloenzymes are controlled by the metal ion's electronic and magnetic properties, which are tuned by the coordinated ligands, and subtly altered by H–bonding interactions within the protein, or interactions with nearby metal ions.20 The amino-acid hetero-atoms which connect metal ions to proteins can have a dramatic influence on key properties such as redox potential, metal ion Lewis acidity,21 electron-transfer rates, and HOMO/LUMO orbital energies. For example, replacing the NHis ligand in hemoglobin with a Scys in cytochrome P450, dramatically alters the function from that of a dioxygen carrier, to one which activates O2 and catalyzes the oxidation of unactivated hydrocarbons.22 A correlation between structure, key properties, and function can be most readily obtained by building small molecular analogues of these sites.3 Multidentate ligands are generally required to maintain a relatively rigid structure. However, the synthesis of multidentate ligands incorporating thiolates can be challenging23 given the relative instability of the thiolate group towards both electrophilic attack and oxidative damage. Furthermore, higher-valent transition–metal thiolates can be difficult to synthesize since they are prone to autoreduction and disulfide formation. The synthesis of complexes containing an open-coordination site, to allow for the possible binding of biologically relevant substrates, is especially difficult, and requires the use of steric bulk, non–coordinating solvents, and strict avoidance of potentially coordinating counterions.24–27
Recently we reported a rare example of a coordinatively unsaturated thiolate-ligated FeII complex [FeII(SMe2N4(tren))](PF6) (1), that reacts with HO2 to afford six-coordinate [FeIII(SMe2N4(tren))(MeCN)]2+ via a peroxide intermediate [FeIII(SMe2N4(tren))(OOH)]+ (2).28,29 Thiolate–ligated 1 maintains a five coordinate structure, despite being crystallized from a coordinating solvent (MeCN). Considering the relative scarcity of five–coordinate complexes containing first–row transition-metals in a heteroleptic thiolate/nitrogen coordination environment,23 and in particular Cu(II) thiolates, we decided to prepare a series of first–row transition–metal complexes derived from our [SMe2N4(tren)]1− ligand. Herein we describe synthetic procedures affording the series of complexes [MII(SMe2N4(tren))]+ (MII = MnII, CoII, NiII, CuII, and ZnII), and examine and compare their structural, electronic, and magnetic properties.
Experimental
General Methods
All reactions were performed using standard Schlenk techniques under an atmosphere of dinitrogen. Reagents were all obtained from Aldrich Chemical Company and used without further purifications. Solvents were purified through standard procedures.30 3-methyl-3-mercapto-2-butanone was prepared according to a published procedure.31 NMR spectra were recorded on a Bruker DPX 500 FTNMR spectrometer and referenced to the residual protio solvent. Temperatures were determined using van Geets’ methods.321H NMR chemical shifts (δ) are reported in parts per million (ppm) and coupling constants (J) are reported in Hz. IR spectra were obtained as KBr pellets and are recorded on a Perkins Elmer 1700 FTIR. EPR spectra were obtained using a Varian CW-EPR spectrometer at 4 K equipped with an Oxford helium cryostat. Cyclic voltammograms were recorded in MeCN solutions with Bu4N(PF6) (0.100 M) as the supporting electrolyte, using a EG&G Princeton Applied Research potentiostat with a glassy carbon working electrode, an SCE reference electrode, and a platinum auxiliary electrode. Electronic absorption spectra were recorded using a Hewlett-Packard 8453 diode array spectrometer. Magnetic moments were determined via Evans’ Method corrected for superconducting solenoids at 301.5 MHz33,34 or by using a Quantum Design SQUID Mangnetometer. Elemental analyses were performed by Galbraith Laboratories (Knoxville, TN) and Atlantic Microlab Inc (Norcross, GA).
Preparation of of [NiII(SN4(tren)–RSdang)]2·4H2O (3; “dang”= dangling, uncoordinated thiolate)
Nickel(II) acetate tetrahydrate (0.75 g, 3 mmol) in methanol (20 mL) was added slowly, via cannula, to a stirred solution (20 mL) of 2,5 dihydroxy- 2,5 dimethyl- 1,4 dithiane35 (0.54 g, 3 mmol) in methanol at 0°C. The solution immediately turned red/brown and, by the end of the addition, began to deposit a dark red-brown microcrystalline solid. After an hour of stirring at 0°C, sodium hydroxide (0.24 g, 6 mmol) in 10 mL. of methanol was added, followed by tris(2-aminoethyl)amine (tren) (0.44 g, 3 mmol), also in 10 mL of methanol. This caused the precipitate to dissolve, and the solution to turn dark olive-brown. The reaction mixture was then cooled overnight (5°C), and the volume of the resulting deep blue-green solution was reduced under vacuum to ca. 15 mL. This resulted in the formation of a light blue precipitate that was then redissolved by the addition of 15 mL of H2O, with warming. Diethyl ether was then added until the solution was saturated, and the resulting solution was placed in a −25°C freezer for 2–3 days to afford 438 mg (40%) of dark blue crystals of 3. Elemental Analysis for Ni2C24H48N8S4·2H2O Calcd: C, 38.99; H, 7.09; N, 15.15. Found: C, 37.90; H, 6.29; N, 13.77. μeff(D2O)/Ni = 2.89 μB. μeff(MeOD)/Ni = 3.24 μB. Absorption spectrum (MeOH): λmax(εM) 584(32), 884(30) nm. Diffuse reflectance spectrum (solid state): λmax(εM) 402, 578, 883 nm. Absorption spectrum (H2O): λmax(εM) 570(41), 808(25) nm. Diffuse reflectance spectrum (solid state): λmax 402, 578, 883 nm. IR(neat solid): 1670(νC=N), 1616(νC=N) cm−1.
Preparation of [NiII(SMe2N4(tren))](PF6) (4)
Sodium methoxide (216 mg, 4.00 mmol), 3-methyl-3-mercapto-2-butanone (472 mg, 4.00 mmol), and tren (585 mg, 4.00 mmol) were dissolved in 50 mL of MeOH and cooled to −40° C. Anhydrous NiCl2 was then slurried in 10 mL of cold MeOH and the ligand solution was then added dropwise to the metal resulting in a green solution. This was allowed to rise to room temperature and stir for 4 days and the solution became a dark rust color. The solids were removed by passing the solution through a plug of Celite, the methanol was removed under reduced pressure, and the solution dissolved in an MeCN solution of NaPF6 (678 mg, 4.10 mmols) and stirred for 5 hours. The resulting NaCl was removed via filtration through Celite, the MeCN was concentrated to ~ 10 mL and diethyl ether (30 mL) was layered over top of the solution. After six days at −40° C red crystals of 4 had formed (1.04 g, 57.0% yield). Electronic Absorption Spectrum (MeCN): λmax (nm) (ε (M−1 cm−1)): 275 (2394), 410 (330). IR: νC=N: 1605 cm−1. Redox potential (MeCN vs. SCE): Epa(NiIII/II)= +325 mV (irreversible). Magnetic Moment (302.1 K, MeCN solution): 2.86 μB. Elemental Analysis for NiC11H25N4SPF6 Calcd: C, 29.24; H, 5.61; N, 12.48. Found: C, 29.00; H, 5.71; N, 12.63.
Preparation of [MnII(SMe2N4(tren))](PF6) (5)
Manganese sulfate monohydrate (676 mg, 4.00 mmol) was slurried in methanol (10 mL). To this was added 3-methyl-3-mercapto-2-butanone (472 mg, 4.00 mmol) and sodium methoxide (216 mg, 4.00 mmol) in methanol (25 mL). While stirring, tris(2-aminoethyl)amine (tren) (585 mg, 4.00 mmol) was added dropwise. After 5 minutes, sodium hexafluorophosphate (672 mg, 4.00 mmol) was added. The solution was stirred overnight at room temperature, the solvent evaporated, and the residue redissolved in acetonitrile (20 mL). The solution was stirred for 2 hours and filtered again through Celite. Ether (10 mL) was carefully layered on top and the colorless solution was cooled to −40°C. Overnight, pale colorless needles of 5 had formed which were suitable for x-ray diffraction (527 mg, 29.6% yield). Electronic Absorption Spectrum (MeCN): λmax (nm) (ε(M−1cm−1): 240 (2911). IR: νC=N: 1647 cm−1. Redox Potential (MeCN vs. SCE): Ep,a(MnIII/II) = + 270 mV (quasi-reversible), E1/2(MnIV/III) +705 mV. Magnetic Moment (solid state): 6.01 μB.
Preparation of [CoII(SMe2N4(tren))](PF6) (6)
Sodium hydroxide (120 mg, 3.00 mmols) was dissolved in 120 mL of MeOH and 3-methyl-3-mercapto-2-butanone (335 mg, 3.00 mmols) was added dropwise. To this, cobalt(II) chloride hexahydrate (714 mg, 3.00 mmols) in 15 mL of MeOH was added dropwise. After stirring for 10 min, tris(2-aminoethyl)amine (tren) (440 mg, 3.00 mmols) was added dropwise, followed by the addition of 680 mg of KPF6 (3.60 mmols). The solution was stirred at room temperature for 24 hours, at which time the solids were filtered through a plug of Celite. The volatiles were removed under reduced pressure and the resulting green powder was dissolved in 15 mL of MeCN. Diethyl ether (50 mL) was layered on top of this green solution, and the two layers were allowed to diffuse together over several days affording 1.26 g of 6 (93.1% yield). Electronic Absorption Spectrum (MeCN): λmax (nm) (ε (M−1 cm−1)): 195 (12500), 363 (950), 465 (160), 600 (130). IR: νC=N: 1646 cm−1. Redox Potential (MeCN vs. SCE): Ep,a(CoIII/II)= +270 mV (irreversible). Magnetic Moment (MeCN, 294 K): 4.05 μB. Elemental Analysis for CoC11H25N4SPF6 Calcd: C, 29.40; H, 5.61; N, 12.47. Found: C, 28.93; H, 5.56; N, 12.49.
Preparation of [CuII(SMe2N4(tren))](PF6) (7)
Sodium methoxide (216 mg, 4.00 mmols) was dissolved in 20 mL methanol followed by the addition of 472 mg of 3-mercapto-3-methyl-2-butanone (4.00 mmols) and 585 mg of tren (4.00 mmols). Copper (II) acetate monohydrate (799 mg, 4.00 mmols) was then dissolved in 10 mL of methanol and added dropwise to the ligand solution. After stirring for 10 min, 678 mg of sodium hexaflurophosphate (4.01 mmols) in 10 mL of methanol was added, and the solution was stirred overnight at room temperature. The resulting dark green solution was filtered to remove all insoluble products, the methanol was removed, and the green powder dissolved in 10 mL of MeCN. This was filtered and layered with 40 mL of diethyl ether. The two layers were allowed to diffuse together overnight at room temperature to afford dark green crystals of 7 (687 mg, 33.7% yield). Electronic Absorption Spectrum (MeCN): λmax (nm) (ε (M−1 cm−1)): 216 (3280), 236 (3400), 382 (3340), 618 (315). EPR spectrum (MeOH/EtOH glass): g|| = 2.17 (A||(Cu)= 150 × 10−4 cm−1), g⊥ = 2.07. IR: νC=N: 1659 cm−1. Redox potential (MeCN vs. SCE): E1/2 (CuII/I)= −703 mV (quasi-reversible). Epa(CuIII/II)= +690 mV (irreversible). Magnetic Moment (302.1 K, MeCN solution): 1.97 μB. Elemental Analysis for CuC11H25N4SPF6 Calcd: C, 29.11; H, 5.55; N, 12.34. Found: C, 29.02; H, 5.54; N, 11.97.
Preparation of [ZnII(SMe2N4(tren))](PF6) (8)
This was prepared in an analogous manner as 7 except 541 mg of zinc (II) chloride was used in place of the copper (II) acetate. This afforded 8 as large yellow tinted crystals (1.79 g, 98.4% yield). 1H NMR (MeCN-d3): 3.55 (2 H, t, J = 8 Hz, CH2-N=C), 3.28 (2 H, t, J = 8 Hz, CH2-N(CH2)2), 2.94 (m, 4H, (CH2)2NCH2), 2.86 (m, 2H, (HCH)NH2, 2.74 (m, 2H, (HCH)NH2), 2.20 (s, 3H, CH3), 1.61 (s, 6H, (CH3)2C). Electronic Absorption Spectrum (MeCN): 219 (4 800), 231 (sh), 269 (394). IR νC=N: 1653 cm−1. Elemental Analysis for ZnC11H25N4SPF6 Calcd: C, 28.99; H, 5.53; N, 12.29. Found: C, 29.08; H, 5.55; N, 12.41.
X-ray Crystallography
A dark blue 0.25 × 0.30 × 0.35 mm block of 3 was mounted on a glass capillary with epoxy. Data was collected at 25 °C on an Enraf–Nonius CAD4 (MoKα, λ= 0.71073 Å). Twenty-five reflections in the range 2θ= 30–40° wer found and centered to determine the cell constants and orientation matrix. The data were corrected for Lorentz and polarization effects using MOLen, and an empirical absorption correction, based on a set of ψ scans (μ= 14.30 cm–1), was applied. The crystal showed no signs of decay. E–statistics (XPREP, SHELXTL PLUS) suggested the centric triclinic space group P1 (bar), and this space group was confirmed by satisfactory refinement of the structure to low error indices. The structure was solved by locating the Ni atom in a Patterson map, and confirmed using a direct methods solution. The remaining non-hydrogen atoms were located from successive difference Fourier map calculations. Atom scattering factors were taken from a standard source.36 Hydrogen atoms positions were determined from a difference map, and refined at fixed positions using a riding model. The 4128 observed reflections with F>4σ(F) were used in the refinement of 3, and the final full–matrix least-squares refinement converged at R(Rw)= 0.0458 with 185 parameters. A final difference map showed no peaks larger than 0.75 e−/Å3. The asymmetric unit of 3 consists of one half of a Ni2 dimer and two H2O solvent molecules, one full occupancy, and one H2O 1/4 occupancy.
A red 0.23 × 0.16 × 0.08 mm plate of 4 was mounted on a glass capillary with oil. Data was collected at −137 °C on a Nonius Kappa CCD diffractometer. The crystal-to-detector distance was set to 30 mm and exposure time was 20 seconds per degree for all data sets with a scan width of 1.8°. The data collection was 40.8% complete to 28.68° in ϑ. A total of 52638 partial and complete reflections were collected covering the indices, h = −22 to 22, k = −9 to 9, l = −26 to 26. 3519 reflections were symmetry independent and the Rint = 0.065 indicated that the data was average (average quality = 0.07). Indexing and unit cell refinements indicated a monoclinic C lattice in the space group C2/c (No. 15).
A clear 0.59 × 0.26 × 0.26 mm needle of 5 was mounted on a glass capillary with oil. Data was collected at −143 °C. on a Nonius Kappa CCD diffractometer. The crystal-to-detector distance was set to 30 mm and exposure time was 30 seconds per degree for all data sets with a scan width of 1°. The data collection was 86.5% complete to 25° in ϑ. A total of 148720 partial and complete reflections were collected covering the indices, h = −8 to 9, k = −15 to 15, l = −21 to 21. 2733 reflections were symmetry independent and the Rint = 0.0658 indicated that the data was average (average quality = 0.07). Indexing and unit cell refinements indicated a orthorhombic P lattice in the space group P212121 (No. 19). Although the sample was sufficiently large it showed a high mosaicity causing the diffraction for higher theta angles to be difficult to separate by the numerical algorithm employed in the KAPP CCD spectrometer. As a result, more reflections at higher angle in theta turned out to be too much overlapping to be counted in the data statistics. The total diffraction angle 2theta is sufficiently high to insure proper evaluation of thermal parameters.
A red/green crystal prism of 6, cut down to 0.30 × 0.38 × 0.5 mm, was mounted on a glass capillary with epoxy. The crystals were strongly pleochroic, changing from red-brown to grass green. Data was collected at −145 °C on a Nonius Kappa CCD diffractometer. The crystal-to-detector distance was set to 40 mm and exposure time was 30 seconds per degree for all data sets with a scan width of 1.8°. The data collection was 77.6% complete to 28.26° in ϑ. A total of 5046 partial and complete reflections were collected covering the indices, h = −6 to 10, k = −10 to 12, l = −21 to 21. 3241 reflections were symmetry independent and the Rint = 0.047 indicated that the data was good. Indexing and unit cell refinements indicated a orthorhombic P lattice in the space group P212121 (No. 19).
A green crystal block of 7, cut down to 0.22 × 0.19 × 0.10 mm, was mounted on a glass capillary with oil. Data was collected at −143 °C. The crystal-to-detector distance was set to 30 mm and exposure time was 15 seconds per degree for all data sets with a scan width of 1.9°. The data collection was 93.8% complete to 26.34° in ϑ. A total of 13700 partial and complete reflections were collected covering the indices, h = 0 to 10, k = 0 to 13, l = −22 to 22. 3509 reflections were symmetry independent and the Rint = 0.0619 indicated that the data was of average quality. Indexing and unit cell refinements indicated a orthorhombic P lattice in the space group P212121 (No. 18).
A clear crystal block of 8, cut down to 0.48 × 0.43 × 0.31 mm, was mounted on a glass capillary with oil. Data was collected at −143 °C on a Nonius Kappa CCD diffractometer. The crystal-to-detector distance was set to 30 mm and exposure time was 15 seconds per degree for all data sets with a scan width of 1 °. The data collection was 96% complete to 28.26° in ϑ. A total of 12740 partial and complete reflections were collected covering the indices, h = 0 to 10, k = 0 to 16, l = −22 to 23. 4221 reflections were symmetry independent and the Rint = 0.0492 indicated that the data was of better than average quality. Indexing and unit cell refinements indicated a orthorhombic P lattice in the space group P212121 (No. 19).
The data for 4, 5, 6, 7, and 8 were all integrated and scaled using hkl-SCALEPACK, and an absorption correction was performed using SORTAV. Solution by direct methods (SIR97; default 4) produced a complete heavy atom phasing model consistent with the proposed structure. All non-hydrogen atoms were refined anisotropically by full-matrix least-squares methods, while all hydrogen atoms were located using a riding model. Crystal data for 4–8 is presented in Table 1. Selected bond distances and angles are assembled in Table 2.
Table 1.
Crystal Data, Intensity Collectionsa and Structure Refinement Parameters for [NiII(SN4(tren)–RSdang)]2·4H2O (3), [NiII(SMe2N4(tren))]Cl (4), [MnII(SMe2N4(tren))](PF6) (5), [CoII(SMe2N4(tren))](PF6) (6), [CuII(SMe2N4(tren))](PF6) (7), and [ZnII(SMe2N4(tren))]+ (8).
| 3 | 4 | 5 | 6 | 7 | 8 | |
|---|---|---|---|---|---|---|
| Formula | Ni2C24H52N8S4O2.5 | NiC11H25ClN4S | MnC11H25N4F6PS | CoC11H25N4F6PS | CuC11H25N4F6PS | ZnC11H25N4F6PS |
| MW | 738.4 | 339.57 | 445.32 | 449.31 | 453.92 | 455.75 |
| T, K | 295 | 161(2) | 130(2) | 153(2) | 130(2) | 130(2) |
| unit cell | triclinic | monoclinic | orthorhombic | Orthorhombic | orthorhombic | orthorhombic |
| a, Å | 8.936(2) | 19.1240(3) | 8.194(4) | 8.1770(6) | 8.1120(5) | 8.1600(3) |
| b, Å | 10.152(2) | 7.7534(3) | 12.831(5) | 12.7180(6) | 12.7400(4) | 12.7280(4) |
| c, Å | 10.458(2) | 21.3505(7) | 18.198(8) | 17.977(1) | 18.031(1) | 17.9620(5) |
| α, deg | 61.71(3) | 90 | 90 | 90 | 90 | 90 |
| β, deg | 81.80(3) | 100.139(2) | 90 | 90 | 90 | 90 |
| γ, deg | 87.39(3) | 90 | 90 | 90 | 90 | 90 |
| V, Å3 | 826.2(4) | 3116.3(2) | 1913(1) | 1869.5(2) | 1863.4(2) | 1865.5(1) |
| Z | 1 | 8 | 4 | 4 | 4 | 4 |
| d(calc), g/cm3 | 1.484 | 1.448 | 1.546 | 1.596 | 1.618 | 1.572 |
| space group | P1 bar | C2/c | P212121 | P212121 | P212121 | P212121 |
| R | 0.0458b | 0.0420b | 0.0583b | 0.0375b | 0.0417b | 0.0280 b |
| Rw | 0.064c | 0.1221c | 0.1592c | 0.1030c | 0.0916d | 0.0691 d |
| GOF | 1.22 | 0.789 | 1.04 | 1.09 | 0.995 | 1.01 |
Table 2.
Selected Bond Distances (Å) and Bond Angles (deg) for Five Coordinate [MnII(SMe2N4(tren))]+ (5), [FeII(SMe2N4(tren))]+ (1), [CoII(SMe2N4(tren))]+ (6), [NiII(SMe2N4(tren))]+ (4), [CuII(SMe2N4(tren))]+ (7), and [ZnII(SMe2N4(tren))]+ (8).
| 5 | 1 | 6 | 4 | 7 | 8 | |
|---|---|---|---|---|---|---|
| M-S(1) | 2.412(3) | 2.3281(9) | 2.297(1) | 2.256(1) | 2.254(1) | 2.3220(6) |
| M-N(1) | 2.166(8) | 2.091(3) | 2.037(3) | 2.000(3) | 1.977(3) | 2.074(1) |
| M-N(2) | 2.334(8) | 2.268(3) | 2.239(3) | 2.097(3) | 2.082(3) | 2.301(2) |
| M-N(3) | 2.201(8) | 2.131(3) | 2.087(3) | 2.048(3) | 2.068(3) | 2.083(2) |
| M-N(4) | 2.198(8) | 2.117(3) | 2.081(3) | 2.077(3) | 2.171(4) | 2.075(2) |
| N(1)-C(#)imine | 1.295(11) | 1.277(4) | 1.272(5) | 1.292(4) | 1.275(5) | 1.272(3) |
| S(1)-M-N(1) | 81.9(3) | 84.02(8) | 85.8(1) | 85.37(8) | 86.3(1) | 85.35(6) |
| S(1)-M-N(2) | 158.8(2) | 163.02(7) | 165.39(8) | 159.34(9) | 170.27(9) | 163.88(5) |
| S(1)-M-N(3) | 115.0(3) | 108.81(8) | 107.0(1) | 115.22(8) | 103.29(9) | 106.86(6) |
| S(1)-M-N(4) | 109.1(2) | 107.86(9) | 107.8(1) | 101.04(8) | 100.68(9) | 108.74(6) |
| N(1)-M-N(3) | 125.0(3) | 124.9(1) | 125.9(1) | 158.7(1) | 135.9(2) | 126.41(8) |
| N(1)-M-N(4) | 109.5(3) | 110.3(1) | 111.3(1) | 96.2(1) | 111.5(1) | 110.83(8) |
| N(3)-M-N(4) | 112.2(3) | 115.2(1) | 113.6(1) | 99.31(1) | 108.8(2) | 113.32(8) |
| τ-value | 0.564 | 0.635 | 0.658 | 0.011 | 0.573 | 0.624 |
Results and Discussion
Gem-dimethyls Prevent Dimerization
Monomeric metal thiolates are non–trivial to synthesize, since they are prone to dimerization and auto-redox processes affording disulfides. Oligomerization is especially prevalent with coordinatively unsaturated complexes. Steric bulk can, however, be used to circumvent these problematic side–reactions.37,38 For example, by incorporating gem–dimethyls adjacent to the thiolates of [FeII(SMe2N4(tren))](PF6) (1),29 [FeIII(S2Me2N3(Pr,Pr))]+,31 [FeIII(S2Me2N3(Et,Pr))]+,39 and [FeII(Py2S(X)] (X= Cl −, Br −),23 monomeric five-coordinate structures are obtained. The tripodal tris(2-aminoethyl)amine (tren) ligand was used as a scaffold in 1 so as to favor a pyramidal five coordinate structure likely to be reactive. Schrock has shown, for example, that a bulky aromatic substituted deprotonated tren-derivative affords a four coordinate molybdenum complex that reacts with dinitrogen to afford a functional nitrogenase model.40 By appending a gem-dimethyl protected thiolate arm onto the tren ligand scaffold we have been able to generate a functional SOR model containing Fe2+.28,41 In the absence of these gem-dimethyls, dimerization was found to occur with the analogous Ni2+ complex to afford [NiII(SN4(tren)–RSdang)]2 (3; “dang”= dangling, uncoordinated thiolate; Figure 1), obtained via a Ni2+ templated Schiff–base condensation between tris(2-aminoethyl)amine (tren) and α-thioacetone (CH3C(O)CH2SH). Isolation of 3 requires the use of protic solvents (Figure S–1), the reason for which became apparent upon inspection of the packing diagram (Figure S–2). Each half of the dimeric unit contains a “dangling” thiolate (S(2) and S(2a)) that remains uncoordinated to the metal ion. This is quite unusual. Although dangling thioethers42,43 and dangling protonated thiols44 have been observed in transition-metal chemistry, we know of no other examples of structures containing a dangling thiolate. Hydrogen bonds involving co–crystallized water molecules (Figure S–1; Table S–4; S(2) ···H2O(1)= 2.504 Å; S(2)–H–O(1)= 154.7°; S(2) ···H2O(1a)= 2.524 Å; S(2)–H–O(1a)= 159.1°), as well as intra–and intermolecular N–H protons (Figure S–2), stabilize the anionic charge of the “dangling” thiolates S(2) and S(2a) (Figure 1). Ligand constraints, involving a alkyl thiolate connected to a conformationaly-restricted sp2 hybridized carbon C(8) (Figure 1), appear to be responsible for the dangling thiolate arms. Although the dangling arm is free to rotate about the C(9)-C(8) bond, restricted rotation about the imine C(8)=N(3) π-bond prevents coordination to the apical site (occupied by bridging S(1a)), and restricts binding to the equatorial site already occupied by S(1). In other words, the two thiolates S(1) and S(2) compete for binding to the same equatorial site.
Figure 1.
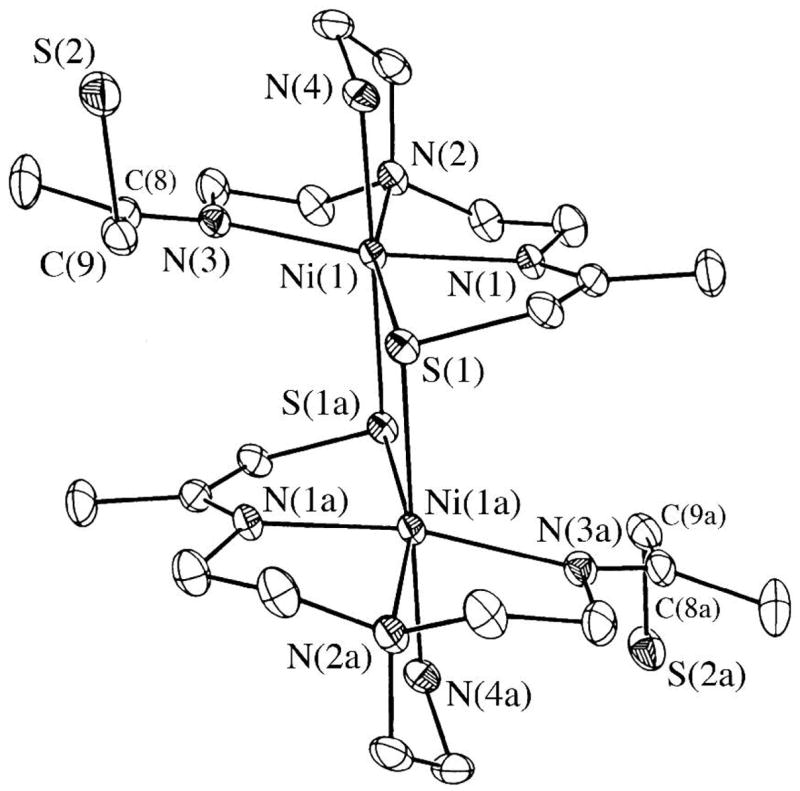
ORTEP of dimeric [NiII(SN4(tren)–RSdang)]2 · 2H2O (3) showing the atom labeling scheme. All hydrogen atoms have been removed for clarity.
The two halves of dimeric 3 are related by a crystallographic center of symmetry, with a Ni(1)....Ni(1a) separation of 3.606 Å. The Ni+2 ions of 3 are six-coordinate in distorted octahedral sites comprised of two thiolates (S(1) and S(1a)), both of which are bridging, two imines N(1) and N(3), and two amines N(2) and N(4), one of which (N(4)) is trans to the apical thiolate S(1a) (Figure 1). The Ni-S distances (Table 2) fall in the normal range (2.41– 2.54 Å) for six-coordinate Ni-thiolates,45–48 and the Ni(1)-S(1a) bridging distance (2.470(1) Å) is only 0.04 Å longer than the chelated Ni(1)-S(1) distance (2.434(1) Å), indicating that the bridge connecting the two halves of the dimer is moderately strong. In fact, the Ni(1) ion sits 0.037 Å below the S(1)N(1)N(2)N(3) plane towards S(1a). The imines of 3 (N(3) and N(1); Figure 1) are differentiated by their adjacent thiolate arms (metal-bound vs dangling and uncoordinated), and have noticeably different metrical parameters. The Ni–N(3) imine bond (2.192(3)Å) is exceptionally long relative to Ni-N(1) (2.073(2) Å), and most six-coordinate Ni–N(imine) bonds (usual range: 1.98– 2.08Å).49–53 Signs of strain within the dangling C(8)=N(3) imine bond, include its distorted bond angles (range 111.1°–132.5° deviates significantly from ideal 120°), long bond distance (1.303(4) Å (C(8)=N(3)) versus 1.278(5) Å (chelated C(2)=N(1)) and low νC=N stretching frequency (1670 cm−1 (chelated imine) versus 1616 cm−1 (dangling imine); KBr pellet). Since a co-crystallized H2O appears to be essential for the stabilization of the uncoordinated thiolate S(2), it may be that the packing forces required for the inclusion of H2O puts strain on the imine.
Based on its and electronic spectral properties in the solid state (diffuse reflectance data in experimental section) versus solution, it appears the dimeric structure of 3 persists in solution. However, the magnetic data (vide infra) indicate that the two Ni2+ ions are weakly coupled at ambient temperatures. Also, the close contact (3.69 Å) between C(3) and C(8a) (Figure 1), and C(1) and the midpoint between C(6a) and C(7a) (Figure 2), as well as the slight bending of the C(3) methyl group away from C(8a) in structure 3, suggested that if one were to insert a bulkier substituent on either the C(3) or C(1) positions (Figure 1), then dimerization would be prevented. With this in mind, we synthesized the gem-dimethyl derivative of α-thioacetone, 3-thio-3-methyl-2-butanone (CH3C(O)C(CH3)2SH).31 Condensation of this bulkier thioketone with tren at a Ni2+ template afforded monomeric [NiII(SMe2N4(tren))]+ (4; Figure 3), which resembles the active site of nickel-containing superoxide dismutase (Ni-SOD).11,12 This method also afforded monomeric derivatives containing Mn2+, Fe2+, Co2+, Cu2+, and Zn2+, which are isostructural to one another (Table 1), but structurally distinct from the Ni2+ complex.
Figure 2.
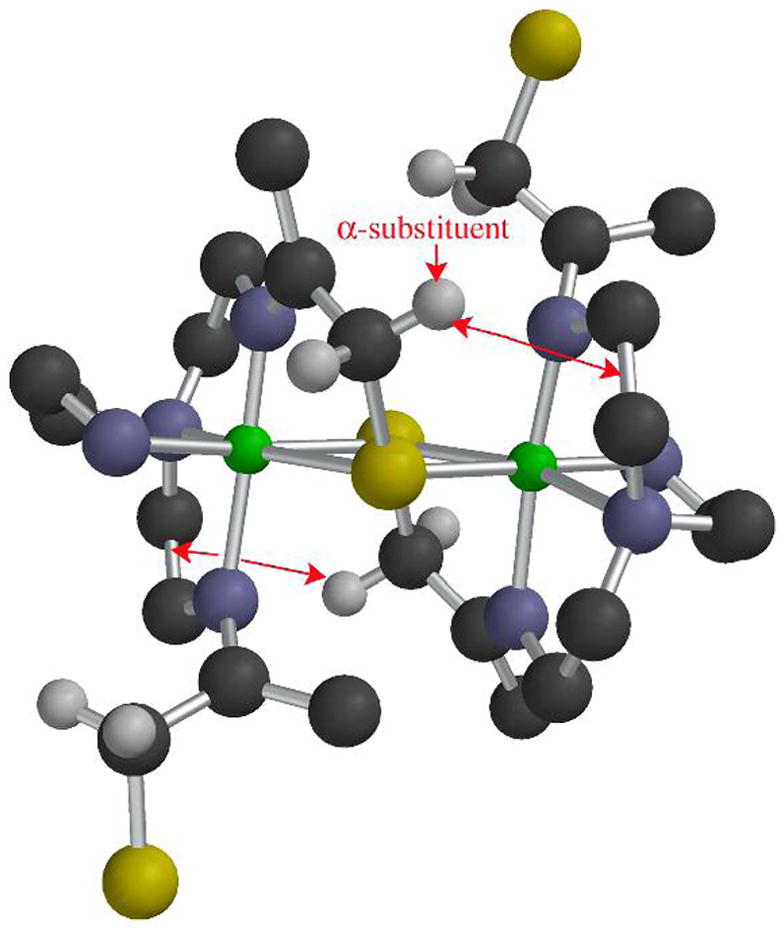
Ball-and-stick diagram of dimeric [NiII(SN4(tren)–RS dang)]2 (3) showing the intramolecular contacts between the α-substituent adjacent to the sulfur, and the midpoint between a C–C bond on the other half of the dimer. The model suggests that replacement of the α-hydrogens with α-methyls would create steric crowding that disfavors dimer formation.
Figure 3.

ORTEP of the cation of monomeric [NiII(SMe2N4(tren))]Cl (4), [MnII(SMe2N4(tren))](PF6) (5), [CoII(SMe2N4(tren))](PF6) (6) and [ZnII(SMe2N4(tren))](PF6) (8) showing the atom labeling scheme. All hydrogen atoms, with the exception of the amine hydrogens, have been removed for clarity.
Synthesis of [MII(SMe2N4(tren))]+, M= Ni(4), Mn(5), Co(6), Cu(7), Zn(8)
A metal–templated Schiff–base condensation, identical to that used previously to synthesize [FeII(SMe2N4(tren))](PF6) (1),29 was used to synthesize [MII(SMe2N4(tren))](PF6) (M = Mn (5), Co (6), Ni(4), Cu (7), and Zn (8)). The Cu2+ and Zn2+ derivatives form readily at room temperature regardless of the order of reagent addition (amine, thioketone, and M2+ salt). The Mn2+ and Fe2+ complexes are extremely oxygen sensitive, and colorless solutions develop a purple or red color, respectively, in an inert atmosphere dry box with as little as, or greater than, 2 ppm of O2, as determined using a Teledyne O2 analyzer. Although less sensitive to O2, the Ni2+ and Co2+ complexes 4 and 6 were found to be extremely sensitive to the order of reagent addition, and/or temperature. In order to obtain reasonable yields of 6, the metal salt, CoCl2·6H2O, must be combined with the thioketone prior to the addition of the amine. This initially affords an insoluble purple precipitate that redissolves upon the addition of tren to afford a green homogeneous solution. In order to avoid black intractable solids in the preparation of 4, reactions must be done at low temperatures ≤ −40° C. If the NiCl2 salt is added to the ligand, as opposed to adding the ligand to the NiCl2 solution, then significant amounts of black insoluble solids form. Extreme sensitivity to reaction conditions was also observed in the preparation of [NiII(S2Me2N3(Pr,Pr))]+, a water–soluble nickel thiolate complex previously reported by our group.54 Isolation of single crystals of the Mn2+, Co2+, Cu2+, and Zn2+ derivatives allowed us to determine their structures by X-ray crystallography. The structure of the Fe2+ derivative had been previously reported.29
A Rare Example of a Stable CuII–Thiolate
The copper complex [CuII(SMe2N4(tren))]+ (7; Figure 4) is one of few reported examples of synthetic monomeric CuII–thiolate complexes.55–59 Copper(II)–thiolates are typically unstable, and readily convert to disulfides and Cu+1 unless significant amounts of steric bulk are incorporated.58–60 The first structurally characterized example, [CuII(SC6F5)(HB(3,5-iPr2pz)3)] (9),55,56 incorporates bulky isopropyl groups, and is four coordinate in non-coordinating solvents. Kitajima’s model 9 nicely reproduces the properties, of blue copper proteins (cupredoxins).61–63 Blue copper proteins are electron transport proteins which facilitate rapid electron transfer via a highly covalent Cu(II)–SR bond and rigid trigonal structure.64 Key properties reflecting this structure/function relationship include an intense low-energy CysS → Cu(II) CT band near 600(ε≥ 3500 M−1cm−1) nm, and an unusually small hyperfine coupling constant (A||(Cu) < 80 × 10−4 cm−1) in the axial g|| > g⊥ (S= 1/2) EPR signal. The small A|| reflects an extensive delocalization of odd spin density onto the thiolate ligand.5,6 Pseudo-tetrahedral 9 mimics all of these key properties.55,56 A three-coordinate example, [LCu(II)(SCPh3)] (L= bulky β-diketiminate),57 mimics the lower coordinate laccase blue copper site.65 Copper(II)-protein complexes resembling cupredoxins have been assembled using a combinatorial approach.59 Although approximately 30% of these display the intense low-energy LMCT associated with a Cu(II)-thiolate, this spectral feature bleaches within minutes of formation. The most stable Cu(II)-thiolate-protein complexes display blue-shifted σS→Cu(II) CT bands (near ~400 nm) characteristic of a distorted tetragonal site.59 Higher coordination numbers have been shown to stabilize the Cu(II) oxidation state.63,66 Synthetic Cu(II)-thiolate peptide complexes are most stable when assembled in environments that are accessible to solvent, favoring solvent-bound derivatives with higher coordination numbers.58
Figure 4.
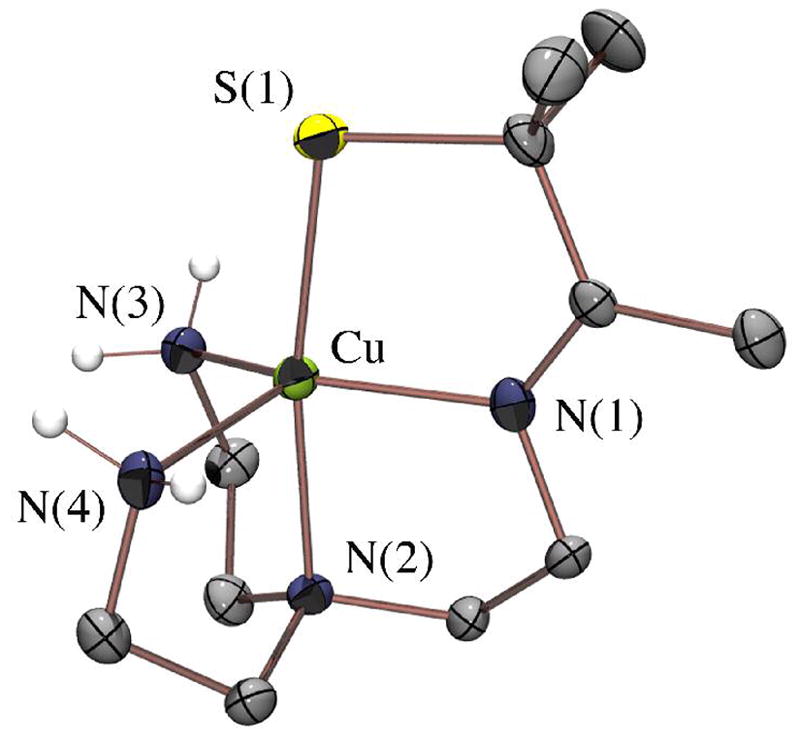
ORTEP of the cation of [CuII(SMe2N4(tren))](PF6) (7) showing the atom labeling scheme. All hydrogen atoms, with the exception of the amine hydrogens, have been removed for clarity.
Given the trigonal symmetry of the blue copper site, we reasoned that the tripodal [(tren)N4SMe2]1− ligand would be the ideal environment in which to stabilize a blue-copper-like Cu(II)-SR with a higher coordination number (five). The gem-dimethyls adjacent to the thiolate, as well as the higher coordination number of 7 do indeed appear to provide significant stabilization, since 7 was found to be indefinitely stable both in solution (H2O, MeOH, MeCN) and in the solid state, and shows no signs of bleaching due to Cu(II) reduction. The ligand’s preferential distortion towards a tetragonal geometry (vide infra) results in a perturbed Cu(II)-SR site closer to that of the green copper enzyme nitrite reductase.67
Structure of [MII(SMe2N4(tren))]+, M= Ni(4), Mn(5), Co(6), Cu(7), and Zn(8)
As shown in the ORTEP diagrams of Figures 3 and 4, the metal ions of 5–8 are all contained in geometrically similar tetragonally distorted (τ (range): 0.564 – 0.658)68 trigonal bipyramidal (tbp) environments. The Ni2+ ion of 4, on the other hand, is contained in an approximately idealized square pyramidal (sq pyr) (τ = 0.011)68 environment. The distortion of 5–8 from idealized geometry (τ(ideal tbp) = 1.0; τ(ideal sq pyr) = 0.0) is most likely caused by ligand constraints. A similar distortion is observed with Goldberg’s five coordinate [FeII(Py2S(X)] (X= Cl−, Br−) complexes.23 With for 4–8 the S(1)–M–N(2) angle would remain approximately linear for both idealized tbp and sq pyr geometries, lying in the basal plane of the square pyramid, and forming the apex of the trigonal bipyramid. The distortion in 5–8 involves the opening of the N(1)–M–N(3) angle from 120° towards 180° (range: 124.9° in 1 to 135.9 ° in 7 and 158.7 in 4), and the compression of the N(4)–M–N(3) and N(4)–M–N(1) angles from 120° towards 90° (range: 115.2 in 1 to 108.8° in 7 and 96.2 ° in 4). Molecular mechanics calculations (using SPARTAN) consistently minimize to a structure with τ~0.6, showing that ligand constraints are responsible for the observed distortions. A square pyramidal τ ~ 0 structure is only obtained when the angles are constrained, indicating that the geometry of the nickel complex must be reflect the geometric preferences of the metal ion driven by a net stabilization of populated antibonding d-orbitals (see Figure S-4). Although the interconversion between idealized trigonal bipyramidal and square pyramidal structures is usually quite facile, this is only when all of the ligands are identical and monodentate.69 In an asymmetric environment, the barrier to interconversion may be high,69,70 and with multidentate ligands, ligand constraints might favor one isomer over the other. In the absence of the condensed thiolate arm, the tripodal tren ligand used in this study would usually favor a trigonal bipyramidal structure.
Periodic Trends in Bonding
Bond lengths in 4–8 follow expected periodic trends, decreasing from left to right across the periodic table (from Mn2+ to Zn2+; Table 2) with exceptions occurring upon the occupation of antibonding eg(σ)* orbitals. The experimentally determined M–S thiolate bond lengths (blue diamonds) are plotted versus dn- configuration in Figure 5, and compared with Shannon covalent radii for high–spin M2+ (pink squares).71 Metal–sulfur bond lengths are shortest in the Cu2+ complex 7 (2.254(1) Å), and longest in the Mn2+ complex 5 (2.412(3) Å). The short Cu–S bond fits with previous observed trends in M–S(thiolate) covalency,72 and occurs because there is a better energy match between the sulfur π–orbitals and metal ion d-orbitals for a late transition-metal (such as Cu2+) versus an earlier transition-metal (such as Mn2+). The short, highly covalent, Cu(II)–SR bond of 7 is significant, given that this characteristic has been suggested to facilitate rapid electron transfer in blue copper electron transport proteins.5,6,61 This bond (Table 2) is slightly longer than that of the three/four–coordinate (type 1) blue copper proteins (range: 2.1 – 2.2 Å) as well as four-coordinate Cu(II)(SC6F5)(HB(3,5-iPr2pz)3) (2.176(4) Å),55 and significantly longer than that of three-coordinate [LCu(II)(SCPh3)] (2.124 Å),57 reflecting the higher coordination number of 7. At first glance, the plot of Figure 5 appears to indicate that the Cu2+–S thiolate bond of 7 is significantly more covalent than predicted. However, one needs to take into account the fact that Shannon radii are determined for six-coordinate octahedral coordination environments, whereas herein we are looking at a five-coordinate ~trigonal bipyramidal (or ~square pyramidal) environment. In a trigonal bipyramidal environment, only one σ* orbital (dz2*) is significantly destabilized, versus two (dz2* & dx2-y2*) in an octahedral geometry (Figure S-3). This orbital is singly occupied for Mn2+, Fe2+, Co2+, Ni2+ Cu2+, but doubly occupied with Zn2+, and the dramatic increase in M–S and M–N(2) bond lengths with M2+ = Zn2+ versus M2+ = Co2+, Ni2+ , Co2+, (Figure 5 and Table 2) reflects this. The significantly higher ionization potential associated with Zn2+ results in a shorter Zn(II)–S versus Mn(II)–S bond, however, despite its doubly occupied antibonding sigma orbital.
Figure 5.
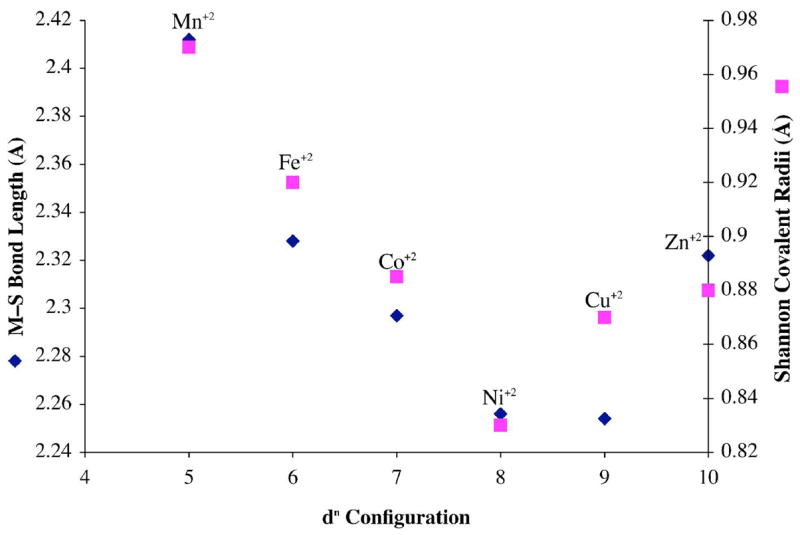
Plot of experimentally determined M–S bond lengths (Å) for the series [MII(SMe2N4(tren))] (M= Mn, Fe, Co, Ni, Cu, Zn) (blue diamonds) and Shannon covalent radii (Å) (pink squares) versus dn–configuration.
Electronic Properties of [MII(SMe2N4(tren))]+, M= Fe(1), Ni(4), Mn(5), Co(6), Cu(7), Zn(8)
In contrast to the oxidized Fe(III) thiolate derivatives of the [(tren)N4SMe2]1− ligand, which are low–spin and intensely colored,29,73 the M(II) derivatives of this series are high–spin and colorless to moderately colored. The weaker color occurs because the intense S → M+n CT (charge transfer) bands which dominate the visible spectrum of the Fe(III) derivatives74 fall in the UV region for the early metal M(II) derivatives (Table 3), and/or have lower extinction coefficients due to poorer orbital overlap. Weaker d-d bands are seen at lower energies between 400–802 nm (Table 3). The d10 Zn2+ and d5 Mn2+ complexes of course lack d-d transitions, since either the d-orbitals are filled, or the transitions are both Laporte– and spin–forbidden. The high energy UV transition associated with the Zn2+ complex must be ligand centered (possibly imine π → π*), since the d-orbitals are completely occupied. As the metal ion d–orbitals fall in energy closer to those of the sulfur, either upon metal ion oxidation,74 or moving to the right in the periodic table, the S → M+n CT bands move into the visible region. These trends are also observed for [MII{HB(3,5-iPr2pz)3}(SC6F5)] (M= Mn, Fe, Co, Ni, Cu, Zn).72 Why the CuII–SR bond of our complex [CuII(SMe2N4(tren))]+ (7) appears to be less covalent than one would expect will be the topic of a separate paper.75 For each compound in the series, [MII(SMe2N4(tren))]+ (M= Mn (5), Fe (1), Co (6), Ni (4), Cu (7), Zn (8)), the electronic absorption spectra (Figure 6, and Figures S-5 – S-7) are solvent-independent, and identical in coordinating (MeCN, pyridine, MeOH), and non-coordinating (CH2Cl2) solvents, indicating that solvents do not bind to the metal. This is surprising, given their electron deficient nature, and the scarcity of five-coordinate structures. In contrast, the alkoxide and amine derivatives readily bind solvents and/or coordinating anions implying that the thiolate sulfur decreases the metal ion’s Lewis acidity.21
Table 3.
Electronic absorption transitions for the series [MII(SMe2N4(tren))]+ (M= Mn, Fe, Co, Ni, Cu, Zn)
| λmax (ε) (nm (M−1cm−1)) | λmax (ε) (nm (M−1cm−1)) | λmax (ε) (nm (M−1cm−1)) | |
|---|---|---|---|
| [MnII(SMe2N4(tren))]+ | 240 (2910) | ||
| [FeII(SMe2N4(tren))]+ (1) | 262 (4700) | 357 (sh) | 410 (sh) |
| [CoII(SMe2N4(tren))]+ (6) | 236(4570)
358(990) |
454(215) | 600 (100) |
| [NiII(SMe2N4(tren))]+ (4) | 275 (2394) | 410 (330) | |
| [CuII(SMe2N4(tren))]+ (7) | 216 (3280);
236 (3400) |
380 (3340) | 618 (315) |
| [ZnII(SMe2N4(tren))]+ (8) | 219 (4800) |
Figure 6.
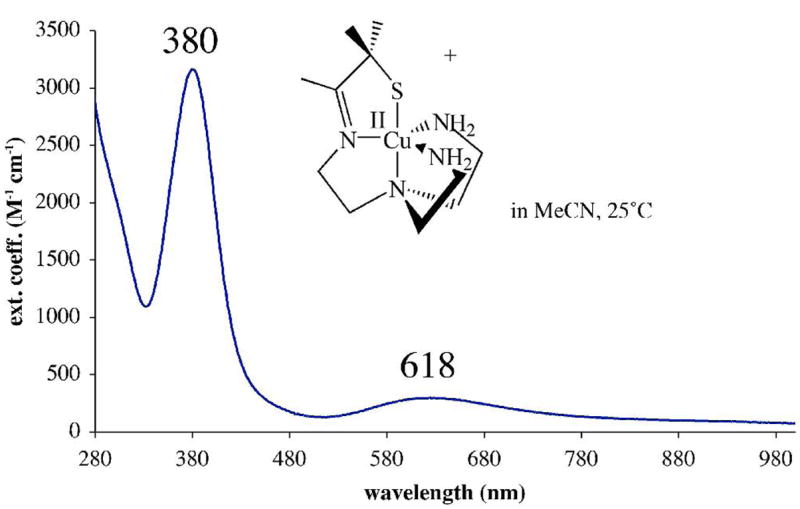
Electronic absorption spectrum of [CuII(SMe2N4(tren))](PF6) (7) in MeCN at ambient temperature.
A Perturbed Green Copper Protein Site Analogue
Solutions of [CuII(SMe2N4(tren))]+ (7) are sensitive to H–bonding solvents. In MeCN the complex is green with an intense band at 380(3340) nm, and a weaker band, at 618(315) (Figure 6, Table 3). In water complex 7 is turquoise blue, and these bands blue–shift to 358(1719) and 640(126) (Figure S–8). This is most likely caused by H–bonding interactions between the protic solvent and thiolate lone-pairs.76 The relative intensities of the two bands in Figure 6 are in contrast to the electronic absorption properties of the cupredoxins (blue copper proteins). Cupredoxins display a significantly more intense transition (ε ~ 3000 – 6000 M−1cm−1) near 600 nm, and a much less intense (ε < 1000 M−1cm−1) transition near 400 nm.5,64 In fact, the absorption spectrum (and color) of 7 more closely resemble that of the highly perturbed “green Cu” sites of nitrite reductase,67 77 stellacyanin, and plantacyanin.78 The green color of these biological Cu sites arises from the combination of a blue (~600(~1800) nm) πS→Cu(II) transition and yellow (~400(~3000) nm) σS→Cu(II) transition. Structural and electronic perturbations suggested to account for the differences between blue and green copper proteins, include an increased ligand–field strength, an increase in coordination number,62 a longer Cu(II)-SR bond, and a reorientation of the sulfur π-orbitals (relative to the metal d-orbitals) that reduces the extent of π-overlap in the Cu-SR bond. Consistent with one of these explanations, when the fourth ligand of nitrite reductase is replaced with a non-coordinating ligand, the intensity of the blue πS→Cu(II) band (near 600 nm) increases while that of the yellow πS→Cu(II) band (near 400 nm) decreases.77 The significantly weaker intensity of the lower energy band (Figure 6, Table 3) indicates that there is much less π–overlap in five–coordinate 7, relative to 3–4 coordinate blue copper proteins (ε ~5000 M−1cm−1),64 as well as Tolman’s 3-coordinate Cu(II)–SR (ε =5800 M−1cm−1).57 The origin of the electronic absorption spectral bands for 7, in relation to the blue, green, and red Cu protein sites,5 will be the subject of a separate paper.75
Magnetic Properties of [MII(SMe2N4(tren))]+, M= Fe(1), Ni(4), Mn(5), Co(6), Cu(7), Zn(8)
The ligand-field provided by the pentadentate thiolate-containing [(tren)N4SMe2]1− ligand is not strong enough to pair more electrons relative to the free M+2 ion. This is true even for the square pyramidal Ni2+ complex, which is S=1 as opposed to S=0 (Figures S-9 and S–11). One might have anticipated that a driving force for the geometry change with Ni2+ involved a spin-state change S=1 → S=0, however, 4 was found to be S= 1 over the entire temperature range examined (T= 4 – 200 K; Figure S-11). The ambient temperature magnetic moment of dimeric [NiII(SN4(tren)–RSdang)]2 (3; Figure 1) in H2O (μeff(H2O) = 2.89 μB/Ni) is also consistent with an S=1 high-spin state, with very little coupling between the Ni2+ ions. This is not too surprising given that there are other examples of weakly coupled dimeric Ni2+ complexes.46 Solution magnetic moments for 1 and 5–7 are consistent with a sequential drop in the number of unpaired electrons from five to one as one proceeds through the series Mn2+ (μeff = 6.01 μB Fe2+ (μeff = 5.39 μB),79 Co2+ (μeff = 4.05 μB Ni2+(μeff = 2.86 μB Cu2+ (μeff = 1.97 μB), and in the solid state, the temperature–dependent inverse magnetic susceptibility plots are linear (Figures S-10 and S–11) and obey the Curie law. Although rare for a five–coordinate first-row metal ion, thiolates have been shown to stabilize low–spin five-coordinate metal ions, but this typically involves the metal in a higher oxidation state.31,39 Upon oxidation, the iron29 and cobalt80 complexes 1 and 6 convert to low–spin (S= 1/2, 0, respectively) six-coordinate structures. X-band EPR verified that the odd electron systems possess a high-spin ground-state (Figures 7, 8, and S–12). For example, the EPR spectrum of Figure 7, demonstrates that the manganese complex 5 is S= 5/2. The 90 G separation between the six main features of the multi-line spectrum is consistent with a mononuclear structure,81,82 and arises due to coupling between the unpaired electrons and the I= 5/2 55 Mn (100% natural abundance) nucleus. The additional inner features arise due to zero-field splitting.83–85 The near axial EPR signal of 7(Figure 8), with g|| > g⊥ (g|| = 2.17 , and g⊥= 2.07), is typical for Cu(II) and consistent with a singly occupied and highly delocalized dx2-y2 HOMO in a distorted site. Although the complexity of the spectrum and slight rhombicity requires higher resolution Q-band data to obtain more accurate fits,75 the estimated hyperfine coupling to the I= 3/2 Cu ion of 7 (A||(Cu) = 150 × 10−4 cm−1) is significantly larger than that of blue copper proteins (A||(Cu) reported range: 37 × 10−4 −63 × 10−4 cm−1),64 Kitajima’s Cu-(SC6F5)(HB(3,5- iPr2pz)3) (A||(Cu)= 54 × 10−4 cm−1; g|| = 2.30) and Cu(SCPh3)(HB(3,5-iPrpz)) (A||(Cu)= 74 × 10−4 cm−1; g|| =2.23),55 and Tolman’s 3-coordinate [LCu(II)(SCPh2(CH2OCH3))] (A|| = 111 × 10−4 cm−1; g|| = 2.17; g⊥ = 2.04),57 but less than that of five-coordinate Cu(II) complexes lacking thiolate ligands.86 This indicates that the Cu(II)-SR bond in 7 is significantly less covalent than that of the blue copper proteins, but more covalent than Cu(II)-O or Cu(II)-N bonds.
Figure 7.
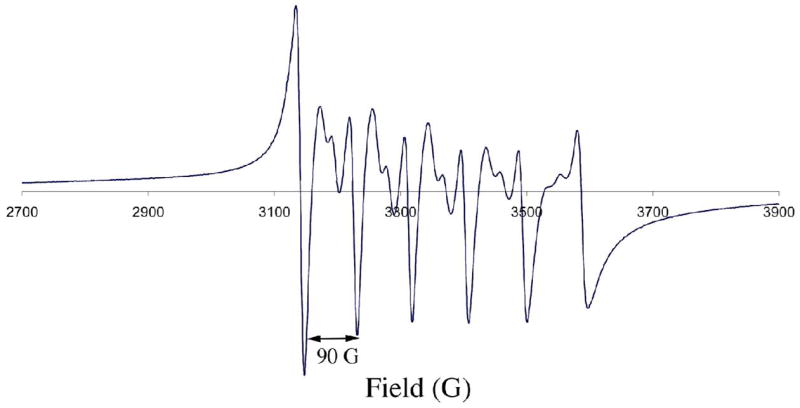
X-band EPR spectrum of [MnII(SMe2N4(tren))](PF6) (5) at 5 K in MeOH/EtOH (9:1) glass. Microwave frequency, 9.42 GHz.
Figure 8.
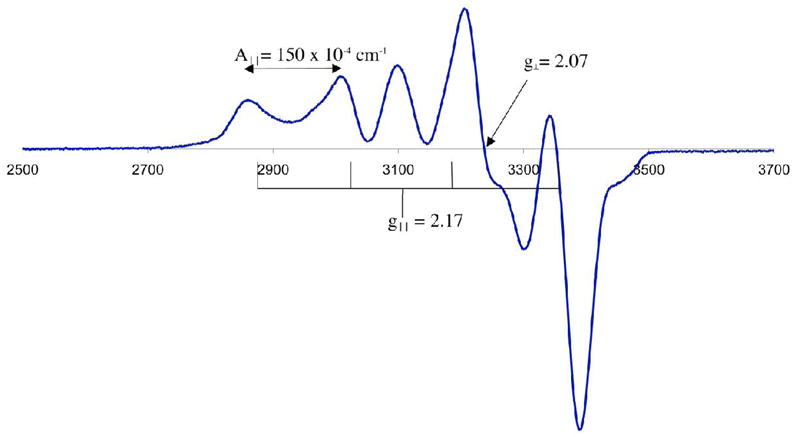
X-band EPR spectrum of [CuII(SMe2N4(tren))](PF6) (7) at 77 K in MeOH/EtOH (9:1) glass. Microwave frequency, 9.39 GHz.
Redox Properties of [MII(SMe2N4(tren))]+, M= Fe(1), Ni(4), Mn(5), Co(6), Cu(7), Zn(8)
As shown by the redox potentials of Table 4, the [(tren)N4SMe2]1− ligand stabilizes the late first row metal ions Ni2+ and Cu2+ in the +2 oxidation state. The high coordination number imposed by the ligand makes the reduced Cu+1 state accessible only at a fairly negative potential (Figure S–14; E1/2= −703 mV vs SCE). This is in contrast to the lower-coordinate trigonal Cu(II) center of type 1 blue copper proteins, which are reduced in the potential range of +680 mV (rusticyanin) to +240 mV (auracyanin) vs NHE.64 Although the +3 oxidation state is accessible with relatively mild oxidants for the earlier transition-metals (manganese, iron and cobalt (Figure 9)) in this ligand environment, the waves are quasi–reversible (to irreversible) because the structures change upon oxidation to accommodate the binding of a sixth ligand.29,80 Re-reduction of the solvent-bound cobalt(III) complex is also irreversible on the CV time-scale because MeCN solvent loss occurs upon reduction. The Co3+ → Co2+ re-reduction potential (Ep,c= −729 mV vs SCE; Figure 9) is dramatically shifted relative to the Co2+ → Co3+ oxidation potential (Ep,a= +270 mV vs SCE), reflecting the increased stability of the six-coordinate, solvent-bound Co3+ ion. Ferrocenium-induced oxidation of 1 and 6 in MeCN affords the corresponding solvent–bound [MIII(SMe2N4(tren))(MeCN)]2+ (M= Fe,29 Co80) complexes. The structure of the iron(III) complex was reported previously.29 Irreversible oxidation of 4 (Figure S-13) and 7 (Figure S-14) to Ni3+ and Cu3+ occurs at potentials of +325 mV, and +690 mV vs SCE, respectively. The +4 oxidation state is accessible for the manganese complex at a slightly higher, reversible potential of +705 mV vs SCE (Figure 10). An irreversible ligand–centered oxidation, apparent in the zinc complex (Figure S–15), occurs at potentials greater than +1.0 V.
Table 4.
Redox potentials for the series [MII(SMe2N4(tren))]+ (M= Mn, Fe, Co, Ni, Cu, Zn)
| E1/2 (vs SCE) Oxidation | E1/2 (vs SCE) reduction | |
|---|---|---|
| [MnII(SMe2N4(tren))]+ (5) | +140 mV (quasi-rev)
+705 mV |
|
| [FeII(SMe2N4(tren))]+ (1) | −150 mV (quasi-rev)79 | |
| [CoII(SMe2N4(tren))]+ (6) | +270 mV (irrev, Epa) | |
| [NiII(SMe2N4(tren))]+ (4) | +325 mV (irrev, Epa) | |
| [CuII(SMe2N4(tren))]+ (7) | +690 mV (irrev, Epa) | −703 mV (quasi-rev) |
| [ZnII(SMe2N4(tren))]+ (8) | +1.10 V (irrev, Epa) |
Figure 9.
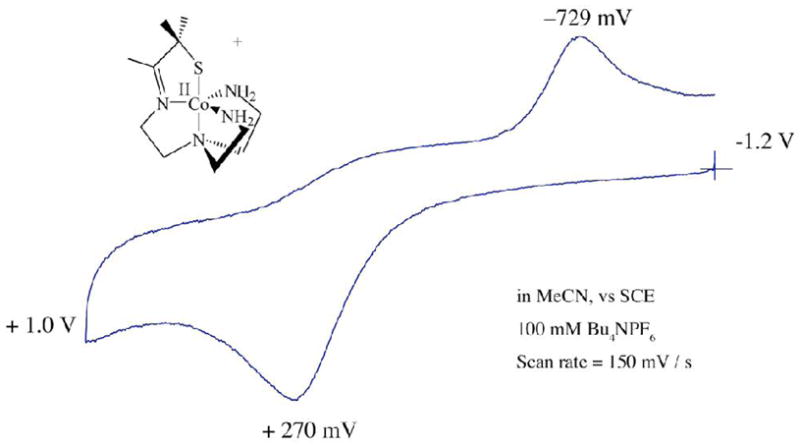
Cyclic voltammogram of [CoII(SMe2N4(tren))](PF6) (6) in MeCN at 298 K (0.1 M (Bu4N)PF6, glassy carbon electrode, 150 mV/sec scan rate). Peak potentials versus SCE indicated.
Figure 10.
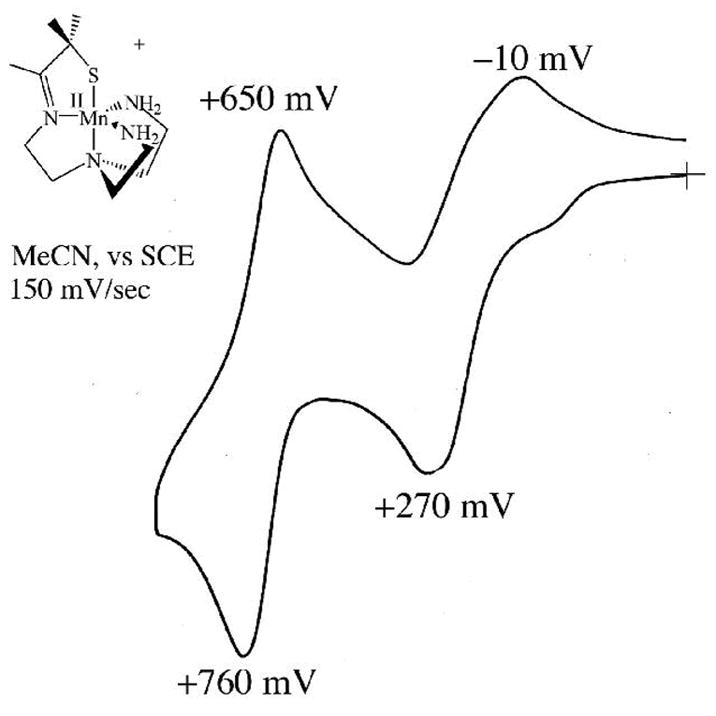
Cyclic voltammogram of [MnII(SMe2N4(tren))](PF6) (5) in MeCN at 298 K (0.1 M (Bu4N)PF6, glassy carbon electrode, 150 mV/sec scan rate). Peak potentials versus SCE indicated.
Summary and Conclusions
A comparison of the electronic, magnetic, and structural properties of a series of five–coordinate first–row transition–metal ions (M2+; M= Mn, Fe, Co, Ni, Cu, Zn) ligated by a tripodal thiolate–containing ligand [(tren)-N4SMe2]1− demonstrates an adherence to expected periodic trends with exceptions occurring upon the occupation of σ* orbitals. Gem-dimethyls adjacent to the sulfur prevent dimerization. All of the complexes, with the exception of [NiII(SMe2N4(tren)]+, are isostructural and adopt a tetragonally distorted trigonal bipyramidal geometry favored by ligand constraints. Geometric preferences of the metal ion driven by a net stabilization of populated antibonding d-orbitals control the structure of the Ni2+ complex. The M–SR bond lengthes decrease as one proceeds across the periodic table, and are most covalent in the Cu(II)–SR and Ni(II)–SR complexes. The copper complex electronically resembles the green copper proteins, and represents a rare example of a stable Cu(II)-thiolate. The nickel complex resembles the active site of nickel superoxide dismutase. In contrast to the intensely colored, low–spin Fe(III)-thiolates, the M(II)-thiolates described herein are colorless to moderately colored, and high–spin, reflecting the poorer energy match between the metal d– and sulfur–orbitals upon reduction of the metal ion. As the d–orbitals drop in energy proceeding across the periodic table, the sulfur-to-metal charge transfer transition moves into the visible region, and the M–SR bonds become more covalent. The redox potentials cathodically shift across the series M2+ (M= Mn, Fe, Co, Ni, Cu), and the reduced M+1 oxidation state is only accessible with the later transition metal copper, and the more oxidized M+4 oxidation state is only accessible for the early transition-metal manganese. Future work will include reactivity studies involving dioxygen, and the characterization of higher-valent derivatives.
Supplementary Material
Supporting information conatins crystallographic data for complexes 1– 5, and electronic absorption spectra for 1 – 5.
Acknowledgments
This work was supported by the NIH (Grant Number: GM 45811). We thank Vickie DeRose and Brandon Green for helpful discussion. We thank Santiago Toledo, Erika Shaffer, and Morgan Gleaves for experimental assistance.
References
- 1.Krishnamurthy D, Kasper GD, Namuswe F, Kerber WD, Sarjeant AN, Moënne-Loccoz P, Goldberg DP. J Am Chem Soc. 2006;128:14222–14223. doi: 10.1021/ja064525o. [DOI] [PubMed] [Google Scholar]
- 2.Kovacs JA. Chem Rev. 2004;104:825–848. doi: 10.1021/cr020619e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holm RH, Kennepohl P, Solomon EI. Chem Rev. 1996;96:2239–2314. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 4.Lippard SJ, Berg JM. Principles of Bioinorganic Chemistry. University Science; Mill Valley: 1994. [Google Scholar]
- 5.Solomon EI. Inorg Chem. 2006;45:8012–8025. doi: 10.1021/ic060450d. [DOI] [PubMed] [Google Scholar]
- 6.Gray HB, Malström B, Williams RJP. J Biol Inorg Chem. 2000;5:551 – 559. doi: 10.1007/s007750000146. [DOI] [PubMed] [Google Scholar]
- 7.Rao PV, Holm RH. Chem Rev. 2004;104:527–560. doi: 10.1021/cr020615+. [DOI] [PubMed] [Google Scholar]
- 8.Rodrigues JV, Abreu IA, Cabelli D, Teixeira M. Biochemistry. 2006;45:9266–9278. doi: 10.1021/bi052489k. [DOI] [PubMed] [Google Scholar]
- 9.Brines LM, Kovacs JA. Eur J Inorg Chem. 2007:29–38. [Google Scholar]
- 10.Kurtz DM., Jr Acc Chem Res. 2004;37:902–908. doi: 10.1021/ar0200091. [DOI] [PubMed] [Google Scholar]
- 11.Fiedler AT, Bryngelson PA, Maroney MJ, Brunold TC. J Am Chem Soc. 2005;127:5449–5462. doi: 10.1021/ja042521i. [DOI] [PubMed] [Google Scholar]
- 12.Wuerges J, Lee JW, Yim YI, Yim HS, Kang SO, Carugo KD. Proc Natl Acad Sci USA. 2004;101:8569–8574. doi: 10.1073/pnas.0308514101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shearer J, Long LM. Inorg Chem. 2006;45:2358–2360. doi: 10.1021/ic0514344. [DOI] [PubMed] [Google Scholar]
- 14.Denisov IG, Makris TM, Sligar SG, Schlichting I. Chem Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 15.Ogata H, Hirota S, Nakahara A, Komori H, Shibata N, Kato T, Kano K, Higuchi Y. Structure. 2005;13:1635–1642. doi: 10.1016/j.str.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 16.Rajagopalan PTR, Yu XC, Pei D. J Am Chem Soc. 1997;119:12418–12419. [Google Scholar]
- 17.Chang S, Karambelkar VV, diTargiani RC, Goldberg DP. Inorg Chem. 2001;40:194–195. doi: 10.1021/ic001079b. [DOI] [PubMed] [Google Scholar]
- 18.Lindahl PA. Biochemistry. 2002;41:2097–2105. doi: 10.1021/bi015932+. [DOI] [PubMed] [Google Scholar]
- 19.Rao PV, Bhaduri S, Jiang J, Hong D, Holm RH. J Am Chem Soc. 2005;127:1933–1945. doi: 10.1021/ja040222n. [DOI] [PubMed] [Google Scholar]
- 20.Kovacs JA. Science. 2003;299:1024–1025. doi: 10.1126/science.1081792. [DOI] [PubMed] [Google Scholar]
- 21.Brines LM, Villar-Acevedo G, Kitagawa T, Swartz RD, Lugo-Mas P, Kaminsky W, Benedict JB, Kovacs JA. Inorg Chim Acta. 2007 doi: 10.1016/j.ica.2007.07.038. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adachi SI, Nagano S, Ishimori K, Watanabe Y, Morishima I, Egawa T, Kitagawa T, Makino R. Biochemistry. 1993;32:241–252. doi: 10.1021/bi00052a031. [DOI] [PubMed] [Google Scholar]
- 23.Krishnamurthy D, Sarjeant AN, Goldberg DP, Caneschi, Totti F, Zakharov LN, Rheingold AL. Chem Eur J. 2005;11:7328–7341. doi: 10.1002/chem.200500156. [DOI] [PubMed] [Google Scholar]
- 24.Yandulov DV, Schrock RR. J Am Chem Soc. 2002;124:6252–6253. doi: 10.1021/ja020186x. [DOI] [PubMed] [Google Scholar]
- 25.Smith JM, Sadique AR, Cundari TR, Rodgers KR, Lukat-Rodgers G, Lachicotte RJ, Flaschenriem CJ, Vela J, Holland PL. J Am Chem Soc. 2006;128:756–769. doi: 10.1021/ja052707x. [DOI] [PubMed] [Google Scholar]
- 26.O'Keefe BJ, Breyfogle LE, Hillmyer MA, Tolman WB. J Am Chem Soc. 2002;124:4384–4393. doi: 10.1021/ja012689t. [DOI] [PubMed] [Google Scholar]
- 27.Brown SD, Betley TA, Peters JC. J Am Chem Soc. 2003;125:322–323. doi: 10.1021/ja028448i. [DOI] [PubMed] [Google Scholar]
- 28.Shearer J, Scarrow RC, Kovacs JA. J Am Chem Soc. 2002;124:11709–11717. doi: 10.1021/ja012722b. [DOI] [PubMed] [Google Scholar]
- 29.Shearer J, Nehring J, Kaminsky W, Kovacs JA. Inorg Chem. 2001;40:5483–5484. doi: 10.1021/ic010221l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perrin DD, Armarego WLF, Perrin DR. Purification of Laboratory Chemicals. 2. Pergamon Press; Elmsford, NY: 1980. [Google Scholar]
- 31.Ellison JJ, Nienstedt A, Shoner SC, Barnhart D, Cowen JA, Kovacs JA. J Am Chem Soc. 1998;120:5691–5700. [Google Scholar]
- 32.Van Geet AL. Anal Chem. 1968;40:2227–2229. [Google Scholar]
- 33.Evans DA. J Chem Soc. 1959:2005. [Google Scholar]
- 34.Live DH, Chan SI. Anal Chem. 1970;42:791. [Google Scholar]
- 35.Hromatka O, Engel E. Monatsh Chem. 1948;78:29. [Google Scholar]
- 36.Cromer DT, Waber JT. International Tables for X-ray Crystallography. IV Kynoch; Birmingham, England: 1974. [Google Scholar]
- 37.Maelia LE, Millar M, Koch SA. Inorg Chem. 1992;31:4594–600. [Google Scholar]
- 38.Millar M, Lee JF, Fikar R. Inorg Chim Acta. 1996;243:333–338. [Google Scholar]
- 39.Schweitzer D, Shearer J, Rittenberg DK, Shoner SC, Ellison JJ, Loloee R, Lovell SC, Barnhart DKJA. Inorg Chem. 2002;41:3128–3136. doi: 10.1021/ic0109187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yandulov DV, Schrock RR. Science. 2003;301:76–78. doi: 10.1126/science.1085326. [DOI] [PubMed] [Google Scholar]
- 41.Theisen RM, Kovacs JA. Inorg Chem. 2005;44:1169–1171. doi: 10.1021/ic048818z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar M, Day RO, Colpas GJ, Maroney MJ. J Am Chem Soc. 1989;111:5974–6. [Google Scholar]
- 43.Ragunathan KG, Bharadwaj PK. J Chem Soc Dalton Trans. 1992:2417–2422. [Google Scholar]
- 44.Hsieh TC, Gebreyes K, Zubieta J. J Chem Soc, Chem Comm. 1984:1172–1174. [Google Scholar]
- 45.Rosenfeld SG, Berends HP, Gelmini L, Stephan DW, Mascharak PK. Inorg Chem. 1987;26:2792–2797. [Google Scholar]
- 46.Baidya N, Olmstead M, Mascharak PK. Inorg Chem. 1991;30:929–937. [Google Scholar]
- 47.Osakada K, Yamamoto T, Yamamoto A, Takenaka A, Sasada Y. Acta Cryst. 1984;83:47. [Google Scholar]
- 48.Yamamoto T, Sekine Y. Inorg Chim Acta. 1984;83:47. [Google Scholar]
- 49.Boge EM, Mockler GM, Sinn E. Inorg Chem. 1977;16:467. [Google Scholar]
- 50.DiVaira M, Orioli PL, Sacconi L. Inorg Chem. 1971;10:553. [Google Scholar]
- 51.Seleborg M, Holt SL, Post B. Inorg Chem. 1971;10:1501. [Google Scholar]
- 52.Healy PC, Mockler GM, Freyberg DP, Sinn E. J Chem Soc, Dalton Trans. 1975:691. [Google Scholar]
- 53.Choudhury SB, Ray D, Chakravorty A. Inorg Chem. 1991;30:4354. [Google Scholar]
- 54.Shoner SC, Olmstead MM, Kovacs JA. Inorg Chem. 1994;33:7–8. [Google Scholar]
- 55.Kitajima N, Fujisawa K, Tanaka M, Moro-oka Y. J Am Chem Soc. 1992;114:9232–9233. [Google Scholar]
- 56.Kitajima N, Fujisawa K, Moro-oka Y. J Am Chem Soc. 1990;112:3210–3212. [Google Scholar]
- 57.Holland PL, Tolman WB. J Am Chem Soc. 1999;121:7270–7271. [Google Scholar]
- 58.Daugherty RG, Wasowicz T, Gibney BR, DeRose VJ. Inorg Chem. 2002:2623–2632. doi: 10.1021/ic010555a. [DOI] [PubMed] [Google Scholar]
- 59.Schnepf R, Horth P, Bill E, Wieghardt K, Hildebrandt P, Haehnel W. J Am Chem Soc. 2001;123:2186–2195. doi: 10.1021/ja001880k. [DOI] [PubMed] [Google Scholar]
- 60.Thompson JS, Marks TJ, Ibers JA. J Am Chem Soc. 1979;101:4180–4192. [Google Scholar]
- 61.Adman ET, Turley S, Bramson R, Petratos K, Banner D, Tsernoglou D, Beppu T, Watanabe H. J Biol Chem. 1989;264:87–99. [PubMed] [Google Scholar]
- 62.Pierloot K, De Kerpel JOA, Ryde U, Olsson MHM, Roos BO. J Am Chem Soc. 1998;120:13156–13166. [Google Scholar]
- 63.Randall DW, George SD, Hedman B, Hodgson KO, Fujisawa K, Solomon EI. J Am Chem Soc. 2000;122:11620–11631. [Google Scholar]
- 64.Solomon EI, Baldwin MJ, Lowery MD. Chem Rev. 1992;92:521–542. [Google Scholar]
- 65.Ducros V, Brzozowski AM, Wilson KS, Brown SH, Ostergaard P, Schneider P, Yaver DS, Pedersen AH, Davies GJ. Nature Struct Biol. 1998;5:310–316. doi: 10.1038/nsb0498-310. [DOI] [PubMed] [Google Scholar]
- 66.Rorabacher DB. Chem Rev. 2004;104:651–697. doi: 10.1021/cr020630e. [DOI] [PubMed] [Google Scholar]
- 67.LaCroix LB, Shadle SE, Wang Y, Averill BA, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 1996;118:7755–7768. [Google Scholar]
- 68.Addison AW, Rao TN, Reedijk J. J Chem Soc Dalton Trans. 1984:1349. [Google Scholar]
- 69.Muetterties EL. Acc Chem Res. 1970;3:266–273. [Google Scholar]
- 70.Sacconi L, Bertini I. J Am Chem Soc. 1968;90:5443–5446. [Google Scholar]
- 71.Shannon RD. Acta Cryst. 1976;A32:751–767. [Google Scholar]
- 72.Gorelsky SI, Basumallick L, Fura-Weis J, Sarangi R, Hodgson KO, Hedman B, Fujisawa K, Solomon EI. Inorg Chem. 2005;44:4947–4960. doi: 10.1021/ic050371m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shearer J, Fitch SB, Kaminsky W, Benedict J, Scarrow RC, Kovacs JA. Proc Natl Acad Sci, USA. 2003;100:3671–3676. doi: 10.1073/pnas.0637029100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kennepohl P, Neese F, Schweitzer D, Jackson HL, Kovacs JA, Solomon EI. Inorg Chem. 2005;44:1826–1836. doi: 10.1021/ic0487068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xie X, Brines LM, Shearer J, Kovacs JA, Solomon EI. manuscript in preparation. [Google Scholar]
- 76.Jackson HL, Shoner SC, Rittenberg D, Cowen JA, Lovell S, Barnhart D, Kovacs JA. Inorg Chem. 2001;40:1646–1653. doi: 10.1021/ic001271d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Basumallick L, Szilagyi RK, Zhao Y, Shapleigh JP, Scholes CP, Solomon EI. J Am Chem Soc. 2003;125:14784–14792. doi: 10.1021/ja037232t. [DOI] [PubMed] [Google Scholar]
- 78.LaCroix LB, Randall DW, Nersissian AM, Hoitink CWG, Canters GW, Valentine JS, Solomon EI. J Am Chem Soc. 1998;120:9621–9631. [Google Scholar]
- 79.Theisen RM. PhD thesis. University of Washington; 2005. [Google Scholar]
- 80.Swartz RD, Kovacs JA. manuscript in preparation. [Google Scholar]
- 81.Howard T, Telser J, DeRose VJ. Inorg Chem. 2000:3379–3385. doi: 10.1021/ic0000247. [DOI] [PubMed] [Google Scholar]
- 82.Reed GH, Markham GD. Biological Magnetic Resonance. Vol. 6. Plenum Press; New york: 1984. pp. 73–135. [Google Scholar]
- 83.Allen BT. J Chem Phys. 1965;43:3820–3826. [Google Scholar]
- 84.Morrissey SR, Horton TE, DeRose VJ. J Am Chem Soc. 2000;122:3473–3481. [Google Scholar]
- 85.Vogt M. Texas A&M. 2004. [Google Scholar]
- 86.Marlin DS, Olmstead MM, Mascharak PK. Inorg Chem. 2001;40:7003–7008. doi: 10.1021/ic010523n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information conatins crystallographic data for complexes 1– 5, and electronic absorption spectra for 1 – 5.