

Abstract
To identify residues and segments in the central region of apolipoprotein A-I (apoA-I) that are important for the protein structure and stability, we studied the effects of four double charge ablations, D102A/D103A, E110A/E111A, R116V/K118A, and R160V/H162A, and two deletion mutations, Δ(61-78) and Δ(121-142), on the conformation and stability of apoA-I in the lipid-free state and in reconstituted discoidal phospholipid:cholesterol:apoA-I particles (rHDL). The findings suggest that D102/D103 and E110/E111 located in helix 4, and segment(s) between residues 61 and 78 are involved in maintenance of the conformation and stability of apoA-I in both the lipid-free state and in rHDL. R116/K118 located in helix 4 are essential for the conformation and stabilization of apoA-I in rHDL, but not vital for the lipid-free state of the protein. The R160V/H162A substitutions in helix 6 lead to a less compact tertiary structure of lipid-free apoA-I without notable effects on the lipid-free or lipid-bound secondary conformation suggesting involvement of R160/H162 in important inter-helical interactions. The results on the Δ(121-142) mutant, together with our earlier findings, suggest disordered structure of a major segment between residues 121 and 143, likely including residues 131-143, in lipid-free apoA-I. Our findings provide the first experimental evidence for stabilization of rHDL by electrostatic inter-helical interactions, in agreement with the double belt model. The effects of alterations in the conformation and stability of the apoA-I mutants on in vitro and in vivo functions of apoA-I and lipid homeostasis are discussed.
Apolipoprotein A-I (apoA-I) is the major protein component of high density lipoprotein (HDL) that plays a key role in the biogenesis and atheroprotective functions of HDL and in reverse cholesterol transport (1-3). Lipid-free apoA-I secreted by cells can interact functionally with ATP binding cassette transporter 1 (ABCA1) to promote efflux of cholesterol and phospholipids from cells. This process leads to the lipidation of apoA-I and formation of discoidal HDL (4, 5). ApoA-I in discoidal HDL activates the enzyme lecithin:cholesterol acyltransferase (LCAT) converting cholesterol to cholesterol ester and thereby promoting the maturation of HDL particles from discoidal to spherical. ApoA-I bound to discoidal and spherical HDL also interacts functionally with scavenger receptor class B type I (SR-BI) (4, 6, 7). On binding HDL, SR-BI mediates selective uptake of cholesterol esters and other lipids from HDL by cells and net efflux of excess cholesterol (4, 6). In the course of apoA-I metabolism, the protein function is modulated by alterations in its structure and stability (5, 8-15). Therefore, detailed understanding of the lipid-free and lipid-bound conformation and stability of apoA-I is important for understanding changes in the protein function.
ApoA-I has a unique structure and a conformational flexibility that may underlie its functions. ApoA-I contains 22- and 11-amino acid repeats, which are organized in amphipathic α-helices (16-18). X-ray crystallographic analysis of an N-terminally truncated apoA-I, Δ(1-43), crystallized from high-salt solution showed a nearly continuous helical structure punctuated by small or pronounced kinks, with an overall “horseshoe” shape that was thought to mimic the lipid-bound apoA-I conformation (18). However, the limited resolution of this analysis (4Å) precludes detailed structural interpretation, such as specific inter- or intra-helical interactions, and the applicability of this structure to monomeric full-size apoA-I has not been clear (19). A significant number of studies employing fluorescence (20, 21), NMR (22) and EPR (23) spectroscopy, analytical ultracentrifugation (24), and limited proteolysis (24, 25) have been aimed at understanding the lipid-free and lipid-bound structure of apoA-I. Three-dimensional models of the apoA-I lipid-free conformation have been suggested based on fluorescence energy transfer (26) or cross-linking and mass spectrometry data (19). Detailed double belt (27) and hairpin belt (21) models have been proposed for the arrangement of apoA-I on discoidal reconstituted high density lipoprotein (rHDL) and spherical HDL particles (reviewed in (28)). In the double belt model (27), two ring-shaped apoA-I molecules wrap around the edge of a discoidal lipid bilayer in an anti-parallel orientation. This model predicts specific salt-bridge interactions involving the anti-parallel helices of the apoA-I molecules. However, even with this considerable amount of accumulated data, the structural details of the lipid-free and lipid-bound conformation are not fully defined and experimental results leading to definition of residues involved in stabilizing the protein lipid-free and lipid-bound conformations are not sufficient. So far, there have been no reported experimental data demonstrating that electrostatic interactions between specific basic and acidic residues located in two anti-parallel apoA-I molecules on the edge of rHDL are essential for the stabilization of the discs, as predicted by the double belt model (27).
Studies of bioengineered mutant apoA-I forms remain efficient to obtain information that leads to detailed understanding of the conformation and structure-function relationships of apoA-I (5, 7-15, 24, 29, 30). Our earlier study (29) using mutation to probe the conformation and stability of the C-terminus of apoA-I suggested the presence of stable helical structure in the region between residues 165 and 243 and a close proximity of the N- and C-termini in the lipid-free protein. A subsequent study focused on the central region of apoA-I (residues 123-165) and suggested helical structure of the segment of residues 144-165 (helix 6) and a disordered structure of the segment between residues 121 and 142 (helix 5) in the lipid-free protein (Figure 1A), with the latter segment proposed to fold into helix upon lipid binding (30). In the current work, we probe by mutation the central part of apoA-I targeting the region between residues 61 and 162 to further understand structural details of lipid-free apoA-I and apoA-I in rHDL and to identify residues that are important for the protein conformation and stability. Four double charge ablations, D102A/D103A, E110A/E111A, and R116V/K118A in helix 4, and R160V/H162A in helix 6, and two deletion mutations, Δ(61-78) in helixes 1 and 2, and Δ(121-142), which deletes the putative helix 5, (Figure 1A) have been generated and investigated. The Δ(121-142) mutation was designed to test our earlier suggestion that helix 5 is disordered in lipid-free apoA-I (30). The double charge ablation mutations were selected because, according to the double belt model of apoA-I in rHDL (27), most of the mutated residues have been predicted to participate in inter-helical interactions of anti-parallel apoA-I molecules in the discoidal rHDL and therefore, these specific charged residues might be important for the conformation and stability of apoA-I. In addition, the selected charge ablation mutations as well as the Δ(61-78) deletion have been shown to affect important functions of apoA-I and HDL in vitro and in vivo when expressed in apoA-I-deficient mice. The D102A/D103A and R160V/H162A mutations have been shown to affect functional interactions of rHDL and SR-B1 and thereby affect cholesterol efflux (7). The D102A/D103A, E110A/E111A, R160V/H162A, and Δ(61-78) mutations affect the LCAT-activation ability of apoA-I (12, 31, 32). Moreover, the E110A/E111A and Δ(61-78) mutations have been recently shown to affect lipid homeostasis and induce severe hypertriglyceridemia when expressed in A-I-deficient mice (12, 32). We previously reported unique structural characteristics of the apoA-I[E110A/E111A] along with its functional analysis (12). In this paper, the physicochemical properties of the apoA-I[E110A/E111A] are compared to those of newly investigated mutants. The findings reveal new features in apoA-I organization in the lipid-free state and in rHDL, provide experimental evidence for rHDL stabilization by electrostatic interactions, and contribute to our understanding of how the mutations may affect apoA-I functions and triglyceride homeostasis in mice expressing the mutants.
Figure 1.
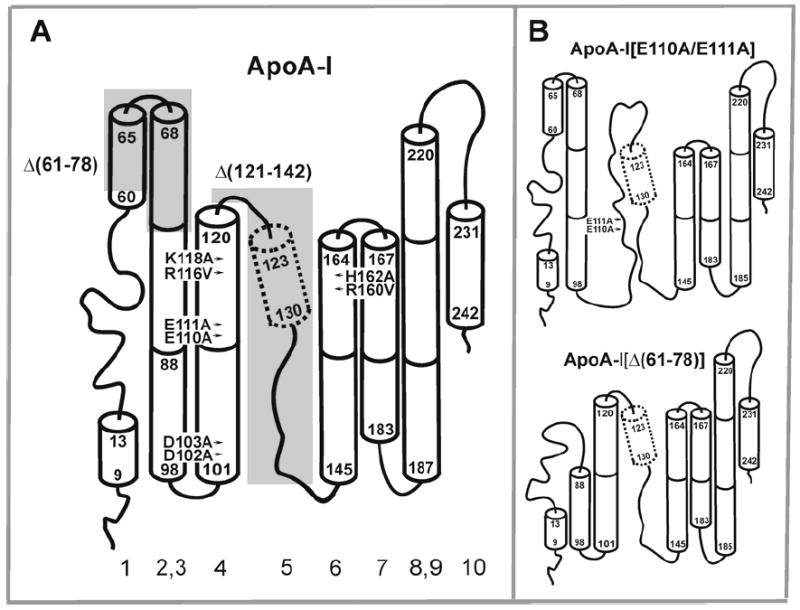
(A) Sites of mutations of apoA-I studied in the current work: (arrows) double charge ablations, (shaded areas) deletion mutations. The secondary structure model of lipid-free apoA-I is adapted from ref 30 with modifications; the model does not address the tertiary conformation. The region between residues 123 and 130 shown by the dotted cylinder and suggested to be disordered in ref. 30 has been proposed to be a ~8-residue helical segment, according to the current analysis. Numbering of helix system is adapted from ref 17. (B) The proposed secondary structure models of the apoA-I[E110A/E111A] and apoA-I[Δ(61-78)] that induce hypertriglyceridemia when expressed in apoA-I-deficient mice.
MATERIALS AND METHODS
Materials
Materials not mentioned in the Experimental procedures have been obtained from sources described previously (12, 30).
Generation of Expression Plasmids, Expression of Recombinant ApoA-I in the Baculovirus System, and Protein Purification
Generation of baculovirus-expressing wild-type apoA-I (WT) and the D102A/D103A, R160V/H162A, and Δ(61-78) mutant forms of apoA-I was performed as described previously (7, 13, 30). The R116V/K118A mutant form of apoA-I was produced in a similar manner. Briefly, the human apoA-I cDNA was mutagenized by polymerase chain reaction, using specific mutagenesis primers, containing the mutation of interest, and a set of flanking universal primers containing the restriction sites BamHI and SalI, using the pBluescript-AI cDNA plasmid as template. The set of universal primers were AIWC-5 and AIWC-3 (5’-ACT CAA GGA TCC GAT GAA CCC CCC CAG AGC CCC TGG GAT–3’) (sense) and (5’- ACT CAA GTC GAC TTT TTC CCA CTT TGG AAA CGT TTA TTC TGA GCA-3’) (anti-sense). Specific mutagenesis primers used for generation of the R116V/K118A mutant were AIMIV2-5 (5’-GAG CTC TAC GTC CAG GCG GTG GAG CCG-3’) and AIMIV2-3 (5’-CGG CTC CAC CGC CTG GAC GTA GAG CTC). The DNA fragment containing the mutation of interest was digested with BamHI and SalI, and cloned into the corresponding sites of `pFASTBAC donor plasmid. The DNA of each mutant in the resulting pFASTBAC-apoA-I constructs was completely sequenced using the Core for DNA sequencing, Department of Biochemistry of Boston University School of Medicine. Cells containing recombinant bacmids were selected by kanamycin, tetracycline and gentamycin resistance as white colonies due to the disruption of LacZ sequence in the recombinant bacmid. Recombinant bacmid DNA was isolated by minipreps and used to transfect a monolayer of Sf-9 insect cells (33). Recombinant viruses were isolated, amplified, titrated, and used to infect larger amounts of Sf-9 cells grown in suspension in Sf-900 II SF medium at 27°C. Isolation of recombinant apoA-I from cell pellet using a Ni2+-nitrilotriacetic acid resin affinity column followed by His tag cleavage by recombinant tobacco-etched viral (rTEV) protease and subsequent purification of His tag-free protein using a second Ni2+-nitrilotriacetic acid resin affinity column was performed as described previously (7, 30). The purified His tag-free WT and mutant proteins were analyzed by 12% SDS-PAGE followed by staining with Coomassie Brilliant Blue R-250 and by Western blotting using monoclonal antibodies against human apoA-I. All the purified proteins used for the experiments were refolded at concentrations <0.1 mg/ml by subsequent dialysis against guanidine hydrochloride (GdnHCl) solutions of 4, 2.5, and 1.25 M in PBS (0.15 M NaCl, 10 mM sodium phosphate, 0.01% EDTA and 0.02% NaN3, pH 7.4), followed by extensive dialysis against the appropriate buffer (10 mM sodium phosphate, 0.02% NaN3, pH 7.4, for (CD) experiments, or PBS for fluorescence measurements and making rHDL particles). Protein concentrations in samples were determined by modified Lowry protein assay (Pierce, Rockford, IL).
Expression of WT and Mutant ProapoA-I Isoforms in the Adenovirus System
The generation of recombinant adenoviruses carrying the genomic sequence for the WT proapoA-I and proapoA-I[E110A/E111A] has been described before (12). The adenovirus expressing proapoA-I[Δ(121-142)] was generated in a similar way. Briefly, the proapoA-I forms were generated by two pairs of polymerase chain reaction primers using the pCA13AIgN plasmid as template. The pCA13AIgN plasmid contained a NotI site at the 5’ end in intron 3 and an XhoI site in the 3’ end of the human apoA-I gene. For the generation of proapoA-I[Δ(121-142)], the genomic apoA-I gene was amplified by a set of specific primers A121F(5’- CTCTACCGCCAGAAGGTGGAGCCACTGGGCGAGGAGATGCGCGAC- 3’) and A121R (5’- GCGCATCTCCTCGCCCAGTGGCTCCACCTTCTGGCGGTAGAGCTC-3’) containing a deletion of interest and a set of flanking primers AINOTF (5’-CCTCCGCGGACAGGCGGCCGCCAGGG-3’) and AISALR (5’-ACATGTCGACCCCCTTTCAGGGCACCTGGCCTTG-3’) containing the restriction sites NotI and SalI. The DNA fragment containing deletion corresponding to residues 121 to 142 of apoA-I was digested by NotI and SalI and subclone into the pCA13AIgN plasmid in which the exon 4 of apoA-I was removed by Not I and XhoI double digestion, thus replacing the WT with the exon sequence containing the deletion. The sequence of proapoA-I[Δ(121-142)] was confirmed by DNA sequencing. For apoA-I production, human HTB13 cells (SW 1783, human astrocytoma) grown to 80 % confluence in Leibovitz’s L-15 medium containing 10% (v/v) fetal bovine serum in roller bottles were infected with adenovirus expressing WT proapoA-I, proapoA-I[E110A/E111A], or proapoA-I[Δ(121-142)] at a multiplicity of infection of 20. After 24 h of incubation, cells were washed twice with serum-free medium and preincubated in serum-free medium for 30 min, then fresh serum-free medium was added. After 24 h the medium was harvested, and fresh serum-free medium was added to the cells. The harvesting was repeated 8-10 times. The proteins were purified from the medium by ion-exchange chromatography followed by gel filtration, as described previously (29). The purified proteins were analyzed by 12% SDS-PAGE and by Western blotting and refolded as described above for the proteins expressed in baculovirus expression system.
Preparation and Analysis of rHDL Containing POPC, Cholesterol, and apoA-I
rHDL particles were prepared by the sodium cholate dialysis method (34), using POPC:cholesterol:apoA-I:sodium cholate in a molar ratio of 100:10:1:100, as previously described (30, 35). The lipid-free apoA-I that was not incorporated in the lipid-protein complexes was removed by extensive dialysis at 4°C against PBS buffer, which was changed at least 3 times, using dialysis tubing of 50,000 Daltons molecular weight cut-off. Gel filtration of the complexes isolated from this preparation on an Amersham Pharmacia Biotech Superose 6HR column, 10/30, driven by Pharmacia FPLC system, showed that all unbound proteins were removed during the dialysis, and the complexes were not contaminated with lipid-free apoA-I. Apolipoprotein-lipid complex formation was verified by analysis with nondenaturing polyacrylamide gradient (8-25%) gel electrophoresis using small-format gels immobilized to plastic backing (Pharmacia Phast gel system, Pharmacia Biotech, Piscataway, NJ) and/or regular format gels. rHDL were observed in the electron microscope and photomicrographs were taken as described previously (30) to analyze the morphology of the complexes.
Circular Dichroism Spectroscopy
CD-measurements were performed on an AVIV 62DS spectropolarimeter (AVIV Associates, Inc.) equipped with a thermoelectric temperature control and calibrated with d-10-camforsulfonic acid, in 5, 2, or 1 mm path length quartz cuvettes, at protein concentration 25-80 μg/mL. Far-UV CD spectra were recorded as described previously (29, 30). For each protein, spectra were recorded at several protein concentrations within the indicated range and then normalized to molar residue ellipticity. The complete superposition of the normalized spectra obtained at various protein concentrations for each recombinant apoA-I is consistent with the absence of protein self-association within the range of protein concentrations used in our experiments. The α-helix content was estimated from the molar residue ellipticity at 222 nm, [Θ222] (36). Protein concentration was determined by modified Lowry protein assay; the protein assay for rHDL was performed in the presence of SDS. Protein concentration in samples determined before and after CD experiments agreed within 5%.
Thermal and GdnHCl-Induced Unfolding of apoA-I Monitored by CD
Thermal and GdnHCl-induced unfolding of lipid-free apoA-I was monitored by ellipticity at 222 nm and the thermodynamic parameters of the transitions were determined as described previously (29, 30). For the heating-induced transitions, midpoint (melting) temperature, Tm, and van’t Hoff enthalpy, ΔHv, were determined from the van’t Hoff analysis of the melting curves (37); melting temperature, Tdm, was also determined as the maximum of the first derivative function d(Θ222)/dT (38). For the GdnHCl-induced transitions, the free energy of denaturation (conformational stability), ΔGDo, the midpoint of denaturation, D1/2, and m value, which reflects the steepness of the denaturation curve in the transition region, were determined using a linear extrapolation method (39).
Thermal unfolding of proteins in rHDL particles was monitored by ellipticity at 222 nm, while samples of rHDL at protein concentrations 10-25 μg/mL were heated from 1 to 97°C at a constant heating rate 0.4 °C /min. The midpoint of thermal unfolding at which denaturation of apoA-I was half complete, T1/2, and the maximum of the first derivative function d(Θ222)/dT, Tdm, were determined from the thermal unfolding curves of lipid-bound apoA-I. Denaturant-induced unfolding of apoA-I in rHDL particles was monitored by ellipticity at 222 nm, following the incubation of aliquots of rHDL at protein concentration 15-135 μg/mL with various concentrations of GdnHCl ranging from 0 to 5 M, at 4 °C for 72 h, as previously described (40, 12). The appropriate blank for each GdnHCl concentration was incubated along with the rHDL samples and the CD signal of the blank was subtracted from the corresponding signal of rHDL to determine mean residue ellipticity at 222 nm at each denaturant concentration. The midpoint of denaturation, D1/2, that is the GndHCl concentration at which denaturation was half completed, was determined graphically from each denaturant-induced unfolding curve. The free energy of denaturation, ΔGDo*, was determined according to the method described by Sparks, Lund-Katz, and Phillips (40) based on the denaturant binding model (41). The free energy of denaturation, ΔGDo and the midpoint of denaturation, D1/2, were determined also using a linear extrapolation method as described previously (12).
Fluorescence Spectroscopy
Fluorescence measurements were performed on a FluoroMax-2 fluorescence spectrometer (Instruments S.A., Inc.) at 25°C using 5 nm excitation and 2.5 nm emission slit widths. 8-Anilino-2-naphthalene-sulfonate (ANS) fluorescence emission spectra were recorded in PBS buffer in the presence of WT apoA-I or one of the mutant apoA-I at a concentration of each protein of 0.05 mg/ml and an ANS concentration of 0.25 mM. ANS fluorescence was excited at a wavelength of 395 nm and the emission spectra were scanned from 400 to 560 nm. For comparison, ANS fluorescence was recorded in the buffer in the absence of any protein and in the presence of 0.05 mg/ml “reference protein”, carbonic anhydrase as a typical globular water-soluble protein, or bovine serum albumin as a protein known to have hydrophobic binding pockets. Trp emission spectra of WT and mutant apoA-I were recorded at protein concentration 0.05 mg/ml, using 295 nm excitation wavelength to avoid Tyr fluorescence; the emission was scanned from 300 to 380 nm. The wavelength of maximum fluorescence (WMF) was determined from uncorrected fluorescence spectra after subtraction of the buffer base line.
RESULTS
Expression and Purification of Recombinant ApoA-I
For proteins expressed in the baculovirus expression system, 15-20 mg of pure recombinant protein was obtained from the cell lysate of 1 L of culture medium after cleavage of the His tag. In the adenovirus-expression system, 1L of culture media yielded ~ 30-80 mg of pure recombinant protein. Western blotting analysis confirmed immunological identity of all the purified proteins to apoA-I. 12% SDS-PAGE analysis showed that the molecular weights of mutant proteins estimated from the gels were in agreement with those predicted from the nature of the mutations (Figure 2). The purity of the proteins studied in this work was more than 95%. WT apoA-I was expressed in both baculovirus and adenovirus expression systems. Figures 3-7 show the data recorded for WT expressed only in the baculovirus expression system, because the data for WT expressed in the two expression systems were similar to each other. However, all the Tables contain the data for WT expressed in both expression systems, and all the observations in this study are based on the comparison of the variant forms of apoA-I to the WT expressed in the corresponding expression system.
Figure 2.

SDS-PAGE (12%) analysis of purified recombinant apoA-I forms. (A) Proteins in lanes 1-5 are apoA-I forms produced by the baculovirus expression system and purified after cleavage of the His tag: lane 1, WT; lane 2, apoA-I[R160V/H162A]; lane 3, apoA-I[R116V/K118A]; lane 4, apoA-I[D102A/D103A]; lane 5, apoA-I[Δ(61-78)]. (B) Proteins in lanes 2-4 are apoA-I forms produced by the adenovirus expression system and purified: lane 2, WT; lane 3, apoA-I[E110A/E111A]; lane 4, apoA-I[Δ(121-142)]. In lane 6, (A), and lane 1, (B), are molecular mass markers (masses in kilodaltons).
Figure 3.
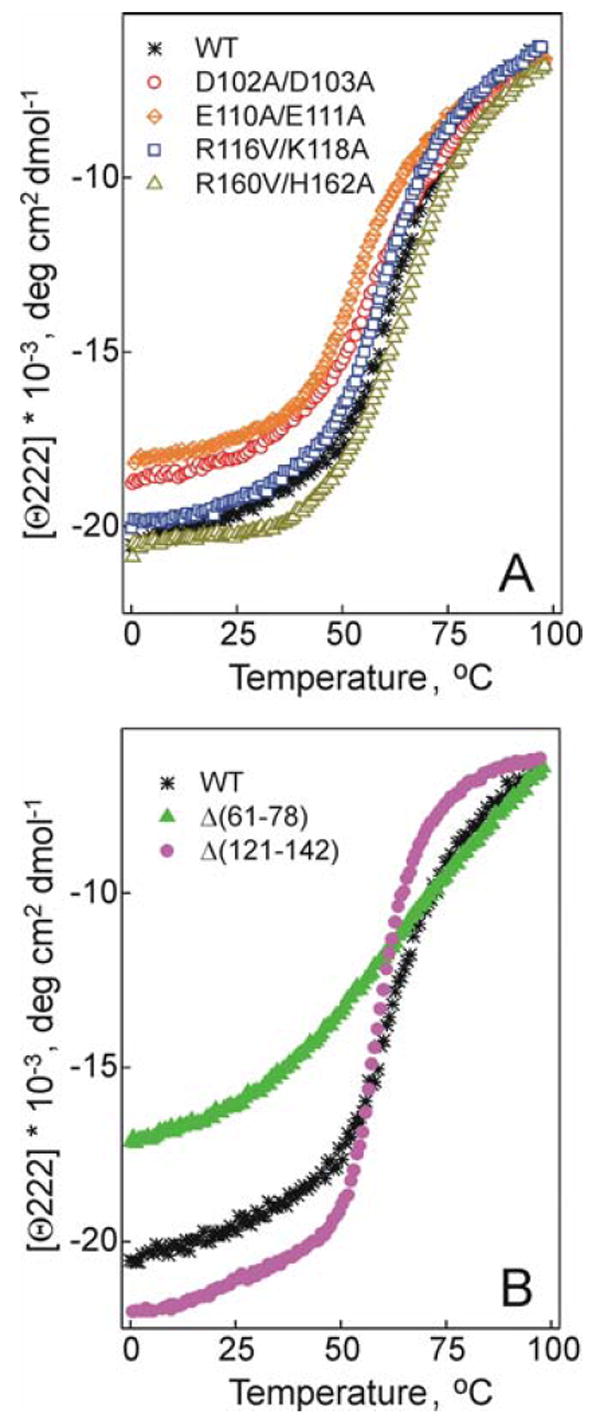
Effect of the double charge ablation mutations D102A/D103A, E110A/E111A, R116V/K118A, and R160V/H162A (A) and deletion mutations Δ(61-78) and Δ(121-142) (B) on thermal unfolding of lipid-free apoA-I. The WT produced by the baculovirus expression system is shown for comparison in each panel.
Figure 7.
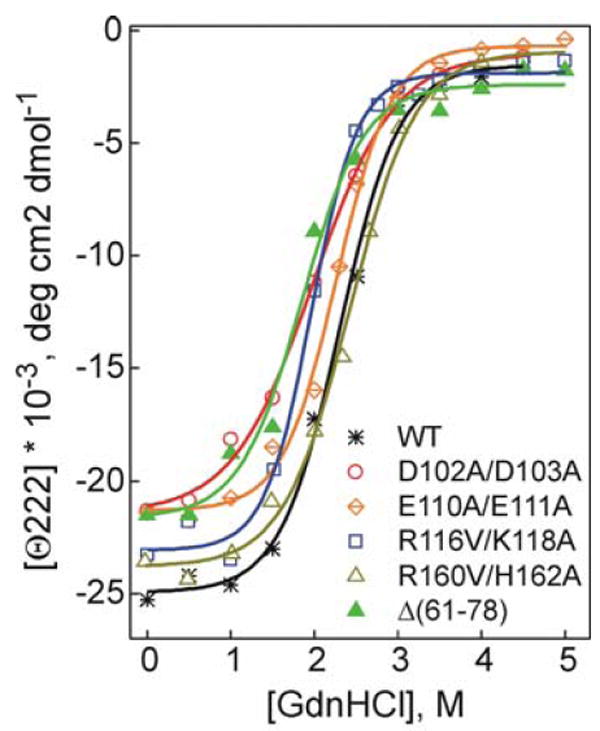
Effect of the mutations on denaturant-induced unfolding of apoA-I in rHDL monitored by the ellipticity at 222 nm.
Analysis of Lipid-Free ApoA-I
Effect of the Mutations on the α-Helical Content
The α-helical content of the lipid-free WT and mutant apoA-I forms (Table 1) was estimated from the normalized far-UV CD spectra (not shown). In the lipid-free state, the E110A/E111A and D102A/D103A double charge ablations caused a ~7% and ~4% reduction, respectively, in the α-helical content, that corresponds to a loss of ~18 and ~10 residues, respectively, in helical conformation. In contrast, two other double charge ablations, the R116V/K118A and R160V/H162A, do not affect significantly the α-helical content. The Δ(61-78) deletion leads to a reduction in the α-helical content by ~8% that corresponds to the loss of ~29 residues in helical conformation. Given that the Δ(61-78) deletion removes 18 residues from apoA-I, this mutation leads to unfolding of helical segments of at least 11 residues long. The other deletion, Δ(121-142) , does not affect significantly the protein percent α-helical content. However, because the number of residues in helical conformation is calculated by multiplying the number of residues in the protein by the α-helical content, this deletion actually results in a loss of residues in the helical conformation of apoA-I. In contrast to the Δ(61-78) deletion, the loss of residues in the helical conformation resulting from the Δ(121-142) deletion (~ 8 residues) is significantly smaller than the number of deleted residues (22 residues).
Table 1.
α-Helical Content and Thermodynamic Parameters of WT and Mutant Forms of Lipid -Free ApoA-I Determined by Far-UV CD a.
| apoA-I | α-helix d (%) | # residues
|
Tdmf (°C) | Tmf (°C) | ΔHvf (kcal/mol) | ΔGDog (kcal/mol) | mg [kcal (mol of A-I)-1 (mol of GndHCl)-1] | D1/2g, (M) | |
|---|---|---|---|---|---|---|---|---|---|
| in protein | in helix e | ||||||||
| WT b | 58 ± 2 | 248 | 144 | 62 ± 0.5 | 60 ± 0.5 | 41 ± 1 | 2.5 ± 0.1 | 2.5 ± 0.1 | 1.0 ± 0.05 |
| apoA-I[D102A/D103A] b | 54 ± 2* | 248 | 134 (-10) | 58 ± 1** | 56 ± 1** | 28 ± 2*** | 2.1 ± 0.1* | 2.2 ± 0.2 | 0.9 ± 0.03 |
| apoA-I[R116V/ K118A] b | 57 ± 4 | 248 | 141 (~0) | 60 ± 0.5* | 58 ± 0.5* | 35 ± 2* | 2.4 ± 0.2 | 2.4 ± 0.1 | 1.0 ± 0.04 |
| apoA-I[R160V/H162A] b | 59 ± 4 | 248 | 146 (~0) | 63 ± 1 | 61 ± 1 | 32 ± 2*** | 2.1 ± 0.2 | 2.2 ± 0.2 | 0.9 ± 0.05 |
| apoA-I[Δ(61-78)] b | 50 ± 3** | 230(-18) | 115 (-29) | - | 47± 1*** | 16 ± 1*** | 1.0 ± 0.2*** | 1.3 ± 0.2*** | 0.7 ± 0.05** |
|
|
|
|
|
|
|
|
|
|
|
| WT c | 59 ± 2 | 249 | 147 | 59 ± 0.5 | 59± 1 | 48 ± 1 | 3.0 ± 0.2 | 3.0 ± 0.2 | 1.0 ± 0.05 |
| apoA-I[E110A/E111A] c | 52 ± 2** | 249 | 129(-18) | 54 ± 1** | 54 ± 0.5** | 30 ± 1** | 2.0 ± 0.2* | 2.0 ± 0.2* | 1.0 ± 0.00 |
| apoA-I[Δ(121-142)] c | 61 ± 1 | 227(-22) | 139(-8) | 60 ± 0.5 | 60 ± 1 | 53 ± 1* | 3.7 ± 0.2* | 3.4 ± 0.2 | 1.1± 0.03 |
Values are the average ± S.D. from 3 to 6 experiments for two or three independent preparations of each protein. Significance of differences from the value for WT:
p<0.05,
p<0.01,
p<0.005.
Proteins expressed in the baculovirus expression system.
Protein expressed in the adenovirus expression system.
Estimated from the value [Θ222] at 25 °C. The values are the average ± S.D. from 10 to 16 measurements for each protein.
The number of residues in the helical conformation as estimated by multiplying the number of residues in the protein by its α-helical content. Values in parentheses show changes in the number of residues as compared to WT.
Parameters determined from the thermal unfolding curves. Tdm is a maximum of the first derivative function d[Θ222]/dT; the melting temperature, Tm, and effective enthalpy, ΔHv, were determined from van’t Hoff analysis of the thermal unfolding curves.
The conformational stability, ΔGDo, midpoint of chemical denaturation, D1/2, and m values were determined by the linear extrapolation method from the GdnHCl-induced unfolding curves.
Stability of the Mutant Forms of apoA-I
The thermal unfolding and denaturant-induced unfolding curves for lipid-free WT and variant forms of apoA-I monitored by ellipticity at 222 nm are shown in Figures 3 and 4, respectively. The values of the maximum of the first derivative d(Θ222)/dT of the melting curves, Tdm, as well as parameters determined from the van’t Hoff analysis, the melting temperature, Tm, and the effective enthalpy, ΔHv, are given in Table 1. The conformational stability, ΔGDo, the midpoint of chemical denaturation, D1/2, and m values determined from the GdnHCl-denaturation curves by the linear extrapolation method are also listed in Table 1. The equilibrium character and reversibility of thermal unfolding and the reversibility of chemical unfolding were verified as we described previously (29). Values of Tdm and Tm determined from the thermal unfolding curves are close to each other, in accordance with reversibility of the thermal unfolding of the lipid-free proteins.
Figure 4.
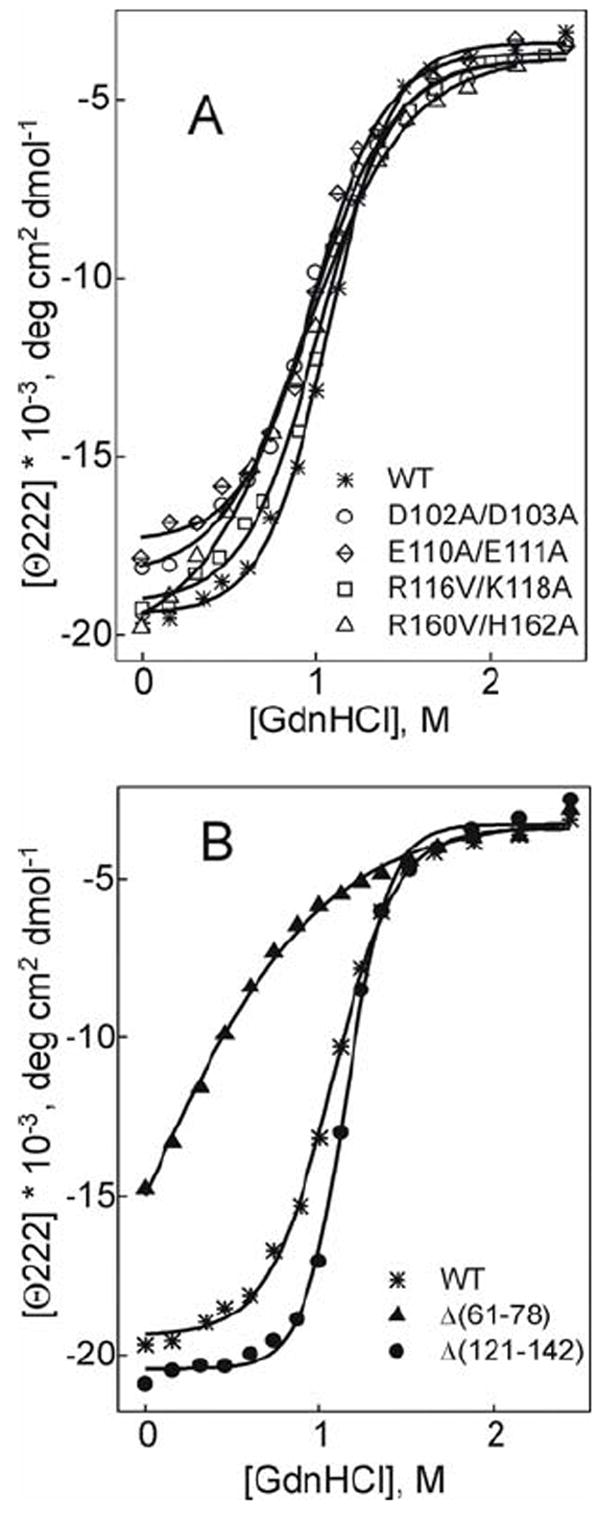
Effect of the double charge ablation mutations D102A/D103A, E110A/E111A, R116V/K118A, and R160V/H162A (A) and deletion mutations Δ(61-78) and Δ(121-142) (B) on denaturant unfolding of lipid-free apoA-I. The WT produced by the baculovirus expression system is shown for comparison in each panel.
The melting curves for the E110A/E111A and D102A/D103A mutants are shifted to lower temperatures compared to the curves for WT apoA-I (Figure 3A), in accordance with a reduction of 5°C (for the E110A/E111A) and a 4°C (for the D102A/D103A) in both Tm and Tdm (Table 1). This indicates a large destabilizing effect of these double charge ablations. The R116V/K118A mutation leads to a smaller (~2°C) reduction in Tm and Tdm indicating a slight destabilization resulted from this double charge ablation. The R160V/H162A mutation does not affect the Tm and Tdm values significantly. All four double charge ablations lead to a reduction in the effective enthalpy ΔHv suggesting lower cooperativity of thermal unfolding. The effect of the double charge ablations on ΔHv is decreased in the order: E110A/E111A> D102A/D103A> R160V/H162A> R116V/K118A and corresponds to an 18, 13, 9, and 6 kcal/mol reduction in ΔHv, respectively. Similar to thermal unfolding, GdnHCl-induced unfolding of the double charge ablation mutants is also affected most notably by the E110A/E111A mutation (Figure 4). This mutation leads to a 1 kcal/mol decrease in the conformational stability ΔGDo, confirming a significant destabilizing effect, and to a 1 kcal (mol of apoA-I)-1(mol of GdnHCl)-1 decrease in the m parameter, indicating reduced cooperativity of unfolding of this mutant. The D102A/D103A double charge ablation leads to a smaller (0.4 kcal/mol) decrease in ΔGDo. The R116V/K118A and R160V/H162A double charge ablations do not alter significantly the parameters of chemical unfolding of apoA-I.
The Δ(61-78) deletion leads to a notable shift of the apoA-I melting curve to lower temperatures and makes the melting curve less steep (Figure 3B). In accordance with this, van’t Hoff analysis shows a 13 °C decrease in Tm indicating a very large destabilization and a 25 kcal/mol decrease in ΔHv suggesting a dramatic reduction in unfolding cooperativity for the Δ(61-78) mutant (Table 1). Because of such a low cooperativity of unfolding reflected in the shape of the melting curves for the Δ(61-78) mutant, the first derivative function d(Θ222)/dT for the curves does not show a well defined peak, which precluded the estimation of the Tdm values for this mutant. In contrast, the shape of the melting curve for the Δ(121-142) deletion mutant suggests highly cooperative unfolding (Figure 3B) that is confirmed by a 5 kcal/mol increase in ΔHv for the mutant compared to WT (Table 1). The effect of the deletion mutations on the GdnHCl- induced unfolding of apoA-I is also opposite for the Δ(121-142) compared to the Δ(61-78) (Figure 4). The Δ(61-78) mutation leads to a 1.5 kcal/mol decrease in the conformational stability ΔGDo and a 0.3 M decrease in the midpoint of chemical denaturation, D1/2, indicating a huge destabilization of apoA-I, in accordance with the observed dramatic reduction in thermal stability. The low α-helical content and the absence of a plateau at low denaturant concentrations indicate that even in the absence of denaturant, the Δ(61-78) mutant is destabilized. The Δ(121-142) mutation leads to 0.7 kcal/mol increase in ΔGDo indicating a stabilizing effect of the deletion. The Δ(61-78) deletion leads to 1.2 kcal (mol of apoA-I)-1(mol of GdnHCl)-1 decrease in the m parameter suggesting lower cooperativity of chemical unfolding compared to WT, in agreement with the observed low cooperativity of thermal unfolding. The Δ(121-142) deletion does not lead to any decrease in the m parameter.
Effect of the Mutations on the ANS and Intrinsic Protein Trp Fluorescence
Fluorescence of ANS in the presence of the lipid-free WT or variant forms of apoA-I was measured to determine if the mutations affect the exposure of hydrophobic cavities or surfaces of apoA-I. The intrinsic fluorescence of the amphipathic dye ANS has been shown to be enhanced and blue-shifted upon binding to hydrophobic surfaces or cavities, while the water-phase dye does not contribute to the emission (42). ANS fluorescence spectra recorded in the presence of WT, mutant apoA-I forms, carbonic anhydrase, or bovine serum albumin, and in buffer alone are shown in Figure 5, WMF and relative intensity values determined from the spectra are listed in Table 2. In the phosphate buffer, ANS fluorescence has a very low intensity and a maximum of emission at 516 nm. In the presence of the “negative control” protein, carbonic anhydrase, ANS fluorescence is not significantly changed, consistent with the compact folding of this globular protein. In contrast, the “positive control”, bovine serum albumin, induces a significant (41 nm) blue shift and almost 10-fold enhancement in ANS fluorescence compared to the dye in the buffer, which is consistent with the known ability of this protein to bind hydrophobic molecules. These data verify the sensitivity of ANS fluorescence to the exposure of hydrophobic surfaces of proteins. In the presence of WT apoA-I expressed in the baculovirus expression system, ANS emission shows a 37 nm blue shift and ~4.5-fold increase in the intensity compared to the emission of the dye in the buffer alone. WT apoA-I expressed in the adenovirus expression system (not shown in Fig 5) induces a very similar effect (Table 2). The double charge ablations D102A/D103A, E110A/E111A, and R160V/H162A induce much higher increase in the fluorescence intensity (6.3 to 6.8-fold) and a larger blue shift (39 to 41 nm) compared to WT, indicating increased exposure of hydrophobic surfaces on these mutant apoA-I. The E110A/E111A mutation induced the largest increase in the fluorescence intensity. In contrast, the R116V/K118A double charge ablation induces a blue shift that is similar to that of WT and an increase in ANS fluorescence intensity (~ 4-fold) that is slightly lower than that of WT. This indicates that, in contrast to other double charge ablations, the R116V/K118A mutation does not increase the exposure of hydrophobic surfaces or cavities on lipid-free apoA-I. In the presence of the Δ(61-78) deletion mutant, ANS fluorescence is greatly enhanced (~7 fold) and blue-shifted (~40 nm), indicating that, of all the mutations studied here, this deletion leads to the greatest exposure of hydrophobic surfaces in lipid-free apoA-I. In contrast, the Δ(121-142) deletion mutant induces a significantly lower increase in ANS fluorescence intensity and a smaller blue shift compared to WT, indicating that the apoA-I[Δ(121-142)] molecule has a more “globular” fold with less hydrophobic surfaces exposed than WT and other apoA-I mutants investigated in this study. Thus, according to the increase in ANS fluorescence intensity, the exposure of hydrophobic surfaces on apoA-I is decreased in the order: apoA-I[Δ(61-78)] ≥ apoA-I[E110A/E111A] > apoA-I[R160V/H162A] ≥ apoA-I[D102A/D103A] ≫ WT > apoA-I[R1116V/K118A] ≫ apoA-I[Δ(121-142)], suggesting a corresponding increase in compact structure of these proteins.
Figure 5.
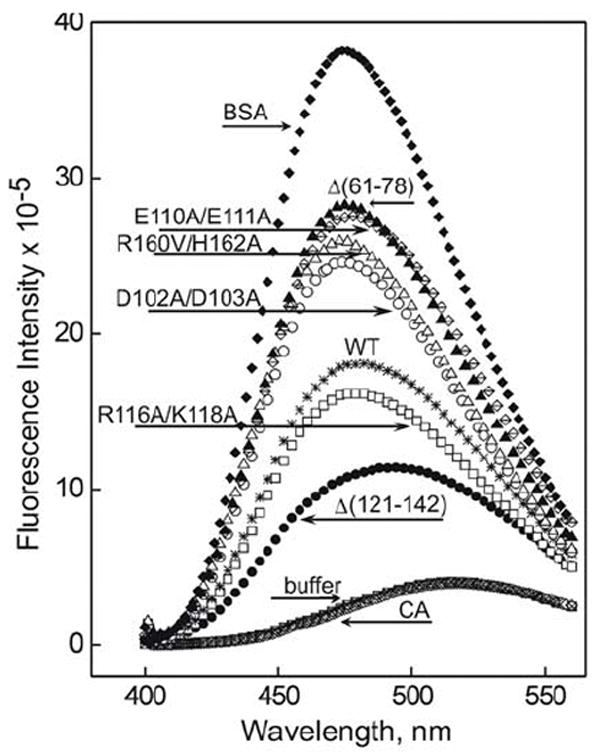
ANS fluorescence spectra obtained in the presence of 50 μg/mL of either WT or one of the mutant forms of apoA-I, bovine serum albumin (BSA), carbonic anhydrase (CA), or in buffer alone.
Table 2.
ANS Fluorescence in the Presence of WT and Mutant Forms of Lipid-Free apoA-I and “Reference Proteins”. Trp Fluorescence of WT and Mutant Forms of Lipid-free apoA-I.
| Protein | WMF c (nm) | I c (relative units d) | I c (relative units e) | WMF f (nm) |
|---|---|---|---|---|
| WT b | 479 (-37)c | 4.5 | 1.0 | 335 |
| apoA-I[D102A/D103A]b | 475 (-41) | 6.3 | 1.3 | 338 |
| apoA-I[R116V/ K118A]b | 479 (-37) | 4.0 | 0.9 | 336 |
| apoA-I[R160V/H162A]b | 475 (-41) | 6.5 | 1.4 | 337 |
| apoA-I[Δ(61-78)]b | 476 (-40) | 7.0 | 1.6 | * |
|
|
|
|
|
|
| WT a | 480 (-36) | 4.3 | 1.0 | 335 |
| apoA-I[E110A/E111A] a | 477 (-39) | 6.8 | 1.5 | 337 |
| apoA-I[Δ(121-142)] a | 492 (-24) | 3.3 | 0.7 | 335 |
|
|
|
|
|
|
| Bovine Serum Albumin | 475 (-41) | 9.5 | 2.1 | N/A |
| Carbonic Anhydrase | 516 | 1.0 | 0.2 | N/A |
| ANS in buffer alone | 516 | 1.0 | 0.2 | N/A |
Proteins were expressed in the adenovirus
Proteins were expressed in the baculovirus expression system.
ANS fluorescence in the presence of 0.05 mg/ml of WT or the mutant forms of apoA-I, carbonic anhydrase or bovine serum albumin, or in PBS buffer alone. Values in parentheses show changes in WMF compared to that for ANS in PBS buffer.
ANS fluorescence intensity in relative units compared to the fluorescence in the buffer alone.
ANS fluorescence intensity in relative units compared to the fluorescence in the presence of WT expressed in the baculovirus expression system.
Intrinsic Trp fluorescence of WT and mutant apoA-I in PBS.
WMF for the apoA-I[Δ(61-78)] was not determined because of the lack of Trp72 in this mutant.
Spectra of the intrinsic Trp fluorescence of WT and mutant forms of apoA-I were recorded and WMF values were determined from the spectra (Table 2) to determine if the mutations affect the hydrophobicity of the environment of Trp residues. The Δ(121-142) mutation appears not to affect WMF of Trp of apoA-I, and the R116V/K118A mutation does not change WMF significantly indicating that environment of Trp residues remains relatively hydrophobic in the Δ(121-142) and R116V/K118A mutants. The D102A/D103A, E110A/E111A, and R160V/H162A mutations lead to a 2-3 nm red shift in WMF, indicating a more polar environment of Trp in the mutant proteins. A value of WMF of tryptophans was not determined for the apoA-I[Δ(61-78)], because the lack of Trp72 in this mutant may affect its tryptophan emission and thus, WMF may not be informative for the assessment of hydrophobicity of the environment of Trp in the Δ(61-78) mutant, compared to that in WT and other mutant forms of apoA-I.
Analysis of ApoA-I in rHDL
Nondenaturating gradient gel electrophoresis of rHDL made from POPC, cholesterol, and either WT or one of the mutant apoA-I verified the formation of the particles; electron micrographs of the rHDL showed discoidal complexes that are stacked on edge (not shown).
Effect of the Mutations on the α-Helical Content
Far-UV CD analysis of rHDL containing WT or one of the mutant forms of apoA-I shows that the R160V/H162A double charge ablation does not affect the secondary conformation of the protein in the lipid-bound state, while other mutations lead to a reduction in the α-helical content (Table 3). A reduction in the α-helical content of apoA-I in rHDL resulted from the double charge ablations is ~9% (the loss of ~22 residues in the helical conformation) for the D102A/D103A mutation, ~4% (the lost of ~10 residues in helical conformation) for the E110A/E111A mutation, and ~5% (the loss of ~12 residues in helical conformation) for the R116V/K118A mutation. The Δ(61-78) deletion leads to a ~8% reduction in the α-helical content of apoA-I in rHDL that indicates a loss of ~31 residues in helical conformation. Given that the Δ(61-78) mutation deletes 18 residues, the data indicate that this deletion results in unfolding of helical segment(s) of at least ~13 residues length in apoA-I bound to rHD.
Table 3.
α-Helical Content and Parameters of Thermal and GdnHCl-Induced Denaturation of WT and Mutant Forms of ApoA-I bound to rHDL Determined by Far-UV CD a.
| apoA-I | α-helix d (%) | # residues
|
T1/2f (°C) | Tdmf (°C) | ΔGDo*g (kcal/mol) | ΔGDog (kcal/mol) | D1/2g (M) | |
|---|---|---|---|---|---|---|---|---|
| in protein | in helix e | |||||||
| WT b | 71 ± 2 | 248 | 176 | 79 ± 0.5 | 91± 0.5 | 4.4 ± 0.2 | 4.1 ± 0.2 | 2.3 ± 0.05 |
| apoA-I[D102A/D103A] b | 62 ± 4* | 248 | 154(-22) | 76 ± 1* | 89 ± 1 | 3.0 ± 0.2*** | 2.4 ± 0.1*** | 1.9 ± 0.07** |
| apoA-I[R116V/ K118A] b | 66 ± 2* | 248 | 164(-12) | 80 ± 1 | 92 ± 1 | 4.2 ± 0.3 | 3.7 ± 0.3 | 2.0 ± 0.1* |
| apoA-I[R160V/H162A] b | 70 ± 4 | 248 | 174 | 78 ± 0.5 | 90 ± 1 | 4.1± 0.3 | 3.5 ± 0.3 | 2.3 ± 0.1 |
| apoA-I[Δ(61-78)] b | 63 ± 3** | 230(-18) | 145(-31) | 76 ± 1* | 89 ± 1 | 3.1± 0.2*** | 2.6± 0.2*** | 1.9 ± 0.1** |
|
|
|
|
|
|
|
|
|
|
| WT c | 67 ± 2 | 249 | 167 | - | - | 4.6 ± 0.2 | 4.1± 0.3 | 2.3 ± 0.0 |
| apoA-I[E110A/E111A] c | 63 ± 2* | 249 | 157(-10) | - | - | 3.9 ± 0.2* | 3.6 ± 0.1* | 2.3 ± 0.1 |
Values are the average ± standard deviation from 3 to 6 experiments for two or three independent preparations of each protein. Significance of differences from the value for WT:
p<0.05,
p<0.01,
p<0.005.
Proteins expressed in the baculovirus expression systems.
Proteins expressed in the adenovirus expression system.
Estimated from the value [Θ222] at 25 °C. The values are the average ± S.D. from 8 to 13 measurements for each protein.
The number of residues in the helical conformation as estimated by multiplying the number of residues in the protein by its α-helial content. Values in parentheses show changes in the number of residues as compared to WT.
Parameters determined from the thermal unfolding curves. T1/2 is a midpoint of thermal unfolding, at which a half of the total change in CD signal is observed; Tdm is a maximum of the derivative function d[Θ222]/dT.
Parameters determined from GdnHCl-induced unfolding curves recorded after 68-72 h incubation of rHDL aliquots with various concentrations of GndHCl. Free energy of denaturation, ΔGDo*, was determined using denaturant binding model as described in ref 40; ΔGDo was determined by the linear extrapolation method (ref 12); the midpoint of denaturation, D1/2, was determined graphically from the curves and by the linear extrapolation method.
Thermal and GdnHCl-Induced Unfolding of Mutant Forms of ApoA-I
The thermal and GdnHCl-induced denaturation curves for WT and mutant apoA-I in rHDL are shown in Figure 6 and 7, respectively; parameters derived from the curves are listed in Table 3. Given that thermal and chemical unfolding of apolipoproteins in rHDL is a kinetically controlled irreversible process (43, 44), the thermal unfolding for all rHDL samples was performed at the same heating rate, 0.4 °C /min. For each protein in rHDL, the midpoint of thermal denaturation, T1/2, significantly from a maximum of the first derivative of the melting curves, Tdm, in accordance with the asymmetric shape of the thermal unfolding curves of the lipid-bound proteins (Figure 6), which confirms the irreversibility of the transitions (43, 44). Denaturant-induced unfolding curves were recorded after 72-hour incubation with GdnHCl to allow apoA-I to achieve complete denaturation (40). The denaturation curves for all rHDL studied are monotonic (Figure 7) indicating that no stable intermediate species are present at any state of the transitions. The denaturation of rHDL made with apoA-I comprises of two processes: a reversible unfolding of lipid-bound apoA-I and irreversible desorption of apolipoproteins from the particle surface (40, 43). It has been shown, however, that the free energy of unfolding determined from the full denaturation curves of rHDL using a denaturant binding model, ΔGDo*, is identical to the free energy of reversible unfolding of lipid-bound apoA-I (40). The values for the free energy of denaturation of lipid-bound apoA-I determined using the denaturant binding model, ΔGDo*, and using the linear extrapolation method, ΔGDo, appear to be in a good agreement (Table 3). The ΔGDo values are slightly lower than ΔGDo *, in accordance with the observation that the linear extrapolation approach gives the lowest estimate for the conformational stability (41). The midpoint of GdnCHl-induced denaturation, D1/2, determined from the denaturation curves (Figure 7) graphically and calculated using the linear extrapolation method resulted in identical values that are shown in Table 3. Thus, the linear extrapolation approach used for characterization of the chemical stability of lipid-free mutant forms of apoA-I (29, 30) appeared to give reasonable parameters to characterize denaturant-induced unfolding of apoA-I in rHDL.
Figure 6.
Effect of the mutations on thermally induced unfolding of apoA-I in rHDL monitored by the ellipticity at 222 nm.
The D102A/D103A mutation leads to a 3°C decrease in the midpoint of thermal unfolding, T1/2, a 0.4 M decrease in the midpoint of GdnHCl denaturation, D1/2, and 1.4-1.7 kcal/mol decrease in the conformation stability, suggesting that the apoA-I[D102A/D103A] is the most unstable double charge ablation mutant apoA-I in rHDL studied here. The E110A/E111A mutation results in 0.5-0.7 kcal/mol decrease in the conformation stability; and the R116V/K118A mutation leads to ~0.3 M decrease in the midpoint D1/2 of GdnHCl denaturation. Thus, all three double charge ablations in helix 4 lead to destabilization of apoA-I in rHDL. The R160V/H162A double charge ablation in helix 6 does not result in significant destabilization of apoA-I in rHDL. The Δ(61-78) deletion leads to a great destabilization of apoA-I in rHDL, as indicated by a 3°C decrease in the midpoint of thermal unfolding T1/2, a 0.4 M decrease in the midpoint D1/2 of GdnHCl denaturation, and 1.3-1.5 kcal/mol decrease in the conformational stability.
DISCUSSION
Studies of lipid-free apoA-I show the complexity and variety of inter-residue and inter-fragment interactions in the protein that are critical for maintaining its integrity. Probing apoA-I conformation by mutation shows that deletion or mutation of some residues disturbs the conformation and stability, while deletion or mutation of others does not affect the overall conformation and stability of the molecule or may even stabilize the protein. For example, deletion of the segment 165-175 has been shown to disrupt dramatically and destabilize the protein structure, while deletion of the segment 187-197 of a similar length does not affect the protein conformation and stability (29), and deletion of the segment 136-143 stabilizes the protein and increases the α-helical content (30). Residues of central helix 6 (residues 144-165) appeared to be involved in tertiary interactions with some parts of the N-terminal half of apoA-I, as indicated by the observed perturbation of the tertiary organization of the N-terminal half of the molecule by a Δ(144-165) deletion. (30). A mutation G185P/G186P located in the C-terminal part of apoA-I affects fluorescence of tryptophans located in the N-terminal half of apoA-I, which suggests a close proximity of the N- and C-termini of apoA-I and their interaction with each other in solution (29). Substitutions L222K/F225K/F229K (29) and E125K/E128K/K133E/E139K (30) stabilize lipid-free apoA-I and increase its α-helical content, while substitutions V156K/A158E (45) and E110A/E111A (12) destabilize the protein and result in a loss of helical structure. In rHDL, electrostatic interactions between acidic and basic residues of two anti-parallel apoA-I molecules via salt bridges have been proposed to be important for disc stability (27). However, no experimental data demonstrating that the proposed salt bridges are essential for the conformation and stability of apoA-I in rHDL have been reported. In the current study, we probe by mutation the central region of apoA-I between residues 61 and 162 in order to further identify important interactions, including possible electrostatic interactions, that determine apoA-I conformation and stability in the lipid-free state and in rHDL.
Two expression systems were used to generate WT and the variant apoA-I forms used in this study: baculovirus expression system for the WT and D102A/D103A, R116V/K118A, R160V/H162A, and Δ(61-78) mutants, and adenovirus expression system for the WT and E110A/E111A and Δ(121-142) mutants. In agreement with previous observations (5, 12, 29, 30), the physicochemical characteristics of WT apoA-I proteins expressed in different expression systems appeared to be slightly different (Tables 1-3). This difference may result from a difference in the propeptide at the N-terminus of the expressed proteins. ApoA-I produced by the baculovirus expression system contains the segment of five residues (Gly-Ala-Met-Gly-Ser-) that corresponds to the recognition sequence of the rTEV protease; apoA-I produced by the adenovirus expression system contains the six-residue propeptide (Arg-His-Phe-Trp-Gln-Gln-). Some studies (29, 46) have reported a difference in the conformation between WT apoA-I expressed in baculovirus expression system or in mammalian C127 cells and apoA-I isolated from human plasma that might also result from the presence of the N-terminus-attached segment in the expressed apoA-I. To exclude any effect of the expression system on the parameters determined in this study, all the conclusions in the study are based on the comparison of characteristics of WT and mutant apoA-I forms generated by the same expression system.
Our earlier analysis of the Δ(136-143) and E125K/E128K/K133E/E139K mutants of apoA-I suggested disordered structure of the segment between residues 121 and 142 in the lipid-free protein (30). To test this suggestion, we studied effects of the Δ(121-142) mutation on the conformation and stability of apoA-I. Although no significant difference in the percent α-helical content between WT and the Δ(121-142) mutant was observed (Table 1), a significant difference between these two proteins in the number of residues in helical conformation (147 for WT vs. 139 for the Δ(121-142) mutant) results because of the different total number of residues in WT and the Δ(121-142) mutant (249 vs. 227). Multiple measurements of the α-helical content for each protein obtained from different purifications consistently resulted in an α-helical content of the Δ(121-142) mutant that was (2 ± 1)% higher than that of WT. This corresponds to a (8 ± 3) residue lower number of residues in helical conformation in the Δ(121-142) mutant compared to that in WT. Thus, the Δ(121-142) deletion leads to a significantly smaller loss of residues in helical conformation compared to the number of deleted residues (22 residues), suggesting that the major part of the deleted segment is not helical in lipid-free apoA-I. However, (8 ± 3) residues of the deleted segment may be in helical conformation. The Δ(121-142) mutant appears to have higher stability and unfolding cooperativity and more compact tertiary structure compared to WT, which agrees with disordered structure of a major part of the deleted segment 121-142. Our earlier analysis of the Δ(136-143) mutant, which is consistent with a disordered structure of the region 136-143 in lipid-free apoA-I (30), together with an expected location of P121 in a loop connecting helixes 4 and 5, suggest that the (8 ± 3) residue helical segment may be located between residues 122 and 135, while the remaining (15± 3) residues of the region 121 -143 are in disordered conformation. It is likely that residues 123 to 130 form a small helix separated from helix 4 by the kink comprised of resides P121 and L122, and the segment of residues 131-143 is disordered (Figure 1A). This disordered segment may serve as “a hinge domain” providing an additional flexibility to the apoA-I molecule, and it appears to overlap with a putative flexible hinge domain of apoA-I in rHDL proposed to exist between residues 130 and 174 that may be either associated or dissociated from rHDL depending on the particle size (50). We have previously reported a ~17-residue gain in helical conformation of lipid-free apoA-I as a result of the E125K/E128K/K133E/E139K mutation (30). This observation is consistent with folding into helix of the (15± 3) residue disordered region located between residues 121 and 143 upon this quadruple-point mutation. Recent findings of Saito et al. (51) also agree with disordered structure of a major region between residues 121 and 143 in lipid-free apoA-I. Based on the observation that the enthalpy of binding of apoA-I to lipid is linearly correlated with the number of amino acids forming α-helix, the authors concluded that the enthalpy of binding of a Δ(123-166) apoA-I mutant to lipid is consistent with disordered structure of a region between residues 123 and 142 in lipid-free apoA-I, which becomes helical upon lipid binding.
We have studied effects of three double charge ablations in helix 4, D102A/D103A, E110A/E111A, and R116V/K118A, and the R160V/H162A double charge ablation in helix 6 on the conformation and stability of apoA-I. Of three double charge ablations made in helix 4, the E110A/E111A mutation has the most significant effect on the protein conformation, stability and unfolding cooperativity of the lipid-free protein. Given that the mutated residues in the apoA-I[E110A/E111A] are located in the middle of helix 4, the loss of ~18 residues in helical conformation caused by this mutation suggests unfolding of a whole helix segment, most likely helix 4 (Figure 1B). This leads to a great reduction in the protein stability and unfolding cooperativity and to a significantly less compact tertiary packing, as indicated by ANS fluorescence measurements. The D102A/D103A mutation located at the N-terminal end of helix 4 leads to a loss of ~10 residues in helical conformation suggesting unfolding of half of a helix segment, most likely, the N-terminal half of helix 4. This also results in a significant reduction in the protein stability and unfolding cooperativity and a looser tertiary packing. However, the changes induced by the D102A/D103A mutation are smaller than those induced by the E110A/E111A mutation. Both the E110A/E111A and D102A/D103A mutations lead to a less hydrophobic environment of Trp residues, consistent with a less compact tertiary packing of the mutant proteins and complete or partial unfolding of helix 4, where one of four Trp residues of apoA-I (Trp 108) is located. The findings suggest that in lipid-free apoA-I, D102/D103 and E110/E111 located in helix 4 are a part of the protein core-folded structure and are crucial for maintenance of the solution conformation and stability of the protein. In contrast, the R116V/K118A mutation located at the C-terminal end of helix 4 does not affect significantly the α-helical content and reduces only slightly the thermal stability and unfolding cooperativity of lipid-free apoA-I. This suggests that R116 and K118 do not play a major role in stabilizing the lipid-free conformation of apoA-I. In contrast to the other double charge ablations, the R116V/K118A mutation leads to a slightly more compact tertiary conformation compared to WT, as indicated by ANS measurements. This may be the result of substitutions of the large charged R116/K118 by relatively small and neutral Ala. The R160V/H162A double charge ablation was made in helix 6 that has been shown to be vital for the structure, stability and important functions of apoA-I (8, 9, 30, 47, 48). From the position in the helix and the nature of the mutation, the R160V/H162A double charge ablation is similar to the R116V/K118A double charge ablation made in helix 4. In the lipid-free protein, the R160V/H162A mutation, similar to the R116V/K118A, does not affect the secondary conformation. However, in contrast to the apoA-I[R116V/K118A], the apoA-I[R160V/H162A] has significantly less compact tertiary structure, consistent with enhanced ANS fluorescence, a red shift of the maximum of Trp fluorescence, and the reduced enthalpy of thermal unfolding. It has been reported that a R160Q mutation also led to a significant red shift of Trp fluorescence of apoA-I (48). The data suggest involvement of R160/H162 in important tertiary (inter-helical) electrostatic interactions in the lipid-free protein.
Most effects of the Δ(61-78) deletion on the characteristics of lipid-free apoA-I appear to be opposite to those of the Δ(121-142) deletion. The Δ(61-78) deletion leads to dramatically reduced stability and unfolding cooperativity, less compact tertiary structure, and a large loss (~29 residues) in helical conformation indicating unfolding of at least an 11- residue helical segment(s) in other part(s) of the molecule that are helical in WT apoA-I. Most likely, the segment of residues 79-88 adjacent to the deletion site is disordered in the apoA-I[Δ(61-78)] (Figure 1B). These significant changes suggest that some or all the residues of the region 61-78 are located in a core-folded structure of lipid-free apoA-I and are important for the protein conformation and stability. This agrees with data showing that the segment of residues 63-73, which overlaps with the 61-78 region, is essential for apoA-I function, such as phospholipid binding (10). Remarkably, of all the mutations studied in this work, the Δ(61-78) and E110A/E111A lead to the least compact tertiary packing and to the most significant increase in disordered conformation in lipid-free apoA-I (Figure 1B). Combined with greatly reduced stability and unfolding cooperativity, this suggests more flexible and less organized structure of the apoA-I[Δ(61-78)] and apoA-I[E110A/E111A] that may facilitate association of these mutants with larger lipoproteins, very low density and intermediate density lipoprotein. In apoA-I deficient mice expressing various mutant forms of apoA-I, two mutants, the apoA-I[Δ(61-78)] and apoA-I[E110A/E111A], were shown to have increased affinity to very low density and intermediate density lipoprotein (12, 32). Similarly in human plasma, one of the apoE isoforms, apoE4 that has the less organized and more flexible structure, is preferably associated with larger very low density lipoprotein, in contrast to apoE2 and apoE3 isoforms that are more structurally organized and preferably bound to smaller HDL particles (49). In apoA-I deficient mice expressing the apoA-I[Δ(61-78)] and apoA-I[E110A/E111A], the increased affinity of these mutant apoA-I to very low density and intermediate density lipoprotein may result in displacement of apoC-II from these lipoproteins making them poorer substrates for lipoprotein lipase and eventually may lead to hypertriglyceridemia (12, 32). Thus, the changes in the secondary and tertiary conformation and stability of apoA-I induced by the Δ(61-78) and E110A/E111A mutations may initiate mechanisms responsible for hypertriglyceridemia in apoA-I deficient mice expressing these mutants. More in vitro experiments are under way to demonstrate that the Δ(61-78) and E110A/E111A mutations increase the affinity of apoA-I to larger lipoprotein particles.
In rHDL, the Δ(61-78) deletion leads to unfolding of helical segment(s) and a great reduction in apoA-I stability, similar to that in the lipid-free state. This indicates that some or all residues of the segment 61-78 are involved in important interactions that are vital for maintenance of the apoA-I conformation and stability in both lipid-free apoA-I and apoA-I bound to rHDL. This agrees with previous findings showing that the segment of residues 63-73, which overlaps with the region 61-78, is important for the lipid-bound conformation and stability of apoA-I (10). All three double charge ablations made in helix 4, D102A/D103A, E110A/E111A, and R116V/K118A, result in reduced α-helical content and stability apoA-I in rHDL suggesting that at least one residue in each mutated pair of residues, D102/D103, E110/E111, and R116/K118, is involved in important interactions that maintain the conformation and stability of lipid-bound apoA-I. Thus, our analysis suggests that D102/D103 and E110/E111 are important for both the lipid-free and lipid-bound conformation of apoA-I; while R116/K118, which do not play a major role in maintenance of the lipid-free conformation, are essential for the conformation and stability of apoA-I in rHDL. The results appear to be in good agreement with the double belt model for rHDL (27). In this model, two apoA-I molecules in anti-parallel orientation form a dimeric structure in which D103, E111, and K118 of one apoA-I molecule form salt-bridges with H162, H155, andE147, respectively, of the other apoA-I molecule. We expected that, similar to the double charge ablations made in helix 4, the double charge ablation R160V/H162A in helix 6 also would affect the conformation of apoA-I in rHDL, because H162 in one apoA-I molecule and D103 in its anti-parallel mate form a salt bridge in the double belt model (27), and the R160V/H162A mutation has been shown to affect in vitro formation of a productive complex between rHDL and SR-B1 (7) and to obliterate LCAT activation ability of apoA-I (31). However, our analysis shows that the R160V/H162A mutation does not alter significantly the α-helical content and stability of apoA-I in rHDL. Similarly, it has been shown that mutations R160L (52) and R160Q (48) do not affect the conformation of apoA-I in rHDL, in agreement with the belt model (27), which predicts that R160 does not form a salt bridge with any residues of anti-parallel apoA-I molecule of the dimer, but instead points toward the surface of the disc. It was proposed that R160, along with residues R149 and R153, is a part of a positively charged cluster of helix 6 residues that interacts directly with LCAT (48). Our analysis of the apoA-I[R160V/H162A] further supports this hypothesis and explains the observed effect of the mutation of R160 on the formation of the productive complex between rHDL and SR-B1. Previous studies suggested that mutation of R160 either alter conformation of apoA-I in rHDL or disrupt direct contacts between the rHDL and SR-B1 (7). Our findings support the latter mechanism. In addition, our analysis suggests that the salt bridge between H162 in one apoA-I molecule and D103 in the anti-parallel apoA-I molecule predicted by the double belt model (27) is not critical for the stabilization of the discs, because the R160V/H162A mutation did not affect significantly the conformation and stability of apoA-I in the discs. It is possible that upon the mutation of H162, D103 may form an intrahelical salt bridge with K106, because in the double belt model, the side chain of K106 does not form a salt bridge with any residue and does not extend out into the aqueous medium. This possible switching of the salt bridge between D103 of one apoA-I molecule and H162 of the anti-parallel apoA-I molecule to an intrahelical salt bridge between D103 and K106 without altering the disc stability and conformation may be physiologically important, if H162 also directly participates in interactions with SR-B1 or LCAT. Such a participation may be predicted from the observed effects of the R160V/H162A mutation on the interaction of rHDL with SR-B1 (7) and from the impaired LCAT activation ability of a natural apoA-I mutant H162Q (53). Interestingly, a resent study by Alexander et al (15) suggests that negatively charge residues of apoA-I helix 6, E146, E147, D150, and D157, also may interact directly with LCAT.
In conclusion, the current study i) provides additional evidence that a major segment between residues 121 and 143, likely including residues 131 to 143, has disordered conformation in lipid-free apoA-I;. ii) identifies residues in the central part of apoA-I that are important for the conformation of apoA-I in the lipid-free state; iii) shows that the specific salt-bridge interactions predicted by the double belt model for apoA-I in rHDL are essential for the conformation and stability of the protein in rHDL; iv) contributes to our understanding of the effects of the mutations on apoA-I functions and induction of hypertriglyceridemia in apoA-I deficient mice following adenovirus-mediated gene transfer of the apoA-I[E110A/E111A] and apoA-I[Δ(61-78)].
Acknowledgments
We thank Donald Gantz for the expert electron microscopy analysis, Amit Gulati and Dr. Margaretha Carraway for sharing with us Sf-9 cells and advice on the protein expression, and Dr. R. Andrew Zoeller for making available for us his Fluoromax-2 fluorescence spectrometer.
Abbreviations
- ABCA1
ATP binding cassette transporter 1
- ANS
8-Anilino-2-naphthalene-sulfonate
- apo
apolipoprotein
- CD
circular dichroism
- GdnHCL
guanidine hydrochloride
- HDL
high density lipoprotein(s)
- LCAT
lecithin:cholesterol acyltransferase
- POPC
palmitoyloleoylphosphatidylcholine
- rHDL
reconstituted high density lipoprotein(s)
- SR-B1
scavenger receptor class B type I
- rTEV protease
recombinant tobacco etched viral protease
- WMF
wavelength of maximum fluorescence
- WT
recombinant human wild type apoA-I
Footnotes
This work was supported by PO1HL 26335 and grant HL 48739 from the National Institute of Heath
References
- 1.Fielding CJ, Fielding PE. Molecular physiology of reverse cholesterol transport. J Lipid Res. 1995;36:211–228. [PubMed] [Google Scholar]
- 2.Meagher EA. Addressing cardiovascular risk beyond low-density lipoprotein cholesterol: the high-density lipoprotein cholesterol story. Curr Cardiol Rep. 2004;6:457–463. doi: 10.1007/s11886-004-0055-2. [DOI] [PubMed] [Google Scholar]
- 3.Asztalos BF. High-density lipoprotein metabolism and progression of atherosclerosis: new insights from the HDL. Atherosclerosis Treatment Study. Curr Opin Cardiol. 2004;19:385–391. doi: 10.1097/01.hco.0000126979.41946.7e. [DOI] [PubMed] [Google Scholar]
- 4.Eck MV, Pennings M, Hoekstra M, Out R, Van Berkel TJ. Scavenger receptor BI and ATP-binding cassette transporter A1 in reverse cholesterol transport and atherosclerosis. Curr Opin Lipidol. 2005;16:307–315. doi: 10.1097/01.mol.0000169351.28019.04. [DOI] [PubMed] [Google Scholar]
- 5.Chroni A, Liu T, Gorshkova IN, Kan H-Y, Uehara Y, von Eckardstien A, Zannis VI. The central helices of apoA-I can promote ATP-binding cassette transporter A1 (ABCA1)-mediated lipid efflux. Amino acid residues 220-231 of the wild-type apoA-I are required for lipid-efflux in vitro and high density lipoprotein formation in vivo. J Biol Chem. 2003;278:6719–6730. doi: 10.1074/jbc.M205232200. [DOI] [PubMed] [Google Scholar]
- 6.Krieger M. Scavenger receptor class B type I is a multiligand HDL receptor that influences diverse physiologic systems. J Clin Invest. 2001;108:793–797. doi: 10.1172/JCI14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu T, Krieger M, Kan H-Y, Zannis VI. The effects of mutations in helices 4 and 6 of ApoA-I on scavenger receptor class B type I (SR-BI)-mediated cholesterol efflux suggest that formation of a productive complex between reconstituted high density lipoprotein and SR-BI is required for efficient lipid transport. J Biol Chem. 2002;277:21576–21584. doi: 10.1074/jbc.M112103200. [DOI] [PubMed] [Google Scholar]
- 8.Sorci-Thomas MG, Currtiss L, Parks JS, Thomas MJ, Kearns MW, Landrum M. The hydrophobic face orientation of apolipoprotein A-I amphipathic helix domain 143-164 regulates lecithin:cholesterol acyltransferase activation. J Biol Chem. 1998;273:11776–11782. doi: 10.1074/jbc.273.19.11776. [DOI] [PubMed] [Google Scholar]
- 9.Sorci-Thomas MG, Thomas M, Curtiss L, Landrum M. Single repeat deletion in apoA-I blocks cholesterol esterification and results in rapid catabolism of Δ6 and wild-type apoA-I in transgenic mice. J Biol Chem. 2000;275:12156–12163. doi: 10.1074/jbc.275.16.12156. [DOI] [PubMed] [Google Scholar]
- 10.Sviridov D, Hoang A, Sawyer WH, Fidge NH. Identification of a sequence of apolipoprotein A-I associated with the activation of lecithin:cholesterol acyltransferase. J Biol Chem. 2000;275:19707–19712. doi: 10.1074/jbc.M000962200. [DOI] [PubMed] [Google Scholar]
- 11.Roosbeek S, Vanloo B, Duverger N, Caster H, Breyne J, De Beun I, Patel H, Vandekerckhove J, Shoulders C, Rosseneu M, Peelman F. Three arginine residues in apolipoprotein A-I are critical for activation of lecithin:cholesterol acyltransferase. J Lipid Res. 2001;42:31–40. [PubMed] [Google Scholar]
- 12.Chroni A, Kan H-Y, Kypreos KE, Gorshkova IN, Shkodrani A, Zannis VI. Substitutions of Glu110 and Glu111 in the middle helix 4 of human ApoA-I by Alanine affect the structure and in vitro functions of apoA-I and induce severe hypertriglyceridemia in ApoA-I-deficient mice. Biochemistry. 2004;43:10442–10457. doi: 10.1021/bi049782p. [DOI] [PubMed] [Google Scholar]
- 13.Chroni A, Liu T, Fitzgerald ML, Freeman MW, Zannis VI. Cross-linking and lipid efflux properties of apoA-I mutants suggest direct association between ApoA-I helices and ABCA1. Biochemistry. 2004;43:2126–2139. doi: 10.1021/bi035813p. [DOI] [PubMed] [Google Scholar]
- 14.Satio H, Lund-Katz S, Phillips MC. Contributions of domain structure and lipid interaction to the functionality of exchangeable human apolipoproteins. Progress in Lipid Res. 2004;43:350–380. doi: 10.1016/j.plipres.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 15.Alexander ET, Bhat S, Thomas MJ, Weinberg RB, Cook VR, Bharadwaj MS, Sorci-Thomas M. Apolipoprotein A-I helix 6 negatively charged residues attenuate lecithin-cholesterol acyltransferase (LCAT) reactivity. Biochemistry. 2005;44:5409–5419. doi: 10.1021/bi047412v. [DOI] [PubMed] [Google Scholar]
- 16.Nolte RT, Atkinson D. Conformational analysis of apolipoprotein A-I and E-3 based on primary sequence and circular dichroism. Biophys J. 1992;63:1221–1239. doi: 10.1016/S0006-3495(92)81698-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Segrest JP, Jones MK, De Loof H, Brouillette CG, Venkatachalapathi YV, Anantharamaiah GM. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J Lipid Res. 1992;33:141–166. [PubMed] [Google Scholar]
- 18.Borhani DW, Rogers DP, Engler JA, Brouillette CG. Crystal structure of truncated human apolipoprotein A-I suggests a lipid-bound conformation. Proc Natl Acad Sci U S A. 1997;94:12291–12296. doi: 10.1073/pnas.94.23.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silva RA, Hilliard GM, Fang J, Macha S, Davidson WS. A three-dimensional molecular model of lipid-free apolipoprotein A-I determined by cross-linking/mass spectrometry and sequence threading. Biochemistry. 2005;44:2759–2769. doi: 10.1021/bi047717+. [DOI] [PubMed] [Google Scholar]
- 20.Tricerri MA, Behling Agree AK, Sanchez SA, Jonas A. Characterization of apolipoprotein A-I structure using a cysteine-specific fluorescence probe. Biochemistry. 2000;39:14682–14691. doi: 10.1021/bi0014251. [DOI] [PubMed] [Google Scholar]
- 21.Tricerri MA, Behling Agree AK, Sanchez SA, Bronski J, Jonas A. Arrangement of apolipoprotein A-I in reconstituted high-density lipoprotein disks: an alternative model based on fluorescence resonance energy transfer experiments. Biochemistry. 2001;40:5065–5074. doi: 10.1021/bi002815q. [DOI] [PubMed] [Google Scholar]
- 22.Cushley RJ, Okon M. NMR studies of lipoprotein structure. Annu Rev Biophys Biomol Struct. 2002;31:177–206. doi: 10.1146/annurev.biophys.31.101101.140910. [DOI] [PubMed] [Google Scholar]
- 23.Oda MN, Forte TM, Ryan RO, Voss JC. The C-terminal domain of apolipoprotein A-I contains a lipid-sensitive conformational trigger. Nat Struct Biol. 10:455–460. doi: 10.1038/nsb931. [DOI] [PubMed] [Google Scholar]
- 24.Rogers DP, Roberts LM, Lebowitz J, Engler JA, Brouillette CG. Structural analysis of apolipoprotein A-I: effects of amino- and carboxy-terminal deletions on the lipid-free structure. Biochemistry. 1998;37:945–955. doi: 10.1021/bi9713512. [DOI] [PubMed] [Google Scholar]
- 25.Roberts LM, Ray MJ, Shih TW, Hayden E, Reader MM, Brouillette CG. Structural analysis of apolipoprotein A-I: limited proteolysis of methionine-reduced and -oxidized lipid-free and lipid-bound human apo A-I. Biochemistry. 1997;36:7615–7624. doi: 10.1021/bi962952g. [DOI] [PubMed] [Google Scholar]
- 26.Behling Agree AK, Tricerri MA, Arnvig McGuire K, Tian SM, Jonas A. Folding and stability of the C-terminal half of apolipoprotein A-I examined with a Cys-specific fluorescence probe. Biochim Biophys Acta. 2002;1594:286–296. doi: 10.1016/s0167-4838(01)00317-x. [DOI] [PubMed] [Google Scholar]
- 27.Segrest JP, Jones MK, Klon AE, Sheldahl CJ, Hellinger M, De Loof H, Harvey SC. A detailed molecular belt model for apolipoprotein A-I in discoidal high density lipoprotein. J Biol Chem. 1999;274:31755–31758. doi: 10.1074/jbc.274.45.31755. [DOI] [PubMed] [Google Scholar]
- 28.Davidson WS, Silva RA. Apolipoprotein structural organization in high density lipoproteins: belts, bundles, hinges and hairpins. Curr Opin Lipidol. 2005;16:295–300. doi: 10.1097/01.mol.0000169349.38321.ad. [DOI] [PubMed] [Google Scholar]
- 29.Gorshkova IN, Liadaki K, Gursky O, Atkinson D, Zannis VI. Probing the lipid-free structure and stability of apolipoprotein A-I by mutation. Biochemistry. 2000;39:15910–15919. doi: 10.1021/bi0014406. [DOI] [PubMed] [Google Scholar]
- 30.Gorshkova IN, Liu T, Zannis VI, Atkinson D. Lipid-free structure and stability of apolipoprotein A-I: probing the central region by mutation. Biochemistry. 2002;41:10529–10539. doi: 10.1021/bi025807d. [DOI] [PubMed] [Google Scholar]
- 31.Chroni A, Duka A, Kan H-Y, Liu T, Zannis VI. Point mutations in apolipoprotein A-I mimic the phenotype observed in patients with classical lecithin:cholesterol acyltransferase deficiency. Biochemistry. 2005;44:14353–14366. doi: 10.1021/bi050962o. [DOI] [PubMed] [Google Scholar]
- 32.Chroni A, Kan H-Y, Shkodrani A, Liu T, Zannis VI. Deletions of helices 2 and 3 of human apoA-I are associated with severe dyslipidemia following adenovirus-mediated gene transfer in apoA-I-deficient mice. Biochemistry. 2005;44:4108–4117. doi: 10.1021/bi047998l. [DOI] [PubMed] [Google Scholar]
- 33.Sorci-Thomas MG, Parks JS, Kearns MW, Pate GN, Zhang C, Thomas M. High level secretion of wild-type and mutant forms of human proapoA-I using baculovirus-mediated Sf-9 cell expression. J Lipid Res. 1996;37:673–683. [PubMed] [Google Scholar]
- 34.Matz CE, Jonas A. Micellar complexes of human apolipoprotein A-I with phosphatidylcholines and cholesterol prepared from cholate-lipid dispersions. J Biol Chem. 1982;257:4535–4540. [PubMed] [Google Scholar]
- 35.Laccotripe M, Makrides SC, Onas A, Zannis VI. The carboxyterminal hydrophobic residues of apolipoprotein A-I affect its rate of phosholipid binding and its association with high density llipoproteins. J Biol Chem. 1987;272:17511–17522. doi: 10.1074/jbc.272.28.17511. [DOI] [PubMed] [Google Scholar]
- 36.Chen Y-H, Yang JT, Martinez HM. Determination of the secondary structures of proteins by circular dichroism and optical rotary dispersion. Biochemistry. 1972;11:1420–1431. doi: 10.1021/bi00772a015. [DOI] [PubMed] [Google Scholar]
- 37.Pace CN, Shirley BA, Thomson JA. In: Protein Structure. Creigton TE, editor. IRL Press; NY: 1989. pp. 311–330. [Google Scholar]
- 38.John DM, Weeks KM. van’t Hof enthalpies without baselines. Protein Science. 2000;9:1416–1419. doi: 10.1110/ps.9.7.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pace CN, Vanderburg KE. Determining globular protein stability: guanidine hydrochloride denaturation of mioglobin. Biochemistry. 1979;18:288–292. doi: 10.1021/bi00569a008. [DOI] [PubMed] [Google Scholar]
- 40.Sparks DL, Lund-Katz S, Phillips M. The charge and structural stability of apolipoprotein A-I in discoidal and spherical recombinant high density lipoprotein particles. J Biol Chem. 1992;267:25839–25847. [PubMed] [Google Scholar]
- 41.Pace CN. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods in Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 42.Lacowicz J. Principles of Fluorescence Spectroscopy. Kluwer Academic/Plenum Publishers; New York: 1999. pp. 71–72. [Google Scholar]
- 43.Reijngoud D-J, Phillips MC. Mechanism of dissociation of human apolipoproteins A-I, A-II, and C from complexes with dimyristoylphosphatidylcholine as studied by thermal denaturation. Biochemistry. 1984;23:726–734. doi: 10.1021/bi00299a022. [DOI] [PubMed] [Google Scholar]
- 44.Mehta R, Gantz DL, Gursky O. Human plasma high-density lipoproteins are stabilized by kinetic factors. J Mol Biol. 2003;328:183–192. doi: 10.1016/s0022-2836(03)00155-4. [DOI] [PubMed] [Google Scholar]
- 45.Han JM, Jeong TS, Lee WS, Choi I, Cho KH. Structural and functional properties of V156K and A158E mutants of apolipoprotein A-I in the lipid-free and lipid-bound states. J Lipid Res. 2005;46:589–596. doi: 10.1194/jlr.M400468-JLR200. [DOI] [PubMed] [Google Scholar]
- 46.Rogers D, Roberts LM, Lebowitz J, Datta G, Anantharamaiah GM, Engler J, Brouillette CG. The lipid-free structure of apolipoprotein A-I: effect of amino-terminal deletions. Biochemistry. 1998;37:11714–11725. doi: 10.1021/bi973112k. [DOI] [PubMed] [Google Scholar]
- 47.McManus DC, Scott BR, Frank PG, Franklin V, Schultz JR, Marcel YL. Distinct central amphipathic alpha-helices in apolipoprotein A-I contribute to the in vivo maturation of high density lipoprotein by either activating lecithin-cholesterol acyltransferase or binding lipids. J Biol Chem. 2000;275:5043–5051. doi: 10.1074/jbc.275.7.5043. [DOI] [PubMed] [Google Scholar]
- 48.Roosbeek S, Vanloo B, Duverger N, Caster H, Breyne J, De Beum I, Patel H, Vandekerckhove J, Shoulders C, Rosseneu M. Three arginine residues in apolipoprotein A-I are critical for activation of lecithin:cholesterol acyltransferase. J Lipid Res. 2001;42:31–40. [PubMed] [Google Scholar]
- 49.Saito H, Dhanasekaran P, Baldwin F, Weisgraber KH, Phillips MC, Lund-Katz S. Effect of polymorphism on the lipid interaction of human apolipoprotein E. J Biol Chem. 2003;278:40723–40729. doi: 10.1074/jbc.M304814200. [DOI] [PubMed] [Google Scholar]
- 50.Maiorano JN, Jandacek RJ, Horace EM, Davidson WS. Identification and structural ramifications of a hinge domain in apolipoprotein A-I discoidal high-density lipoproteins of different size. Biochemistry. 2004;43:11717–11726. doi: 10.1021/bi0496642. [DOI] [PubMed] [Google Scholar]
- 51.Saito H, Dhanasekaran P, Nguyen D, Deridder E, Holvoet P, Lund-Katz S, Phillips MC. Alpha-helix formation is required for high affinity binding of human apolipoprotein A-I to lipids. J Biol Chem. 2004;279:20974–20981. doi: 10.1074/jbc.M402043200. [DOI] [PubMed] [Google Scholar]
- 52.Cho KH, Durbin DM, Jonas A. Role of individual amino acids of apolipoprotein A-I in the activation of lecithin:cholesterol acyltransferase and in HDL rearrangements. J Lipid Res. 2001;42:379–389. [PubMed] [Google Scholar]
- 53.Hoang A, Huang W, Sasaki J, Sviridov D. Natural mutations of apolipoprotein A-I impairing activation of lecithin:cholesterol acyltransferase. Biochim Biophys Acta. 2003;1631:72–76. doi: 10.1016/s1388-1981(02)00357-8. [DOI] [PubMed] [Google Scholar]