

The introduction of HIV-1 protease (HIV-PR) inhibitors has led to a dramatic increase in patient survival; however, these gains are threatened by the emergence of multi-drug resistant strains1. Design of inhibitors that overcome resistance would be greatly facilitated by deeper insight into the mechanistic events associated with binding of substrates and inhibitors, as well as an understanding of the effects of resistance mutations on the structure and dynamic behavior of HIV-PR. We previously reported a series of simulations that provide a model for HIV-PR dynamics, with spontaneous conversions between the bound and unbound crystal forms upon addition or removal of an inhibitor2,3. Importantly, the unbound protease transiently sampled a third fully open state that permits entry to the active site, unlike both crystallographic forms. Recently, a crystal structure of unbound HIV-PR was reported4 for the MDR 769 isolate (PDB: 1TW7); unlike all previous experimental structures, the binding pocket is open (Figure 1). The authors suggest that drug resistance in this strain arises at least in part from inability of inhibitors to induce closing4. We carried out simulations of the MDR 769 HIV-PR mutant, and observe that the reported structure is unstable in solution and rapidly adopts the semi-open conformation observed for the unbound wild-type protease in solution5. Further analysis suggests that the wide-open structure observed for MDR 769 arises not from sequence variation, but instead is an artifact from crystal packing. Thus, despite being the first experimental structure to reveal flap opening sufficient for substrate access to the active site, this structure may not be directly relevant to studies of inhibitor entry or to the cause of HIV-PR drug resistance.
Figure 1.
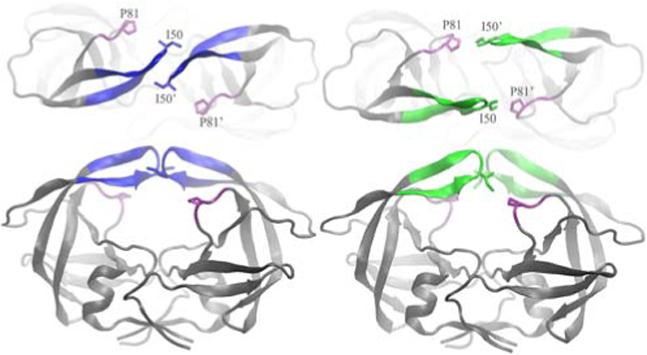
Crystal structures of the wide open (right, 1TW7, flaps in green) and semi-open (left, 1HHP, flaps in blue) forms of HIV-PR. A top view is shown to illustrate the separation of the flaps observed in 1TW7 but not in 1HHP. All molecular images were created with VMD8.
An extensive set of X-ray crystal structures of HIV-PR have been solved6, revealing a C2 symmetric homodimer with a large substrate binding pocket covered by two glycine rich β-hairpins, or flaps7. Consistent structural differences are present between the bound and free states of the protein. In all of the liganded forms, the flaps are pulled in towards the bottom of the active site (“closed” form), while the unbound enzyme is more flexible and adopts an ensemble of “semi-open” conformations with the flaps shifted away from the catalytic residues, but still substantially closed over the active site and a contact between the I50 side chains at the end of each flap tip. A more striking difference between the two forms is that the relative orientation (the “handedness”) of the β-hairpin flaps is reversed3. The “wide open” structure of MDR 769 adopts the closed handedness, but with no direct interactions between the flaps. Instead, this interaction is replaced by a contact between the flap tip I50 side chain and P81' on the other monomer (Figure 1).
The inherent flexibility of the HIV-PR flaps likely makes them sensitive to crystal packing effects; crystallography9 and early simulations10 suggested packing effects as a possible explanation for the difference between closed and semi-open conformations observed in HIV-PR crystal structures. Recently, we reported unrestrained, all-atom molecular dynamics (MD) simulations of HIV-PR in solution that sampled large conformational rearrangements of the active site flaps, with transient sampling of a fully open form3. Removal of the inhibitor from the closed form resulted in conversion to the semi-open structure3, while addition of an inhibitor to the open form resulted in adoption of the closed form2 with an identical hydrogen bond pattern as observed in the crystal structure of the complex11. Each of these transformations is consistent with experimental observations. Using this validated protocol (see supporting information for details), we simulated the MDR 769 sequence starting from the wide-open crystal structure. Both catalytic Asp residues were changed to Asn to be consistent with the crystallographic experiments4. We observed that the open structure from the crystal was unstable and adopted the semi-open structure12 within the first 1 ns (Figure S1). Since it was suggested that a network of hydrogen bonds involving water contributed to the stability of the wide-open flaps in the crystal structure4, we repeated the simulations using an explicit water model, retaining crystallographic waters and solvating the system in a truncated octahedron periodic box with a total of 8572 waters (Figure S2). Once again the crystal structure was unstable and the protease adopted the semi-open conformation12 in under 1 ns (Figure 2). This explicit solvent simulation was extended to 15 ns, during which the semi-open conformation remained stable (1−2 Å RMSD to 1HHP) and the wide-open crystal structure was not revisited (RMSD values of 4.5−5.5 Å to 1TW7). An independent simulation in explicit water showed comparable instability of the wide-open structure.
Figure 2.
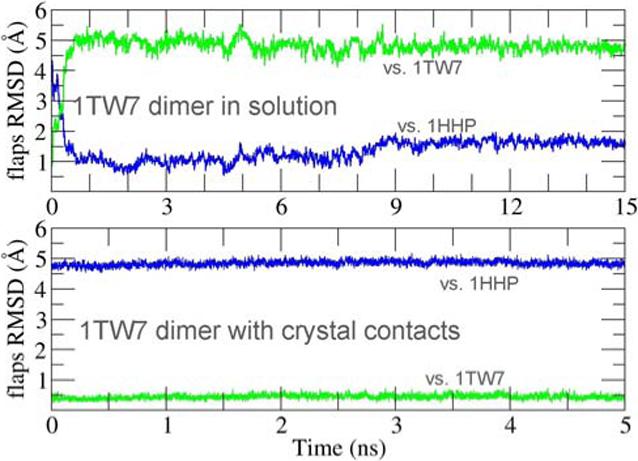
RMSD of the flaps region during simulations, as compared to the semi-open (1HHP, blue) and wide-open (1TW7, green) crystal structures. Top: dimer in explicit solvent; bottom: same as top but also including all crystal symmetry-related protein residues within 15 Å of the dimer.
To explain the apparent contradiction between the results from simulations and crystallography, we examined the interactions between molecules related by crystallographic symmetry. While both 1HHP12 (semi-open) and 1HVR11 (closed) show some crystal contacts in the flaps region, these contacts do not involve the flap tips. In contrast, each flap tip in 1TW7 is buried between the elbow and fulcrum regions of a neighboring dimer (Figure 3), with the unusual P81':I50 contact enclosed by five residues from the symmetry-related neighbor (P39, R41, D60, Q61, I72). Since the conformation of the elbow and fulcrum have been shown to be correlated with flap opening13,14, wedging a flap tip between the fulcrum and elbow could further stabilize the open conformation observed in the crystal. It is interesting to note that this crystal packing actually provides an experimental example of a proposed mechanism of HIV-PR allosteric inhibition.
Figure 3.
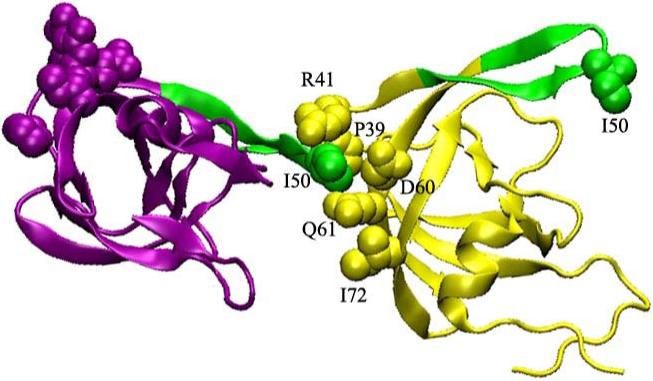
Detailed view of 1TW7 crystal packing interactions involving HIV-PR flaps (shown in green). The flap tip of one monomer (purple) is wedged into the elbow region of a monomer in a neighboring dimer (yellow).
To investigate the influence of these contacts, we repeated our simulations of the dimer in solution, but with inclusion of crystal packing contacts. This was achieved by using the 1TW7 crystal structure to build a system containing one HIV-PR dimer along with all neighboring symmetry-related molecules. Residues farther than 15Å from the central dimer were discarded, as were all crystallographic water molecules. This system of a dimer plus crystal contacts was then solvated with 20196 explicit water molecules (Figure S2). Positional restraints were applied to atoms representing the crystal packing environment, while the central dimer and explicit solvent were fully unrestrained. This open structure with crystal contacts was stable during 5 ns MD with flaps RMSD of only ∼0.5Å compared to 1TW7 (Figure 2b).
This behavior provides further evidence that the open structure observed in the crystal is stabilized by crystal packing contacts. However, the crystal structure may indeed be a minor component of the solution ensemble; 0.044 % of the structures sampled in our long unrestrained simulations3 of wild type HIV-PR had flap conformations with flap RMSD values less than 2.0 Å from the 1TW7 wide-open structure, suggesting a free energy of ∼ 4.5 kcal/mol relative to the dominant semi-open form.
The present observations serve as an illustration of the importance of carefully considering crystal packing effects on the conformations of key surface loops, particularly in structure-based drug design where these may compose or directly interact with the binding pocket. Importantly, the data suggests that the drug resistance of the MDR 769 strain arises from a source other than an inability of the active site flaps to close. Future studies will investigate the solution structure of this HIV-PR mutant in greater detail.
Supplementary Material
Acknowledgment
CS thanks Ben Dunn and Alex Wlodawer for helpful discussions and gratefully acknowledges support from the NIH (GM6167803), DOE (DE-AC02-98CH10886) and NCSA (NPACI MCA02N028).
Footnotes
Supporting Information Available: Detailed Methods, Figures S1 and S2, and initial coordinates for crystal packing simulation. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Walensky RP, Paltiel AD, Losina E, Mercincavage LM, Schackman BR, Sax PE, Weinstein MC, Freedberg KA. Journal of Infectious Diseases. 2006;194:11–19. doi: 10.1086/505147. [DOI] [PubMed] [Google Scholar]
- 2.Hornak V, Okur A, Rizzo RC, Simmerling C. Journal of the American Chemical Society. 2006;128:2812–2813. doi: 10.1021/ja058211x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hornak V, Okur A, Rizzo RC, Simmerling C. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:915–920. doi: 10.1073/pnas.0508452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin P, Vickrey JF, Proteasa G, Jimenez YL, Wawrzak Z, Winters MA, Merigan TC, Kovari LC. Structure. 2005;13:1887–95. doi: 10.1016/j.str.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 5.Freedberg DI, Ishima R, Jacob J, Wang YX, Kustanovich I, Louis JM, Torchia DA. Protein Science. 2002;11:221–232. doi: 10.1110/ps.33202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vondrasek J, Wlodawer A. Proteins-Structure Function and Genetics. 2002;49:429–431. doi: 10.1002/prot.10246. [DOI] [PubMed] [Google Scholar]
- 7.Wlodawer A, Miller M, Jaskolski M, Sathyanarayana BK, Baldwin E, Weber IT, Selk LM, Clawson L, Schneider J, Kent SBH. Science. 1989;245:616–621. doi: 10.1126/science.2548279. [DOI] [PubMed] [Google Scholar]
- 8.Humphrey W, Dalke A, Schulten K. Journal of Molecular Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 9.LangeSavage G, Berchtold H, Liesum A, Budt KH, Peyman A, Knolle J, Sedlacek J, Fabry M, Hilgenfeld R. European Journal of Biochemistry. 1997;249:912–912. [Google Scholar]
- 10.York DM, Darden TA, Pedersen LG, Anderson MW. Biochemistry. 1993;32:3196–3196. [Google Scholar]
- 11.Lam PYS, Jadhav PK, Eyermann CJ, Hodge CN, Ru Y, Bacheler LT, Meek JL, Otto MJ, Rayner MM, Wong YN, Chang CH, Weber PC, Jackson DA, Sharpe TR, Ericksonviitanen S. Science. 1994;263:380–384. doi: 10.1126/science.8278812. [DOI] [PubMed] [Google Scholar]
- 12.Spinelli S, Liu QZ, Alzari PM, Hirel PH, Poljak RJ. Biochimie. 1991;73:1391–1396. doi: 10.1016/0300-9084(91)90169-2. [DOI] [PubMed] [Google Scholar]
- 13.Perryman AL, Lin JH, McCammon JA. Biopolymers. 2006;82:272–284. doi: 10.1002/bip.20497. [DOI] [PubMed] [Google Scholar]
- 14.Perryman AL, Lin J-H, McCammon JA. Protein Sci. 2004;13:1108–1123. doi: 10.1110/ps.03468904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.