

Abstract
First examples of stable carbocations are reported from several classes of thia-PAHs with four fused rings, namely benzo[b]naphtho[2,1-d]thiophene (1), and its 3-methoxy-derivative (2), phenanthro[4,3-b]thiophene (3) and its 7-methoxy- (4), 10-methoxy- (5) and 9-methoxy- (6) derivatives, phenanthro[3,4-b]thiophene (7) and its 7-methoxy- (8) and 9-methoxy- (9) derivative, and 3-methoxybenzo[b]naphtha[1,2-d]thiophene (11). In several cases the resulting carbocations were also studied by GIAO-DFT. Charge delocalization modes in the resulting carbocations were probed. A series of S-alkylated onium tetrafluoroborates namely 1Me+, 1Et+, 2Et+, and 7Me+ (from 1, 2 and 7), 10Me+ and 10Et+ (from benzo[b]naphtha[1,2-d]thiophene 10), 12Me+ and 12Et+ (from phenanthro[3,2-b][1]benzothiophene 12), 13Me+ (from 3-methoxyphenanthro[3,2-b]benzothiophene 13), 14Me+ (from phenanthro[4,3-b][1]benzothiophene 14), and 15Me+ (from 3-methoxyphenanthro[4,3-b][1]benzothiophene 15) were synthesized. PAH-sulfonium salts 1Me+, 1Et+, 10Me+, 10Et+, 12Me+, and 14Me+ proved to be efficient akylating agents towards model nitrogen bases (imidazole and azaindole). Facile transalkylation to model nucleophiles (including guanine) is also supported by favorable reaction energies computed by DFT. Ring opening energies in thia-PAH-epoxides from 1, 3, and 7, and charge delocalization modes in the resulting carbocations were also evaluated. The 4-ring fused thia-PAHs 1, 2, 3, 4, 5, 7, 8, and 11 are effectively nitrated under extremely mild conditions. Nitration regioselectivity corresponds closely to protonation under stable ion conditions. Bromination of 4 and 6 are also reported. Comparative mutagenicity assays (Ames test) were performed on 1 versus 1NO2, 5 versus 5NO2 and 11 versus 11NO2. Compound 5NO2 was found to be a potent direct acting mutagen.
Introduction
Studies aimed at understanding the mechanism of activation of thia-PAHs (the sulfur analogs of PAHs) have been quite limited, despite their recognition as widely distributed environmental contaminants with mutagenic and carcinogenic activity.1,2 Synthesis and bioassay of the 4-ring fused systems, namely benzo[b]naphtho[2,1-d]thiphene 1, phenanthro[4,3-b]thiophene 3, and the isomeric phenanthro[3,4-b]thiophene 7 have been reported (see Fig 1).3–8
Figure 1.
Studied thia-PAHs.
Compound 1 can be viewed as a thia-chrysene with two bay-regions. Its 1,2- and 3,4-dihydrodiols have been synthesized.3,4 The metabolism of 1 was suggested to be in part via bay-region diol-epoxide and in part by sulfur activation (sulfoxide and sulfone formation). The 5-nitro-derivative of 1 binds to DNA and the structures of the metabolites indicated both ring oxidation and nitro reduction.5 Compounds 3 and its isomer 7 can be viewed as the sulfur analogs of benzo[c]phenanthrene BcPh. Interestingly, whereas 3 (sulfur in the fjord-region) is not mutagenic, 7 was reported to be as mutagenic as benzo[a]pyrene (BaP).8 Synthesis of the 6,7-dihydrodiol and 8,9-dihydrodiol of 3 and 7 were reported.7,8 Among the 5-ring fused systems, phenanthro[3,2-b][1]benzothiophene 12 and phenanthro[4,3-b][1]benzothiophene 14 (in Fig 1) have been the subject of synthetic and bioassay study.9–11 Compound 12, which could be viewed as the sulfur analog of dibenz[a,h]anthracene DBA, is a strong carcinogen (more active than DBA).9
Synthesis of its 3,4-dihydrodiol was reported.9 Metabolic activation by rodent liver microsomes produced the 3,4-dihydrodiol and the sulfoxide as major metabolites.10 Compound 14 is a benzannulated analog of 3 with a fjord-region. Synthesis of its 3,4-dihydrodiol was reported.11
The majority of polyaromatic heterocyclic compounds studied for their potential as DNA intercalators are aza-PAHs, for which there are numerous literature reports, in particular on diazapyrenium,12 phenanthridinium,13–15 and benzo[c]phenanthridinium salts,16 as analogs of ethidinium. Examples of sulfonium-based DNA intercalators do not appear to exist. Availability of the compounds listed in Figure 1 (parent systems and isomeric methoxy derivatives) provided the impetus for the present multifaceted investigation that combined stable carbocation generation with the synthesis of several S-alkylated onium salts including a study of their transfer-alkylation towards imidazole and azaindole as representative nitrogen nucleophile receptors. Using nitration (and bromination) as model electrophilic reactions, a number of new derivatives were synthesized. DFT was used to model epoxide ring opening in thia-PAHs, and mutagenicity tests (Ames assay) were performed on the nitro-derivatives of 1, 5, and 11.
Results and Discussion
a) Stable carbocations from thia-PAHs 1 and 2 (Fig 2a–2b, Chart S1, Table S1)
Figure 2.
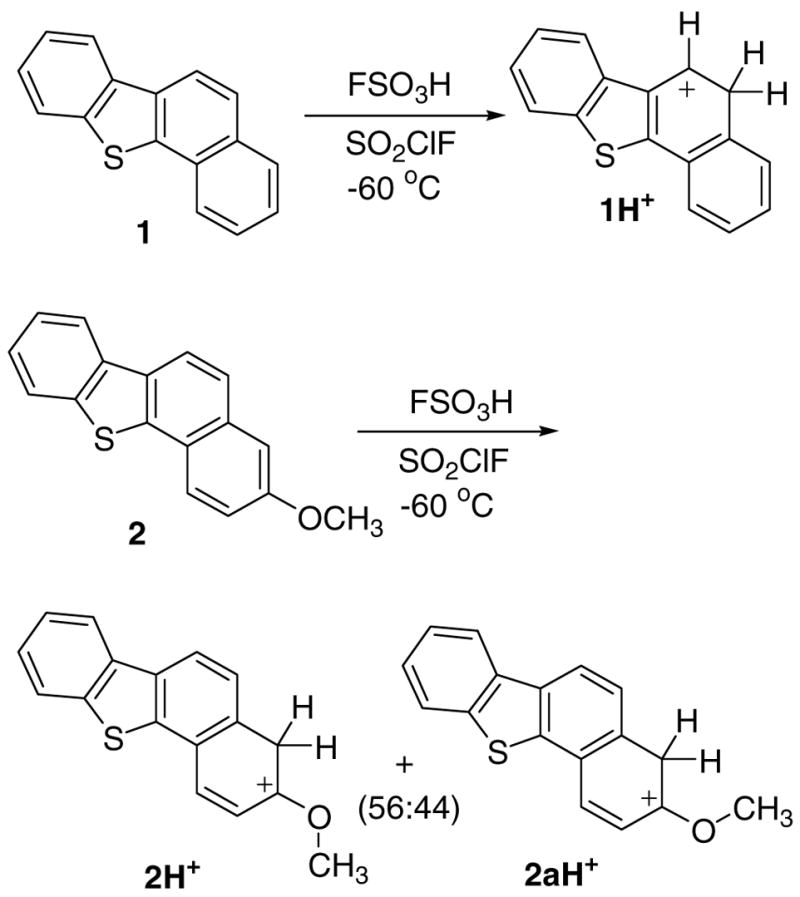

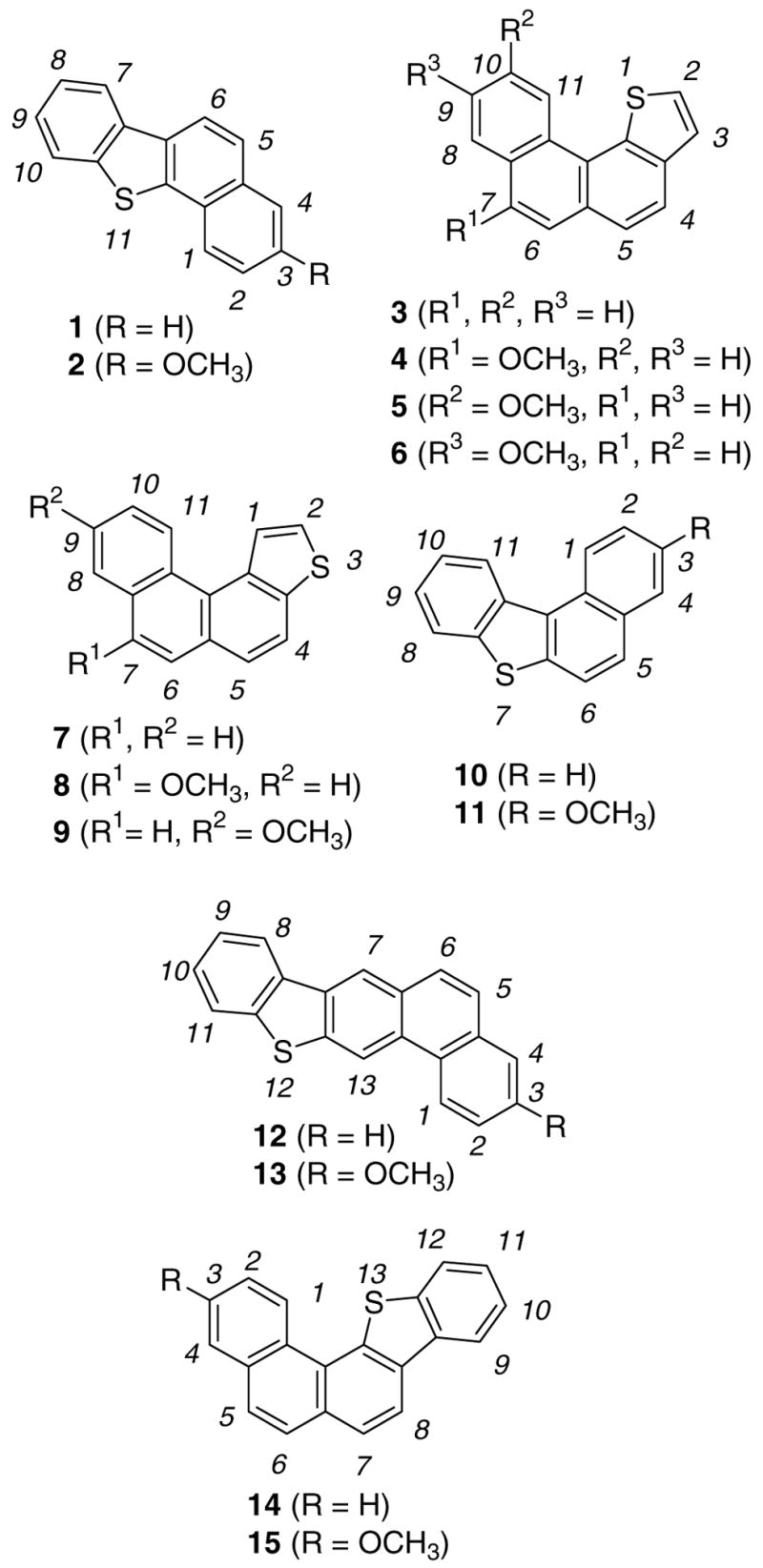
Figure 2a. Protonation of 1 and 2 in FSO3H/SO2ClF.
Figure 2b. Experimental and GIAO/B3LYP/6-31G(d)-derived NMR chemical shifts for 1H+, and 1H22+, Δδ13C values (in parentheses) relative to those for the parent 1 (red circles are roughly proportional to magnitude of Δδ13C values, threshold 5 ppm; a and b designations refer to interchangeable assignments), and changes in the NPA-derived charges (Δq) for 1H+(dark circles are roughly proportional to C Δq and white circle to S Δq, threshold was set to 0.030).
Low temperature protonation of 1 in FSO3H/SO2ClF gave 1H+ by attack at C-5 (Fig. 2a). This outcome agrees with DFT (Table S1) showing that, among all possible protonation sites, the carbocation derived from attack at C-5 has the lowest energy, with S-protonation computed to be significantly less favorable. For comparison, the NMR data for 1H+ (a thia-arenium ion) and 1H22+ (a sulfonium-arenium dication) were computed by GIAO-NMR (Fig. 2b). The experimental NMR data (Fig. 2b) correlate more closely with the GIAO for 1H+. On this basis, significant involvement of 1H22+ was ruled out, although limited equilibrium protonation at sulfur can not be excluded. Charge delocalization in 1H+ is primarily confined to the naphthalenium moiety. Whereas the NPA-derived changes in charges (Δq) agrees with the NMR derived charge map, it also places significant positive charge at sulfur (Fig. 2b).
Strong directive effect of the methoxy substituent becomes evident in low temperature protonation of the 3-methoxy-derivative 2 (Fig. 2a), which on low temperature reaction with FSO3H/SO2ClF gave 2H+ and 2aH+ (as two geometrical isomers) by attack at C-4 (in 56 : 44 ratio). The identity of each conformer was established via NOE experiments. NMR assignments of the two species (in particular those of the quaternary ring junction carbons) were aided by GIAO calculations (see Chart S1). Charge delocalization mode in 2H+ and 2aH+signify strong naphthalenium ion character in these carbocations.
b) Stable carbocations from thia-PAHs 3, 4, 5, and 6 (Fig 3, Chart S2, Tables S1-S2)
Figure 3.
Protonation of 3–6 in FSO3H/SO2ClF
Low temperature protonation of 3 with FSO3H/SO2ClF led to the formation of 3H+ as the sole carbocation (protonation at C-2) (Fig. 3). This underlines the importance of benzylic delocalization as compared to α-sulfur stabilization (protonation at C-3). DFT concurs (Table S1), showing that the carbocation derived from protonation at C-2 is the most stable, with C-3 protonation computed to be 14 kcal/mol less favorable. Chart S2 provides a summary of the experimental NMR assignments for 3H+ and its GIAO-derived data. The charge delocalization mode deduced based on magnitude of Δδ13C is in good agreement with the NPA-derived charge pattern based on Δq (Chart S2), showing relatively extensive delocalization throughout the system with highest charge localization at C-3 in the 5-membered ring. Directive effect of the methoxy-group manifested itself in the protonation of the 7-methoxy-derivative 4, which gave a mixture of two carbocations 4H+ (protonation at C-6; alpha to methoxy) and 4aH+ (protonation at C-2; alpha to sulfur). At the onset, the carbocation ratio was 2:1 in favor of 4H+, changing subsequently to 2:3. According to DFT, 4aH+ is 4.9 kcal/mol more stable (Table S2). Variation in carbocation ratios in solution likely originates from variation in local overheating on contact with the superacid at low temperature.
Experimental and GIAO-derived NMR data for 4H+ and 4aH+ are summarized in Chart S2. Assignment of the relative geometry of the OMe group in 4H+ was based on NOE. Positive charge in 4H+ (a thia-carboxonium ions) is primarily localized at the carboxonium group and the benzannelated ring. In 4aH+, positive charge is mainly localized at the 5-membered ring with limited delocalization throughout the periphery.
An interesting outcome was observed in the low temperature protonation of the 10-methoxy-derivative 5 with FSO3H/SO2ClF, resulting in the formation of 5H+ and 5aH+, as two geometrical isomers (in 4:1 ratio) by protonation in the fjord-region (at C-11). Relative assignments of the two conformers were established by NOE.
Based on the DFT, 5H+ is 1.8 kcal/mol more stable than 5aH+, and protonation at the other alpha-methoxy position (C-9) is 6.4 kcal/mol less favorable relative to 5aH+. Fine-tuning of the experimental NMR assignments for 5H+ and 5aH+ (in particular with respect to ring junction carbons) were assisted by the GIAO-DFT data and are gathered in Chart S2 for comparison.
Low temperature protonation of the 9-methoxy-derivative 6 with FSO3H/SO2ClF resulted in the formation of three carbocations namely 6H+ (protonation at C-2), and 6aH+/6bH+ (two geometrical isomers, formed by protonation at C-8; alpha to methoxy) in 1:4:1 ratio respectively. Relative assignments for 6aH+/6bH+ were derived from NOE, and GIAO trends were used as guide to fine-tune the experimental NMR assignments (Chart S2).
c)Stable carbocations from thia-PAHs 7, 8 and 9 (Fig 4a–4b, Chart S3, Table S1)
Figure 4.


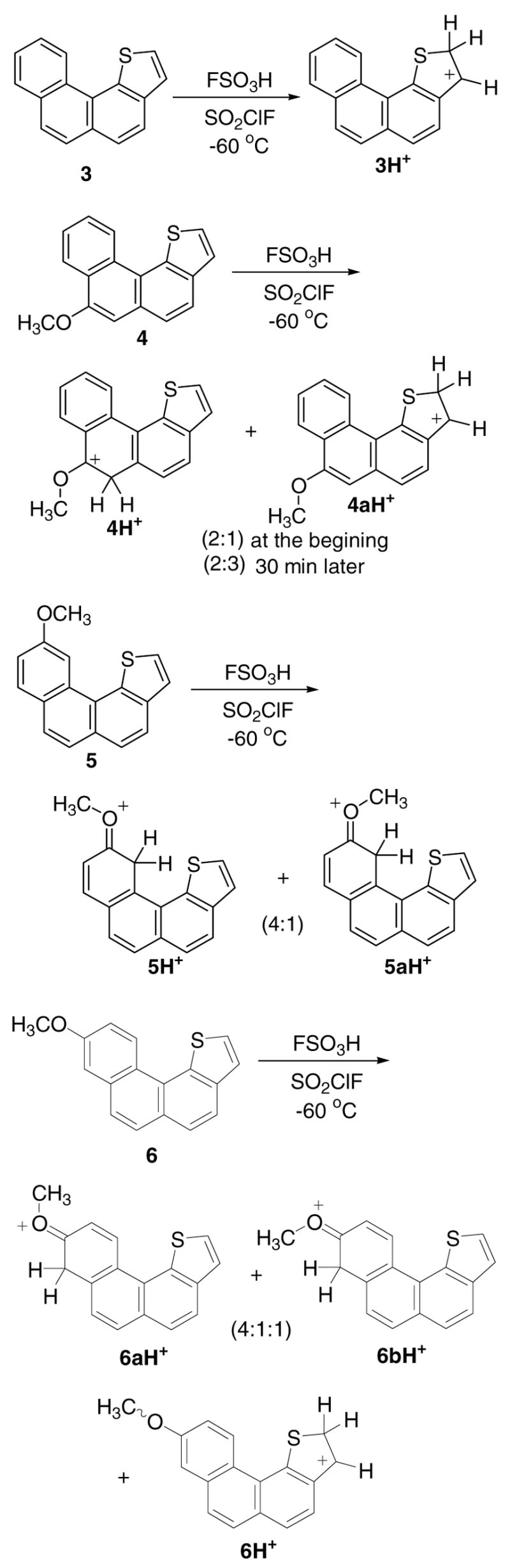
Figure 4a. Protonation of 7–9 in FSO3H/SO2ClF
Figure 4b. Experimental and GIAO/B3LYP/6-31G(d)-derived NMR chemical shifts for 7H+, and Δδ13C values (in parentheses) relative to those for the parent 7 (red circles are roughly proportional to magnitude of Δδ13C values, threshold 5 ppm; a and b designations refer to interchangeable assignments; nd = not detected), and changes in the NPA-derived charges (Δq) for 7H+(dark circles are roughly proportional to C Δq and white circle to S Δq, threshold was set to 0.030).
Low temperature reaction of 7 with FSO3H/SO2ClF resulted in protonation at C-2 to give 7H+ as the sole carbocation (Fig 4a). This outcome agrees with DFT, showing that among all possible protonation sites C-2 protonation was best, with C-5 protonation as the next best possibility, lying 4.4 kcal/mol higher (Table S1). Experimental and GIAO NMR data for 7H+ are collected in Fig. 4b for comparison. Whereas the Δδ13C-derived charge delocalization map indicates extensive delocalization throughout the thia-arenium ion, highest charge localizations are at C-1, C-3a, and C-5. This agrees with the charge map derived via NPA-derived Δq values (Fig. 4b).
Protonation regisoselectivity observed in 7 changed entirely when methoxy-substituents were introduced at C-7 (compound 8) and at C-9 (compound 9), whereby carbocation 8H+ was generated from 8 as the sole carbocation by attack at C-6 (alpha to OMe), and 9H+/9aH+ were obtained from 9 as geometrical isomers (in 4:1 ratio), by protonation at C-9 (alpha to OMe) (Fig 4a). Experimental and GIAO NMR data for 8H+ and for 9H+/9aH+ are gathered in Chart S3 for cross comparison. Charge delocalization in 8H+ is mainly confined to the carboxonium center and the benzannelated ring. Similarly, limited charge delocalization is noted in 9H+/9aH+, with positive charge mainly localized in the methoxy-bearing ring and on one conjugated ring carbon.
d) Protonation of thia-PAH 11 (Fig 5, Chart S4)
Figure 5.
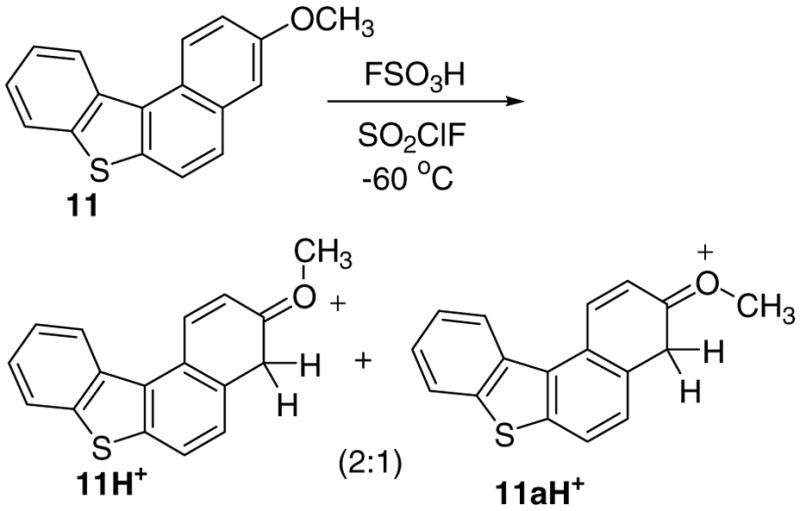
Protonation of 11.
Whereas low temperature reaction of parent 10 with FSO3H/SO2ClF resulted in poorly resolved spectra that could not be analyzed, the 3-methoxy-derivative 11 gave relatively improved spectra showing the formation of 11H+/11aH+ as geometrical isomers (in 2:1 ratio) (Fig 5), for which partial NMR assignments could be made (Chart S1). This outcome once again underscores the influence of the OMe substituents in directing electrophilic attack.
e) Synthesis of onium salts by S-alkylation (Fig 6, Chart S5)
Figure 6.
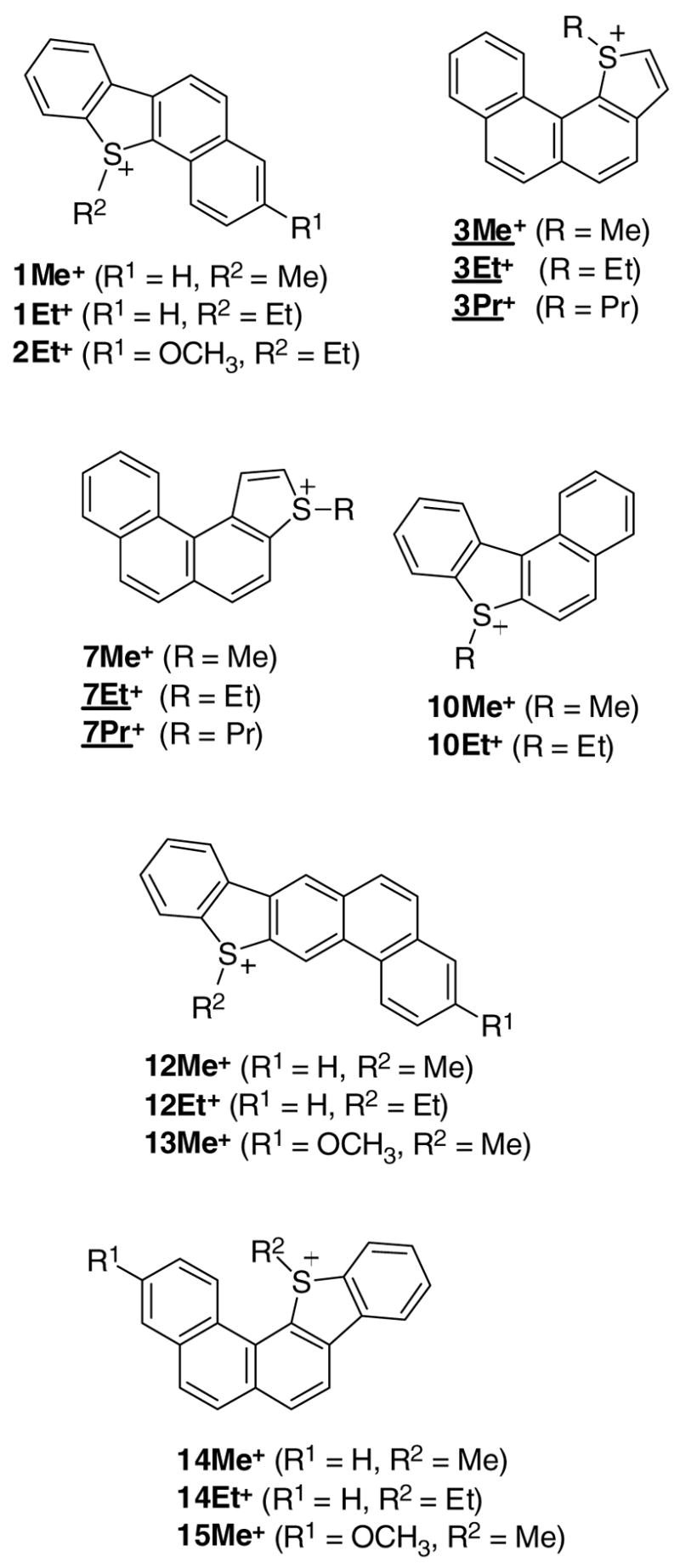
Synthesis of S-alkylated onium salts from thia-PAHs (all sulfonium salts listed were synthesized, except those that are underlined which were only studied computationally; see further).
As mentioned in the introduction, in the context of the present study and in order to further develop the electrophilic chemistry of thia-PAHs, we focused on synthesis and characterization of model sulfonium salts derived from compounds listed in Fig 1, and examined their potential as alkylating agents in model reactions. Whereas trialkyl(aryl)sulfonium salts are readily prepared by reaction with a variety of alkylating agents,17a initial studies aimed at developing suitable alkylating systems for thia-chrysene 1, proved to be unexpectedly challenging. Thus attempted alkylation with EtOTf either without an added Lewis acid or in the presence of various promoters such as AgOTf, BF3/Et2O, and Sc(OTf)3 at r.t. in CH2Cl2 or under reflux in 1,2-dichloroethane (DCE) proved unsuccessful, with 1H NMR monitoring of the reactions indicating no conversion!
Similar attempts using EtI/AgOTf in DCE under reflux also proved unsuccessful. Fortunately, S-alkylation occurred when AgOTf was replaced with AgBF4. The alkylating system RI/AgBF4/DCE was therefore selected for the entire study. In their early work on synthesis of S-alkylated thiophenium salts, Acheson and Harrison18 experienced the same difficulties and selected the RI/AgBF4/DCE system for their study.
Six examples of four-ring fused sulfonium salts namely 1Me+, 1Et+, 2Et+, 7Me+, 10Me+, and 10Et+, and six examples of five-ring fused sulfonium salts namely 12Me+, 12Et+, 13Me+, 14Me+, 14Et+, and 15Me+ were synthesized via this route (Fig 6).
Specific NMR assignments were made for the sulfonium ions 1Me+, 1Et+, 10Me+, 12Me+, and 12Et+ with the aid of 2D-NMR techniques (Chart S5) (for regular NMR data of all sulfonium salts see experimental). The sulfonium cations 1Me+, 1Et+, and 12Me+ were also calculated by GIAO NMR for comparison, and as an aid to fine-tuning the experiment-derived assignments (included in Chart S5). The sulfonium salts were also studied by electrospray-MS (in experimental).
In contrast to their carbocations, the sulfonium salts exhibit limited charge delocalization into the aromatic system. A notable feature in the 1H NMR spectra of the ethylated salts is the diasterotopic nature of –SCH2CH3 protons giving rise to a pair of doublets. Figure 7 shows the DFT-optimized structure of 1Et+. Pyramidalization at sulfur creates an asymmetric center. Figure S1 depicts the computed sulfur orbitals by NBO analysis, which infer sp3 hybridization at sulfur.
Figure 7.
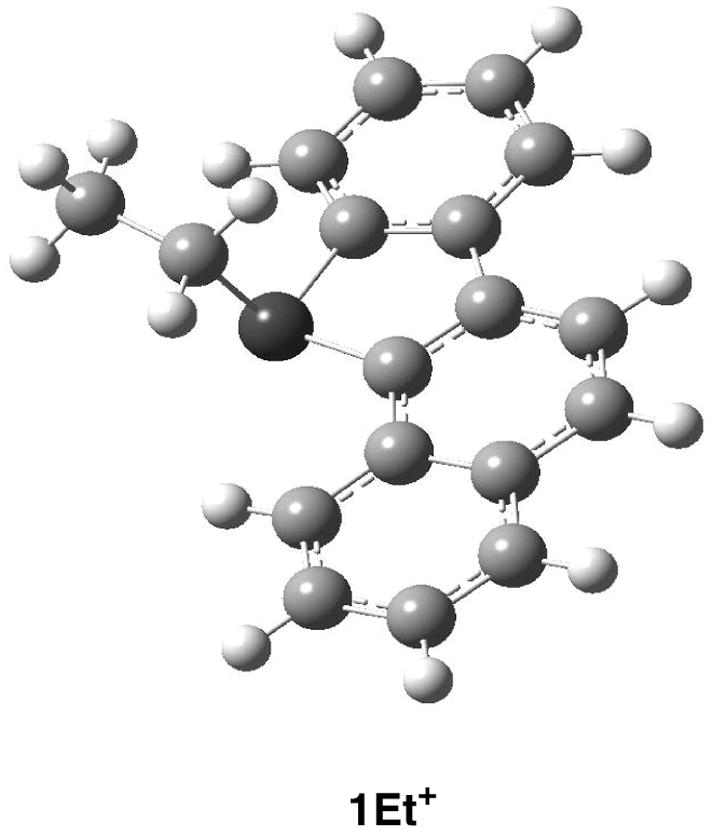
Optimized structure of 1Et+ by B3LYP/6-31G(d).
Sulfonium salts 14Me+, 14Et+, and 15Me+ represent novel examples of S-alkylation in the crowded fjord-region.
The NMR spectrum of 1Me+ salt dissolved in FSO3H/SO2ClF indicated only small variations relative to CDCl3 and was not indicative of ring protonation to form a sulfonium-arenium dication. Attempts to generate a dication by dissolving 1Me+ salt in higher acidity superacid FSO3H/SbF5(1:1)/SO2ClF led to side reactions and degradation of the onium salt.
f) Transfer-alkylation to model nitrogen nucleophile receptors (Scheme 1, Table S3)
Scheme 1.


Facile transferalkylation to model nitrogen nucleophiles.
Facile alkylation of imidazole was observed on mixing with sulfonium salts 1Me+ and 1Et+ at r.t. in CDCl3, as evidenced by 1H NMR monitoring experiments showing rapid decrease in the S-Me+ and S-Et+ signals concomitant with the appearance of the imidazolium N-Me+ and N-Et+ signals (Scheme 1). Similar transalkylation reactions were performed with 1Me+, 1Et+, 10Me+, 10Et+, 12Me+, and 14Me+ salts towards azaindole as model, in CD3CN or CDCl3 as solvents. These reactions indicated facile transalkylation (usually complete within 30 minutes), with the formation of N-alkylated azaindole (see Scheme 1).
As a guide, the reaction energies for alkyl transfer to nitrogen nucleophiles (imidazole, azaindole, and adenine) were computed by DFT (Table S3) for the sulfonium salts 1Me+, 3Me+, 3Et+, 3Pr+, 7Me+, 7Et+, 7Pr+, 10Me+, 12Me+, and 14Me+. Reactions were found to be highly favorable, with those for adenine alkylation being the most favored.
Our model studies indicate that thia-PAHs are promising candidates for DNA intercalation/alkylation studies.
g) Model electrophilic aromatic substitution reactions (Scheme 2)
Scheme 2.
model nitration and bromination reactions.
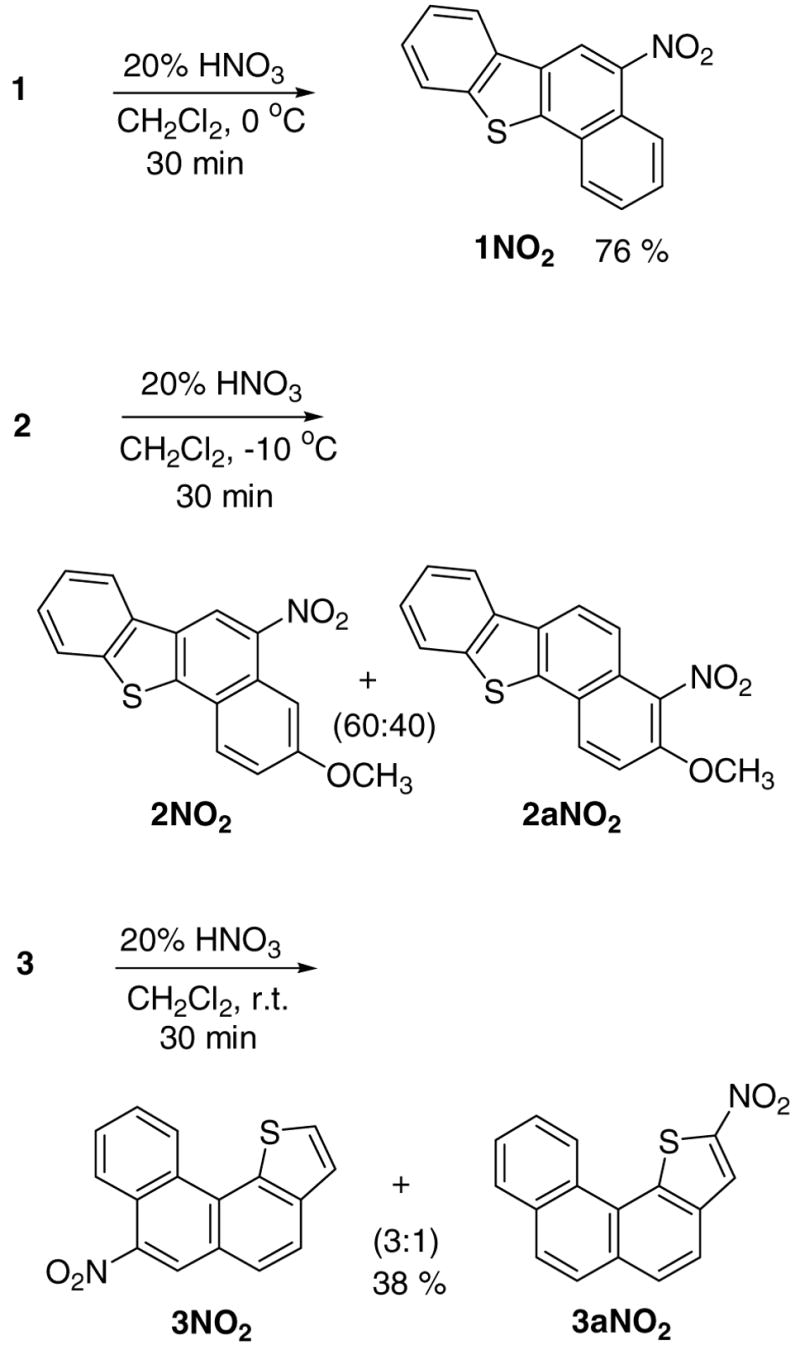
In order to compare the regioselectivities observed in carbocation generation under stable ion conditions with those resulting from conventional electrophilic aromatic substitutions, protic nitration of thia-PAHs 1–5 as well as 8 and 11 were studied. In addition, bromination of 4 and 6 were also examined. Nitration of parent thia-chrysene 1 using mixed acid, and formation of the 5-nitro derivative and its bioassay was reported by King et al.5
In our hands, compound 1 was cleanly nitrated under extremely mild conditions (20% aqueous nitric acid), without the need to use mixed acids, to give 5NO2. Common regioselectivity (attack at C-5) was, therefore, established in protonation and nitration of 1, in concert with DFT. Similar mild nitration of the 3-methoxy-derivative 2 gave a 60:40 mixture of 2NO2 (attack at C-5) and 2aNO2 (attack at C-4, alpha to OMe).
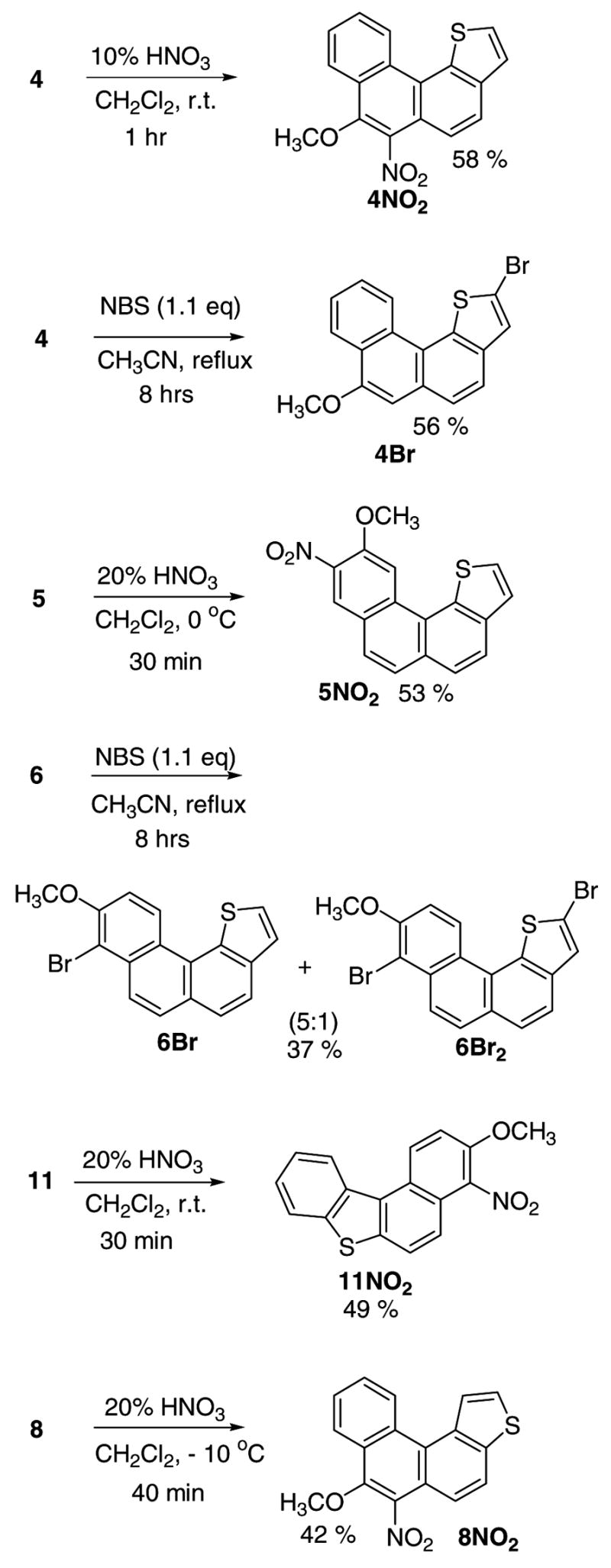
Directive effect of the methoxy-substituent is further manifested in comparative nitrations of compounds 3, 4, and 5. Mild nitration of parent 3 gave two mono-nitrated products, namely 3NO2 and 3aNO2 in 3:1 ratio. The latter matches the regioselectivity of the carbocation (3H+), whereas the former (attack at C-7) corresponds to the next best intermediate (by DFT) lying 6.5 kcal/mol higher (see Table S1). Remarkably, nitration of the 7-methoxy-derivative 4 could be effected in 10% aqueous nitric acid to give 4NO2 in good yield (substitution at C-6). Under stable ion conditions, two carbocations (protonation at C-6 and at C-2) were observed.
Nitration of the 10-methoxy-derivative 5, gave 5NO2 (nitration at C-9), illustrating again the directive influence of the OMe group. As discussed earlier, protonation of 5 occurred at C-11 (fjord-region), which based on DFT was preferred over C-9 protonation by 6.4 kcal/mol (Table S2). In the parent system 3, this energy difference decreased to 5.2 kcal/mol (Table S1). Preferential nitration at C-11 likely originates from differences in steric demand for nitration versus protonation in the congested fjord region.
Mild nitration of 8 resulted in the formation of 8NO2 (attack at C-7), and nitration of 11 gave 11NO2 (attack at C-4), both cases representing common regioselectivities in protonation and nitration.
Finally, bromination of 4 (with NBS) gave 4Br by substitution at C-2. This regioselectivity matches with the carbocation 4aH+ under stable ion conditions. Bromination of the 9-methoxy-derivative 6 gave 6Br as the major product, together with a dibrominated product 6Br2 (in 5: 1 ratio respectively). Bromination regioselectivity in 6Br corresponds to carbocations 6aH+/6bH+ formed under stable ion conditions.
The observed differing regioselectivities in alkylation, taking place at sulfur, versus protonation and nitration reactions, which in agreement with DFT, occurred at ring positions may be rationalized considering the nature of the electrophiles involved. Steric demand of a polarized complex formed via RI/AgBF4 is significantly larger as compared to protonation and nitration electrophiles. S-alkylation is likely an SN2 type displacement process assisted by the counterion (RI/AgBF4 was effective but RI/AgOTf did not work!), and involves coupling of a soft nucleophile with a polarized complex.
h) Modeling thia-PAH epoxide ring opening by DFT (Schemes S1-S2, Chart S6)
As highlighted in the introduction, metabolism of 1 was suggested to be in part via bay-region diol-epoxide and in part by sulfur activation.3,4 The diol epoxides 1a and 1b and their ring opening energies to form benzylic carbocations 1a+ and 1b+ were computed by DFT. As previously observed for aza-PAH epoxides,19 ring opening of the protonated epoxides via thia-PAHs also occurred via barrierless processes. Based on DFT, formation of carbocation 1b+ is 6 kcal/mol more favorable relative to 1a+ (Scheme S1). This focuses attention on the possibility that oxidation at 7,8- and 9,10-positions may have biological significance and calls for synthesis and mutagenicity assay of their dihydrodiols. The NPA-derived changes in charges (Δq) (1a → 1a+ and 1b → 1b+) are illustrated in Chart S6 for comparison. The Δq pattern for the energetically more favored 1b+ implies that the carbocation resulting from 7,8-epoxide ring opening is mainly a carbosulfonium ion 17b (positive charge resides mainly at sulfur and at C-10a).
Focusing on the fjord-region thia-PAHs 3 (not mutagenic) and 7 (mutagenic) (see introduction), the epoxide ring opening energies were computed by DFT, taking into account all possible epoxides (Scheme S2). For comparison, charge delocalization pathways in the resulting carbocations were also deduced based on magnitude of Δq (Chart S6).
Carbocation formed via the 8,9-epoxide ring opening (3d → 3d+) has the lowest energy. Ring opening energies for the K-region epoxide 3b and the fjord-region epoxide 3e are nearly identical (~2 kcal/mol above 3d). That of 6,7-epoxide 3c (whose dihydrodiol has been synthesized)7,8a is an additional 2 kcal/mol higher. Among various epoxides derived from 7, ring opening of 7b (generating 7b+) is most favorable. Comparing the K-region epoxide ring opening energies between 3b and 7b, it can be seen that 7b → 7b+ is 4 kcal/mol more favorable than 3b → 3b+. This is interesting in light of the reported mutagenicity of 7. As for the computed Δq maps (Chart S6), and focusing on the most favored ring openings 3d+ and 7b+, it can be deduced that positive charge in the former is mainly localized on the C/D rings, whereas 7b+ implies a much more delocalized carbocation.
i) Comparative Mutagenicity Assay on selected Nitro-derivatives (Table 1)
Table 1.
Mutagenicity Assay by the Ames Test
| Sample | Average potency (rev/μg) | Average potency (rev/nmol) |
|---|---|---|
| 1 | 29 | 7 |
| 1NO2 | 281 | 78 |
| 1NO2 with S9 | 268 | 75 |
| 5 | 116 | 31 |
| 5NO2 | 5069 | 1566 |
| 5NO2 with S9 | 198 | 61 |
| 11 | 0 | 0 |
| 11NO2 | 0 | 0 |
| 11NO2 with S9 | 20.5 | 6 |
The mono-nitro derivatives 1NO2, 5NO2 and 11NO2 (see section g) and their precursors (1, 5, and 11) were subjected to mutagenicity assay by the Ames test. Among the precursors, compound 5 is most active (it is moderately potent with metabolic activation). Influence of nitration on mutagenic activity can be seen in comparing the nitro derivatives with their respective precursors. Nitration of 11 (→ 11NO2) had little or no bioactivity enhancing effect. Nitration of 1 (→ 1NO2) increased its mutagenic activity to some extent. Nitration of 5, on the other hand, resulted in the formation of an extremely powerful direct acting mutagen. Given the extremely mild conditions under which the nitro-derivatives are formed (see section g and experimental procedure in supporting information), and the proven presence of thia-PAHs in the environment, direct formation of the nitro-derivatives in the environment appears quite feasible and could pose health hazard.
Summary
In the context of the present multifaceted study aimed at developing the electrophilic chemistry of thia-PAHs, a series of novel carbocations were generated from four ring-fused thia-PAHs, and their regioisomeric methoxy -derivatives. Regioselectivity issues and charge delocalization modes were addressed by NMR and by DFT studies. In view of the potential importance of hetero-PAHs and their onium salts as DNA intercalators and alkylating agents, a series of novel S-alkylated salts were synthesized and their potential as powerful alkylating agents towards nitrogen bases were demonstrated in model studies. To gain insight into structure/activity relationships, DFT was used to model epoxide ring opening reactions with the thia-analogs of chrysene and benzo[c]phenanthrene and to examine their charge delocalization pathways as a function of epoxidation site and thia-PAH structure. Using nitration as model electrophilic substitution reactions, a series of nitro-drivatives of thia-PAHs were synthesized under extremely mild conditions. Mutagenicity assays in representative cases demonstrated variable bioactivity, with one example (5NO2) for generation of a potent direct acting mutagen.
Experimental
Synthesis of Onium tetrafluoroborate salts by S-alkylation
For a typical alkylation procedure see supporting information.
1Me+ BF4−: was obtained as a beige solid (contained traces of unreacted 1 by 1H NMR); yield, 2.8 mg (42%); 1H NMR (400 MHz, CDCl3) δ 8.58 (d, J = 8.0 Hz, 1H), 8.33 (d, J = 8.4 Hz, 1H), 8.19 (d, J = 8.0 Hz, 1H), 8.14 (d, J = 8.4 Hz, 1H), 8.08 (d, J = 8.4 Hz, 1H), 8.06 (d, J = 8.4 Hz, 1H), 7.87 (m, 1H), 7.83 (m, 1H), 7.74 (m, 2H), 3.51 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 135.6 (CH), 134.2 (CH), 131.7 (CH), 131.0 (CH), 130.0 (CH), 129.2 (CH), 129.0 (CH), 124.2 (CH), 122.8 (CH), 119.7 (CH), 35.1 (CH3); ES-MS (ESI+) m/z 247.9 [1Me+], 584.1 [1Me+]2[BF4−], 920.2 [1Me+]3[BF4−]2.
1Et+ BF4-: obtained as a beige solid (1H NMR showed ca. 30 % unreacted 1); yield, 3.9 mg (29%); 1H NMR (400 MHz, CDCl3) δ 8.48 (d, J = 8 Hz, 1H), 8.31 (d, J = 8.4 Hz, 1H), 8.16 (d, J = 8.0 Hz, 1H), 8.13 (d, J = 8.4 Hz, 1H), 8.06 (d, J = 8.0 Hz, 1H), 8.04 (d, J = 8.0 Hz, 1H), 7.87 (m, 1H), 7.82 (m, 1H), 7.74 (m, 1H), 7.71 (m, 1H), 4.38 (m, 1H), 4.14 (m, 1H), 0.64 (t, J = 7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 140.8 (C), 140.2 (C), 135.5 (CH), 134.1 (CH), 133.9 (C), 131.5 (CH), 130.9 (CH), 129.9 (CH), 129.2 (CH), 129.0 (CH), 128.9 (C), 127.2 (C),123.8 (CH), 122.9 (CH), 122.2 (C), 119.5 (CH), 44.5 (CH2), 6.5 (CH3); ES-MS (ESI+) m/z 262.7 [1Et+], MS/MS m/z 262.7 → 234.6 (1Et+-C2H4).
2Et+ BF4−: 1H NMR (400 MHz, CDCl3) δ 8.46 (d, J = 8.0 Hz, 1H), 8.16(d, J = 8.0 Hz, 1H), 8.06 (d, J = 8.0 Hz, 1H), 8.02 (d, J = 8.0 Hz, 1H), 7.96 (d, J = 8.0 Hz, 1H), 7.84(t, J = 8.0 Hz, 1H), 7.71 (t, J = 8.0 Hz, 1H), 7.46 (d, J = 8.0 Hz, 1H), 7.33 (d, J = 2.4 Hz, 1H), 4.35 (m, 1H), 4.11 (m, 1H), 3.97 (s, 3H), 0.64 (t, J = 7.2 Hz, 3H); ES-MS (ESI+) m/z 293.2 [2Et+], MS/MS m/z 293.2 → 265.3 (2Et+-C2H4).
7Me+ BF4−: 1H NMR (400 MHz, acetone-d6) δ 8.87 (d, J =8.8Hz, 1H), 8.50 (d, J = 8.4 Hz, 1H), 8.45 (d, J = 8.4 Hz, 1H), 8.40 (d, J = 8.0 Hz, 1H), 8.30 (d, J = 8.0 Hz, 1H), 8.16 (dd, J = 8.0, 1.2 Hz, 1H), 8.05 (d, J = 8.0 Hz, 1H), 8.03 (d, J = 8.0 Hz, 1H), 8.02-7.95 (m, 2H), 7.90-7.85 (m, 2H); 1H NMR (400 MHz, CDCl3) δ 8.83 (d, J = 8.0 Hz, 1H), 8.31 (d, J = 8.0 Hz, 1H), 8.10-7.90 (m, 8H), 3.41 (s, 3H); ); ES-MS (ESI+) m/z 249.2 [7Me+], MS/MS m/z 249.2 → 234.0 (7Me+-CH3).
10Me+ BF4−: obtained as white solid (1H NMR showed traces of unreacted 1); yield, 5.2 mg (52 %); 1H NMR (400 MHz, CD3CN) δ 8.99 (d, J = 8.4 Hz, 1H), 8.91 (d, J = 8.0 Hz, 1H), 8.33 (d, J = 8.0 Hz, 1H), 8.30 (d, J = 8.8 Hz, 1H), 8.21 (d, J = 8.4 Hz, 1H), 8.16 (d, J = 8.8 Hz, 1H), 8.02 (m, 1H), 7.92 (1H, m), 7.86 (1H, m), 7.84 (1H, m), 3.36 (s, 3H); 13C NMR (101 MHz, CD3CN) δ 141.3 (C), 136.8 (3C), 135.1 (CH), 133.5 (CH), 131.9 (C), 131.2 (CH), 131.0 (CH), 130.5 (CH), 130.2 (CH), 130.0 (C), 129.0 (CH), 128.5 (CH), 125.1 (CH), 122.2 (CH), 34.8 (CH3); ES-MS (ESI+) m/z 248.6 [10Me+].
10Et+ BF4−: obtained as brown solid (1H NMR showed ca. 40 % unreacted 10); yield, 7.8 mg (crude, approx. 60%). 1H NMR (400 MHz, CD3CN) δ 8.96 (d, J = 8.8 Hz, 1H), 8.89 (d, J = 7.6 Hz, 1H), 8.28 (d, J = 9.2 Hz, 1H), 8.25 (d, J = 8.8 Hz, 1H), 8.19 (d, J = 8.0 Hz, 1H), 8.08 (d, J = 8.8 Hz, 1H), 8.01 (t, J = 7.6 Hz, 1H), 7.91 (t, J = 6.8 Hz, 1H), 7.87 (t, J = 8.0 Hz, 1H), 7.81 (t, J = 7.6 Hz, 1H), 3.95 (m, 2H), 0.79 (t, J = 7.2 Hz, 3H); ES-MS (ESI+) m/z 263.1 [10Et+]; m/z 263.1 → 235.1 (10Et+-C2H4).
12Me+ BF4−: obtained as white solid (1H NMR showed traces of unreacted 12); yield, 12.7 mg (54%); 1H NMR (400 MHz, CD3CN) δ 9.60 (s, 1H), 8.78 (s, 1H), 8.76 (d, J = 8.0 Hz, 1H), 8.37 (d, J = 8.0 Hz, 1H), 8.24 (d, J = 8.2 Hz, 1H), 8.08 (d, J = 8.8 Hz, 1H), 8.06 (d, J = 8.0 Hz, 1H), 8.01 (d, J = 8.8 Hz, 1H), 7.96 (m, 1H), 7.86 (m, 1H), 7.83 (m, 1H), 7.79 (m, 2H), 3.40 (s, 3H); 13C NMR (101 MHz, CD3CN) δ 140.1 (C), 136.8 (C), 135.9 (C), 135.1 (CH), 133.4 (C), 132.2 (CH), 132.0 (CH), 130.3 (C), 130.0 (CH), 129.5 (CH), 129.1 (CH), 128.6 (CH), 127.4 (CH), 125.8, 125.2 (CH), 124.9 (CH), 124.7 (CH), 123.9 (CH), 35.8 (CH3); ES-MS (ESI+) m/z 299.1 [12Me+], 685.2 [12Me+]2[BF4−]; MS/MS m/z 299.1 → 284.1 (methyl loss).
12Et+ BF4−: obtained as white solid (1H NMR showed ca. 10 % unreacted 12); yield, 5.7 mg (53%). 1H NMR (400 MHz, CD3CN) δ 9.43 (s, 1H), 8.70 (s, 1H), 8.71 (d, J = 8.4 Hz, 1H), 8.31 (d, J = 8.0 Hz, 1H), 8.15 (d, J = 8.0 Hz, 1H), 8.03 (d, J = 8.8 Hz, 1H), 7.96 (d, J = 8.8 Hz, 1H), 8.00 (d, J = 8.0 Hz, 2H), 7.94 (m, 1H), 7.81 (m, 1H), 7.79 (m, 1H), 7.73 (m, 1H), 3.96 (m, 1H), 3.91 (m, 1H), 0.84 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, CD3CN) δ 141.2 (C), 137.6 (C), 135.9 (C), 135.2 (CH), 133.4 (C), 132.1 (CH), 132.0 (CH), 130.2 (C), 129.9 (CH), 129.4 (CH), 129.1 (CH), 129.0 (CH), 127.4 (CH), 125.1 (CH), 125.0 (CH), 124.7 (CH), 123.9 (CH), 46.5 (CH2), 7.3 (CH3); ES-MS (ESI+) m/z 312.5 [12Et+].
13Me+ BF4−: obtained as beige solid (1H NMR showed ca. 50 % unreacted 13); yield, 3.2 mg (crude, approx. 50%); 1H NMR (400 MHz, CD3CN) δ 9.43 (s, 1H), 8.72 (s, 1H), 8.63 (d, J = 9.2 Hz, 1H), 8.35-8.20 (m, 2H), 8.00-7.95 (m, 3H), 7.77 (t, J = 7.8 Hz, 1H), 7.49 (d, J = 2.4 Hz, 1H), 7.45 (dd, J = 9.2 and 2.4 Hz, 1H), 3.98 (s, 3H), 3.38 (s, 3H); ES-MS (ESI+) m/z 329.2 [13Me+], 745.2 [13Me+]2[BF4−], MS/MS m/z 329 → 314.2 (methyl loss), 745.2 → 329.1 [13Me+], 314.1 [13Me+-CH3].
14Me+ BF4−: obtained as brown solid (1H NMR showed ca. 30 % unreacted 14); yield, 10.4 mg (crude, approx. 69 %); 1H NMR (400 MHz, CD3CN) δ 8.74 (d, J = 8.4 Hz, 1H), 8.36 (d, J = 8.4, 1H), 8.32 (d, J = 8.4 Hz, 1H), 8.29 (d, J = 8.4 Hz, 1H), 8.23 (d, J = 8.0 Hz, 1H), 8.11 (d, J = 8.0 Hz, 1H), 7.98 (d, J = 8.8 Hz, 1H), 7.92 (d, J = 8.8 Hz, 1H), 7.95-7.75 (m, 4H), 3.18 (s, 3H); 1H NMR (400 MHz, CDCl3) δ 9.08 (d, J = 8.4 Hz, 1H), 8.64 (d, J = 8.0, 1H), 8.36 (d, J = 8.4 Hz, 1H), 8.27 (d, J = 8.4 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1H), 8.10 (t, J = 8.4 Hz, 1H), 8.03 (d, J = 8.0 Hz, 1H), 7.93 (d, J = 8.8 Hz, 1H), 7.85-7.81 (m, 3H), 7.75 (t, J = 8.0 Hz, 1H), 3.39 (s, 3H); ES-MS (ESI+) m/z 298.9 [14Me+], 685.0 [14Me+]2[BF4−], MS/MS m/z 298.9 → 283.9 (methyl loss), m/z 685 → 583.0, 298.9, and 283.9.
14Et+ BF4−: 1H NMR (400 MHz, CD3CN) δ 8.88 (d, J = 8.4 Hz, 1H), 8.44 (d, J = 8.4, 1H), 8.40 (d, J = 8.4 Hz, 1H), 8.36 (d, J = 8.0 Hz, 1H), 8.23 (d, J = 8.0 Hz, 1H), 8.14 (d, J = 8.0 Hz, 1H), 8.02 (d, J = 8.8 Hz, 1H), 8.00-7.80 (m, 5H), 4.01 (m, 1H), 3.80 (m, 1H), 0.50 (t, J = 7.0 Hz, 3H); ES-MS (ESI+) m/z 312.3 [14Et+], MS/MS m/z 312.3 → 284.2.
15Me+ BF4−: obtained as light-brown solid (1H NMR showed ca. 40 % unreacted 15); yield, 3.8 mg (crude, approx. 60%); 1H NMR (400 MHz, CD3CN) δ 8.62 (d, J = 9.2 Hz, 1H), 8.34 (d, J = 8.4 Hz, 1H), 8.31 (d, J = 7.6 Hz, 1H), 8.28 (d, J = 8.4 Hz, 1H), 8.24 (d, J = 8.0 Hz, 1H), 7.95 (t, J = 8.0 Hz, 1H), 7.91 (s, 2H), 7.88 (d, J = 8.0 Hz, 1H), 7.54 (d, J = 2.8 Hz, 1H), 7.49 (dd, J = 8.8 and 2.8 Hz, 1H), 4.00 (s, 3H), 3.18 (s, 3H); 1H NMR (400 MHz, CDCl3) δ 8.95 (bs, 1H), 8.62 (bs, 1H), 8.29 (d, J = 8.0 Hz, 1H), 8.18 (d, J = 8.0 Hz, 2H), 7.89 (t, J = 8.0 Hz, 1H), 7.82 (s, 2H), 7.75 (bs, 1H), 7.70 (bs, 1H), 7.38 (s, 1H), 4.02 (s, 3H), 3.38 (s, 3H); ES-MS (ESI+) m/z 329.1 [15Me+], 745.2 [15Me+]2[BF4−]; MS/MS: m/z 329 → 314.0 (methyl loss), m/z 745 → 329.1, 314.1, and 270.9.
Model Nitration of thia-PAHs
For a typical procedure see supporting information.
1NO2 (bright yellow solid). yield, 6.4 mg (76 % isolate yield); TLC (Rf 0.57, 20 % CH2Cl2/hexane); mp 197.0–199.0 °C; 1H NMR (500 MHz, CDCl3) δ 8.94 (s, 1H), 8.74 (d, J = 8.5 Hz, 1H), 8.23 (m, 1H), 8.21 (d, J = 8.5 Hz, 1H), 7.97 (m, 1H), 7.77 (t, J = 8.5 Hz, 1H), 7.72 (t, J = 8.5 Hz, 1H), 7.59 (m, 2H); 13C NMR (CDCl3, 125 MHz) δ 144.7 (C),143.9 (C), 139.0 (C), 135.7 (C), 130.4 (C), 129.2 (CH, C), 128.1 (CH), 127.4 (CH), 125.5 (CH), 125.0 (CH), 124.8 (CH), 123.7 (C), 123.1 (CH), 121.8 (CH), 118.5 (CH); IR (KBr) 1533, 1482, 1373, 1143, 1031, 715 cm−1. ES-MS (ESI+) m/z 386.0/387.9 (M+Ag)+.
2NO2 and 2aNO2: the isomeric mixture was isolated after TLC separation (Rf 0.27–0.29, streaks, 20 % CH2Cl2/hexane) as a bright yellow solid; combined yield, 6.8 mg (73%). 2NO2: 1H NMR (500 MHz, CDCl3) δ 9.02 (s, 1H), 8.23 (d, J = 7.5 Hz, 1H), 8,13 (d, J = 9 Hz, 1H), 8.22 (d, J = 2.5 Hz, 1H), 7.95 (d, J = 7.5 Hz, 1H), 7.58-7.52 (m, 2H), 7.37 (dd, J = 9.0 and 2.5 Hz, 1H), 4.02 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 127.1 (CH), 126.7 (CH), 125.5 (CH), 123.0 (CH), 121.6 (CH), 120.4 (CH), 119.4 (CH), 103.8 (CH).
2aNO2: 1H NMR (500 MHz, CDCl3) δ 8.26-8.20 (m, 3H), 7.67 (d, J = 9 Hz, 1H), 7.44 (d, J = 9 Hz, 1H), 7.95 (d, J = 7.5 Hz, 1H), 7.58-7.52 (m, 2H), 4.08 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 128.2 (CH), 126.7 (CH), 125.0 (CH), 122.9 (CH), 122.9 (CH), 121.7 (CH), 117.8 (CH), 113.5 (CH).
ES-MS (ESI+) for 2NO2/2aNO2 m/z 415.6/417.6 (M+Ag)+.
3NO2 and 3aNO2: the isomeric mixture was isolated after TLC separation (Rf 0.6, 50% CH2Cl2/hexane) as a bright yellow solid; 2.9 mg (38% ).
3NO2: 1H NMR (400 MHz, CDCl3) δ 9.33 (d, J = 8.4 Hz, 1H), 8.67 (d, J = 8.4 Hz, 1H), 8.66 (s, 1H), 8.16 (d, J = 8.0 Hz, 1H), 8.00 (d, J = 8.0 Hz, 1H), 7.94 (t, J = 8.4 Hz, 1H), 7.85 (d, J = 5.2 Hz, 1H), 7.84 (t, J = 8.4 Hz, 1H), 7.68 (d, J = 5.2 Hz, 1H).
3aNO2: 1H NMR (400 MHz, CDCl3) δ 9.01 (d, J = 8.4 Hz, 1H), 8.44 (s, 1H), 8.08 (d, J = 8.4 Hz, 1H), 8.08 (d, J = 8.4 Hz, 1H), 7.99 (d, J = 8.4 Hz, 1H), 7.99 (d, J = 8.4 Hz, 1H), 7.93 (d, J = 8.4 Hz, 1H), 7.90 (t, J = 8.4 Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H).
ES-MS (ESI+) for 3NO2/3aNO2 m/z 386.0/387.9 (M+Ag)+.
4NO2. isolated as bright yellow solid; yield, 3.6 mg (58%); TLC (Rf 0.33, 40% CH2Cl2/hexane); mp 125.0–126.0 °C; 1H NMR (500 MHz, CDCl3) δ 9.30 (d, J = 8.5 Hz, 1H), 8.43 (d, J = 8.5 Hz, 1H), 8.15 (d, J = 9 Hz, 1H), 7.98 (t, J = 8.5 Hz, 1H), 7.83 (t, J = 8.5 Hz, 1H,), 7.76 (d, J = 5 Hz, 1H), 7.71 (d, J = 9 Hz, 1H), 7.65 (d, J = 5 Hz, 1H), 4.16 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 145.1 (C), 139.8 (C), 134.9 (C), 130.9 (C), 129.3 (CH), 127.4 (CH), 127.0 (CH), 126.7, 126.6 (CH), 124.7 (2CH), 124.4 (C), 124.2 (CH), 122.0 (C), 64.0 (CH3); IR (KBr) 2925, 1522, 1260, 1092, 805 cm−1; ES-MS (ESI+) m/z 416.1/418.0 (M+Ag)+.
5NO2: isolated as a yellow solid; yield, 3.6 mg (53%); TLC (Rf 0.56, 30% EtOAc/hexane); mp 140.0–142.0 °C; 1H NMR (500 MHz, CDCl3) δ 8.67 (s, 1H), 8.48 (s, 1H), 8.40 (d, J = 8.5 Hz, 1H), 8.20 (d, J = 9.5 Hz, 1H), 8.05 (d, J = 9.5 Hz, 1H), 7.78 (d, J = 5.5 Hz, 1H), 7.68 (d, J = 5.5 Hz, 1H), 7.44 (m, 1H), 4.18 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 132.3 (CH), 126.9 (CH), 124.8 (CH), 124.6 (CH), 124.4 (CH), 120.4 (CH), 118.4 (CH), 126.7 (CH), 107.5 (CH), 55.9 (CH3) (quaternary cabons not detectable due to very low quantity of this product); IR (KBr) 2964, 1522, 1262, 1099, 803 cm−1; ES-MS (ESI+) m/z 416.0/418.0 (M+Ag)+.
8NO2: isolated as a yellow solid; yield, 3.1 mg (42%); TLC (Rf 0.27, 15 % CH2Cl2/hexane); mp 124.0–126.0 °C; 1H NMR (500 MHz, CDCl3) δ 9.15 (d, J = 8.5 Hz, 1H), 8.57 (d, J = 5.5 Hz, 1H), 8.37 (dd, J = 7.0 and 1.0 Hz, 1H), 8.15 (d, J = 8.5 Hz, 1H), 7.86 (t, J = 8.5 Hz, 1H), 7.79 (t, J = 8.5 Hz, 1H), 7.82 (d, J = 5.5 Hz, 1H), 7.63 (d, J = 8.5 Hz, 1H), 4.15 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 145.0 (C), 140.6 (C), 134.7 (C), 131.9 (CH), 128.7 (CH), 128.0 (CH), 127.4 (CH), 127.1 (CH), 126.7 (C), 125.2 (CH), 124.6 (C), 123.9 (CH), 123.4 (CH), 121.8 (C), 117.9 (CH), 63.8 (CH3); IR (KBr) 1528, 1400, 1092, 773, 705 cm−1; ES-MS (ESI+) m/z 416.0/417.9 (M+Ag)+.
11NO2: isolated as a yellow solid; yield, 6.8 mg (49%); TLC (Rf 0.33, 30% CH2Cl2/hexane); mp 193.0–195.0 °C; 1H NMR (500 MHz, CDCl3) δ 9.10 (d, J = 9.5 Hz, 1H), 8.73 (d, J = 8.5 Hz, 1H), 8.02 (d, J = 8.5 Hz, 1H), 8.00 (d, J = 9.0 Hz, 1H), 7.67 (1H, d, J = 9.0 Hz ), 7.62 (t, J = 8.5 Hz, 1H), 7.55 (t, J = 8.5 Hz, 1H), 7.53 (d, J = 9.5 Hz, 1H), 4.09 (s, 3H); 13C NMR (125 MHz, CDCl 3) δ 147.3 (C), 140.3 (C), 137.6 (C), 135.8 (C), 128.9 (C), 126.8 (CH), 125.9 (CH), 125.2 (C, CH), 124.5 (2CH), 124.3 (C), 123.5 (CH), 119.5 (CH), 113.4 (CH), 57.0 (CH3); IR (KBr) 1527, 1358, 1271, 1079, 799 cm−1; ES-MS (ESI+) m/z 416.1/418.1 (M)+.
Model Bromination of thia-PAHs
For a typical procedure see supporting information.
4Br: TLC (Rf 0.56, 40% CH2Cl2/hexane); mp 138.0–140.0 °C; 1H NMR (400 MHz, CDCl3) δ 8.95 (d, J = 8.4 Hz, 1H), 8.52 (dd, J = 8.4 and 0.8 Hz, 1H), 7.86 (d, J = 8.4 Hz, 1H), 7.85 (m, 1H), 7.77 (d, J = 8.4 Hz, 1H), 7.71 (m, 1H), 7.55 (s, 1H), 7.13 (s, 1H), 4.14 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 153.3 (C), 137.6 (C), 136.0 (C), 131.5 (C), 130.0 (C), 127.6 (CH), 127.0 (CH), 126.9 (C), 126.2 (CH), 125.7 (CH), 125.6 (CH), 122.9 (CH), 122.1 (CH), 121.5 (C), 113.2 (C), 103.2 (CH), 55.6 (CH3); IR (KBr) 1604, 1253, 1127, 1104, 847 cm−1; ES-MS (ESI+) m/z 449.1/451.0/453.0 (M+Ag)+.
6Br and 6Br2: the isomeric mixture was obtained after TLC separation (Rf 0.53, 40 % CH2Cl2/hexane) as a white solid (3.8 mg, 37 % combined isolated yield).
6Br (major product): 1H NMR (500 MHz, CDCl 3) δ 9.20 (d, J = 8.5 Hz, 1H), 8.35 (d, J = 9.0 Hz, 1H), 8.04 (d, J = 8.5 Hz, 1H), 7.98 (d, J = 9.0 Hz, 1H), 7.89 (d, J = 8.5 Hz, 1H), 7.69 (d, J = 5.5 Hz, 1H), 7.61 (d, J = 5.5 Hz, 1H), 7.50 (d, J = 8.5 Hz, 1H), 4.12 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 154.0 (C), 139.8 (C), 134.2 (C), 132.8 (C), 129.9 (CH), 126.6 (CH), 126.3 (CH), 126.1 (CH), 125.3 (C), 124.8 (CH), 124.7 (CH), 122.7 (CH), 121.9 (C), 120.0 (C), 112.1 (CH), 110.7 (C), 56.8 (CH3).
6Br2 (minor product): 1H NMR (500 MHz, CDCl3) δ 8.87 (d, J = 8.5 Hz, 1H), 8.34 (d, J = 9.0 Hz, 1H), 7.91 (d, J = 9.0 Hz, 1H), 7.88 (d, J = 8.5 Hz, 1H), 7.83 (d, J = 8.5 Hz, 1H), 7.57 (s, 1H), 7.47 (d, J = 8.5 Hz, 1H), 4.11 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 129.8 (CH), 129.4 (CH), 127.3 (CH), 126.6 (CH), 125.0 (CH), 121.6 (CH), 112.1 (CH), 56.7 (CH3) (quaternary carbons not detectable due to very small quantity of this product); ES-MS (ESI+) m/z 449.1/451.0/452.9 (M+Ag)+.
Supplementary Material
Experimental and GIAO-derived NMR data and computed changes in NPA-derived charges for various carbocations, representative NMR spectra of the carbocations generated in FSO3H/SO2ClF, NMR spectra and specific NMR assignments for the nitro-and bromo-derivatives, ionization energies for the epoxides, reaction energies for transalkylation, carbocation energies, typical experimental procedures for nitration and bromination, carbocation quenching procedure, computational protocols, stable carbocation generation procedure, and experimental procedures for mutagenicity assays. These material are available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
Support of this study under “reactive intermediates of carcinogenesis of PAHs” at KSU by the NCI of NIH (2R15-CA078235-02A1) is gratefully acknowledged. This work was also supported in part by a grant to S. K. (#R826192) by the US-EPA, Washington DC.
References
- 1.Jacob J. Sulfur Analogues of Polycyclic Aromatic Hydrocarbons. Cambridge University Press; Cambridge, UK: 1990. [Google Scholar]
- 2.Harvery RG. In: The Handbook of Environmental Chemistry. Neilson AH, editor. Vol. 1. Springer-Verlag; Berlin: 1998. part 1, and other related references cited therein. [Google Scholar]
- 3.Kumar S, Kim TY. J Org Chem. 2000;65:3883–3884. doi: 10.1021/jo991933k. [DOI] [PubMed] [Google Scholar]
- 4.Misra B, Amin S. Chem Res Toxicol. 1990;3:93–97. doi: 10.1021/tx00014a002. [DOI] [PubMed] [Google Scholar]
- 5.King LC, Kohan MJ, Brooks L, Nelson GB, Ross JA, Allison J, Adams L, Desai D, Amin S, Padgett W, Lambert GR, Richard AM, Nesnow S. Chem Res Toxicol. 2001;14:661–671. doi: 10.1021/tx0001373. [DOI] [PubMed] [Google Scholar]
- 6.Kumar S, Kumar A, Sikka HC. Polycyclic Aromatic Compounds. 2004;24:527–536. [Google Scholar]
- 7.Kumar S, Saravanan S, Reuben P, Kumar A. J Heterocyclic Chem. 2005;42:1345–1355. [Google Scholar]
- 8.(a) Kumar S, Reuben PA, Kumar A. Polycyclic Aromatic Compounds. 2004;24:289–295. [Google Scholar]; (b) Pelroy RA, Stewart DL, Tominaga Y, Iwao M, Castle RN, Lee ML. Mutat Res. 1983;117:31–40. doi: 10.1016/0165-1218(83)90150-7. [DOI] [PubMed] [Google Scholar]
- 9.Kumar S. J Chem Soc, Perkin Trans 1. 2001:1018–1023. [Google Scholar]
- 10.Yuan ZX, Sikka HC, Munir S, Kumar A, Muruganandam AV, Kumar S. Chem Res Toxicol. 2003;16:1581–1588. doi: 10.1021/tx0341310. [DOI] [PubMed] [Google Scholar]
- 11.Kumar S. J Org Chem. 2002;67:8842–8846. doi: 10.1021/jo020493l. [DOI] [PubMed] [Google Scholar]
- 12.(a) Piantanida I, Palm BS, Zinic M, Schneider HJ. J Chem Soc, Perkin Trans 2. 2001:1808–1816. [Google Scholar]; (b) Palm BS, Piantanida I, Zinic M, Schneider HJ. J Chem Soc, Perkin Trans 2. 2000:385–392. [Google Scholar]; (c) Piantanida I, Tomisic V, Zinic M. J Chem Soc, Perkin Trans 2. 2000:375–383. [Google Scholar]; (d) Steiner-Biocic I, Glavas-Obrovac L, Karner I, Piantanida I, Zinic M, Pavelic K, Pavelic J. Anticancer Research. 1996;16:3705–3708. [PubMed] [Google Scholar]
- 13.Tumir LM, Piantanida I, Cindric IJ, Hrenar T, Meic Z, Zinic M. J Phys Org Chem. 2003;16:891–899. [Google Scholar]
- 14.Huber R, Amann N, Wagenknecht HA. J Org Chem. 2004;69:744–751. doi: 10.1021/jo0355404. [DOI] [PubMed] [Google Scholar]
- 15.Ross SA, Pitie M, Meunier B. J Chem Soc, Perkin Trans 1. 2000:571–574. [Google Scholar]
- 16.Nakanishi T, Suzuki M, Mashiba A, Ishikawa K, Yokotsuka T. J Org Chem. 1998;63:4235–4239. [Google Scholar]
- 17.(a) Olah GA, Laali KK, Wang Q, Prakash GKS. Onium Ions. Wiley; New York: 1998. chap 4 [Google Scholar]; (b) Olah GA, Laali KK, Wang Q, Prakash GKS. Onium Ions. Wiley; New York: 1998. chap 7 [Google Scholar]
- 18.Acheson RM, Harrison DR. J Chem Soc (C) 1970:1764–1784. [Google Scholar]
- 19.(a) Borosky GL, Laali KK. Chem Res Toxicol. 2005;18:1876–1886. doi: 10.1021/tx0501841. [DOI] [PubMed] [Google Scholar]; (b) Borosky GL, Laali KK. Org Biomo l Chem. 2005;3:1180–1188. doi: 10.1039/b416429f. [DOI] [PubMed] [Google Scholar]
- 20.Laali KK, Hupertz S, Temu AG, Galembeck SE. Org Biomol Chem. 2005;3:2319–2326. doi: 10.1039/b503084f. [DOI] [PubMed] [Google Scholar]
- 21.Reddy VP, Bellew DR, Prakash GKS. J Fluorine Chem. 1992;56:195–197. [Google Scholar]
- 22.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, Revision B.05. Gaussian, Inc; Pittsburgh, PA: 2003. [Google Scholar]
- 23.Wolinski K, Hinton JF, Pulay P. J Am Chem Soc. 1990;112:8251–8260. [Google Scholar]; Ditchfield R. Mol Phys. 1974;27:789–807. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental and GIAO-derived NMR data and computed changes in NPA-derived charges for various carbocations, representative NMR spectra of the carbocations generated in FSO3H/SO2ClF, NMR spectra and specific NMR assignments for the nitro-and bromo-derivatives, ionization energies for the epoxides, reaction energies for transalkylation, carbocation energies, typical experimental procedures for nitration and bromination, carbocation quenching procedure, computational protocols, stable carbocation generation procedure, and experimental procedures for mutagenicity assays. These material are available free of charge via the Internet at http://pubs.acs.org.