

Abstract
The dynamics of water confined in two different types of reverse micelles are studied using ultrafast infrared pump-probe spectroscopy of the hydroxyl OD stretch of HOD in H2O. Reverse micelles of the surfactant Aerosol-OT (ionic head group) in isooctane and the surfactant Igepal CO 520 (non-ionic head group) in 50/50 weight percent cyclohexane/hexane are prepared to have the same diameter water nanopools. Measurements of the IR spectra and vibrational lifetimes show that the identity of the surfactant head groups affects the local environment experienced by the water molecules inside the reverse micelles. The orientational dynamics (time dependent anisotropy), which is a measure of the hydrogen bond network rearrangement, is very similar for the confined water in the two types of reverse micelles. The results demonstrate that confinement by an interface to form a nanoscopic water pool is a primary factor governing the dynamics of nanoscopic water rather than the presence of charged groups at the interface.
I. Introduction
Many important phenomena take place in nanoscopic water environments. For example, a wide variety of biological processes occur in very crowded aqueous surroundings with the solvating water often playing an important role. Water confined on nanometer length scales is also found in many non-biological situations. The frequent incidence of nanoscopically restricted water brings to the forefront the need to understand the properties of water and aqueous systems in confined environments and near interfaces.
The perturbing effects of confinement, interfaces, and solutes on the dynamics of water have been observed in a number of systems by a variety of techniques. Neutron scattering measurements and MD simulations of water confined in nanoporous silica1–3, alumina4, and Vycor glass5 show slowing of the water dynamics. Surface force measurements using mica surfaces6,7 show that the viscosity and density of very thin films of water are higher than that of bulk water due to structuring of the water near the confining surfaces. Both NMR8,9 and ultrafast IR experiments10 show that the presence of ions in aqueous solutions influences dynamics in the solvation shell of the ions. Magnetic resonance dispersion experiments indicate that the dynamics of water in the first hydration layer of a protein, averaged over the entire protein, are slowed relative to bulk water.11 Ultrafast infrared (IR) pump-probe experiments have demonstrated that the dynamics of water in the nanopores of Nafion fuel cell membranes differ substantially from those of bulk water and vary with size of the pores (hydration level of the membrane).12,13 A series of experiments using ultrafast IR vibrational echo and pump-probe experiments performed directly on water confined in AOT reverse micelles have shown that the water dynamics slow when the radius of the reverse micelle water pool falls below several nanometers.14–19
It is clear from the results obtained on this wide variety of systems that the dynamics of water that is nanoscopically confined or in contact with interfaces or solutes differ from those of bulk water. While the perturbation of the water’s properties may extend only a few hydration layers, in confined environments nearly all the water can fall within this range.14 Most previous studies have focused on a single type of system containing either a charged interface or a neutral hydrophilic interface. In the case of a protein surface, there are both charged and uncharged regions of the interface. Several MD simulations of water in reverse micelles have changed the nature of the interface from hydrophilic to hydrophobic with mixed conclusions as to the effect on the dynamics of water.20,21 A steady state IR experiment compared the line shape of water confined in a nonionic fluorocarbon reverse micelle to water confined in AOT reverse micelles by fitting the water absorption line to multiple Gaussians.22 The authors concluded that the charged groups of AOT were not the only factor governing the properties of water in reverse micelles and “that the size of confinement prevails.” However, the size and shape of the fluorocarbon nonionic reverse micelle are not well characterized, and therefore, direct comparison with AOT reverse micelles is difficult. The studies mentioned above show that the water dynamics in confined systems are different from those of bulk water. However, because the systems are so varied and often poorly characterized in both the topology and the nature of the interfaces, it is difficult to compare them to determine the role that important aspects of the water’s environment play in the modification of its dynamics.
In this paper we address one of the fundamental questions: with all other things being equal, will an interface with charges produce substantially greater changes in water dynamics than a neutral interface? In other words, does nanoscopic confinement alone impact water dynamics or do interfacial charges play a dominant role? Here, we provide strong evidence that water confinement dramatically influences water dynamics even in the absence of charged groups at the interface. The issues addressed here also apply to water within a few nanometers of an interface, such as a protein surface.11,23
The great interest in the nature of confined water in biology, chemistry, and materials science has led to a large number of studies on the properties of water in reverse micelles as model systems.24 Some, but not all, reverse micelle systems have well characterized structures containing nanoscopic pools of water.25,26 Reverse micelles are often described by the parameter w0, which is defined as the number of water molecules per surfactant molecule, w0 = [H2O]/[surf ]. For spherical particles the size of a reverse micelle scales linearly with the w0 parameter. A number of studies have explored the properties of water in a range of sizes and types of reverse micelles including those formed with the ionic surfactants Aerosol-OT (AOT),14–19,27–31 and cetyltrimethylammonium bromide (CTAB),32 as well as several nonionic surfactants.26,33 Frequently, the experiments have employed large probe molecules to characterize the interfacial region or study the solvation dynamics in the water pool.24,25,28 Recently, several groups have significantly enhanced the understanding of the dynamics of water in AOT reverse micelles by directly studying the water using ultrafast IR methods.14–19 However, there have been no direct experimental studies of the dynamics of water in nonionic reverse micelles for comparison.
Below, we present the results of ultrafast infrared pump-probe studies on the dynamics of water confined in two types of reverse micelles, one with an ionic head group surfactant and one with a non-ionic head group surfactant. The ionic system is the well-characterized and widely studied reverse micelle formed with water/AOT/isooctane. AOT, which has an anionic sulfonate head group, forms monodispersed, spherical reverse micelles over a wide range of micelle sizes and temperatures.34,35
There are a large number of nonionic surfactants including Brij, Triton-X, and Igepal. Each of these surfactants can have a range of chain lengths, and the properties of the reverse micelles can be strongly dependant on the composition of the bulk organic phase. Sometimes mixtures of the surfactants with alcohol molecules are used to form reverse micelles. Because of the wide variety of nonionic surfactant/organic phase combinations, no one combination has been as thoroughly studied and characterized as the AOT reverse micelle system.
Of the many combinations, a quite well characterized non-ionic reverse micelle can be made of the surfactant Igepal CO 520 with an organic phase consisting of a 50/50 by weight mixture of cyclohexane and n-hexane.36 This nonionic surfactant, which has an alcohol head group, and organic phase combination was chosen because the resulting reverse micelles have been studied in detail by small-angle neutron scattering (SANS) to determine their structure.36 The stability regime of the reverse micelle phase was mapped out and the water pool sizes were determined at a number of temperatures and w0 values. In addition, the SANS fitting indicated a spherical shape for the reverse micelles and molecular dynamics simulations on a similar system showed only a slight ellipticity.37 The combination of spherical shape and relatively good size characterization make this Igepal system useful for comparison with the AOT reverse micelle system. Because of a lack of similar detailed characterization of other non-ionic reverse micelles, the Igepal system is the only system suitable for these studies.
To answer the fundamental question posed above, the direct method for determining the effect of a charged interface vs. a neutral interface on water dynamics is to prepare reverse micelles with the same size but with different head groups, ionic vs. non-ionic. With size and geometry no longer a variable, it is possible to establish whether the differences in nanoscopic water dynamics from those of bulk water are caused by confinement or the ionic character of the surfactant head group interface. The AOT and Igepal surfactants can be used to address this fundamental question.
II. Materials and Methods
Aerosol OT, Igepal CO 520, isooctane, cyclohexane, n-hexane, D2O and water were obtained from Sigma-Aldrich and were used without further purification. A 0.5 M stock solution of AOT in isooctane and a 0.3 M stock solution of Igepal CO 520 in a 50/50 weight percent mixture of cyclohexane/n-hexane were prepared. The residual water content in the surfactant and oil phase was measured by Karl Fischer titration. To make reverse micelles with the desired w0, precise volumes of water (5% HOD in H2O) were added to measured quantities of each stock solution. The samples were housed in cells containing two CaF2 windows (3 mm thick) and a Teflon spacer with a thickness adjusted to obtain optical densities in the range 0.2–0.4 for all solutions studied. All experiments were conducted at 25°C.
The laser system used in these experiments consists of a Ti:Sapphire oscillator and regenerative amplifier operating at 1 kHz which pump an OPA and difference frequency stage to produce ~70 fs, ~4 μm IR pulses. Pulses are split into pump and probe pulses with the probe’s polarization set to ~45° relative to the pump. After the sample, the components of the probe with polarization parallel and perpendicular to the pump are selected to avoid depolarization effects due to optics in the beam path.38 Polarizers set to parallel and perpendicular are rotated into the beam using a computer controlled wheel. Scans alternate between parallel and perpendicular to reduce effects of long term laser drift. After the polarizers, a half wave plate rotates the polarization of each component to 45° prior to the entrance slit of a 0.25 m monochromator so that both parallel and perpendicular polarization scans experience the same diffraction efficiency from the monochromator grating. The spectrally resolved pump-probe signal is detected by a 32 element HgCdTe detector. Great care is taken to ensure that the probe intensities for both parallel and perpendicular components are the same on a given pixel of the array detector to avoid the consequences of the mildly nonlinear response of HgCdTe with increasing intensity.39
III. Results and Discussion
Igepal surfactants have a head group composed of an alcohol tethered by a polyether chain to the nonpolar tail. AOT, on the other hand, has a sulfonate head group with a sodium counter ion. Empirical formulas relating the size of the AOT and Igepal reverse micelles as a function of w0 were used to determine the amount of water necessary to create micelles of the same size. For AOT reverse micelles with w0 between 2 and 20, the diameter of the water pool is given by the equation: dAOT = (0.29 w0 + 1.1) nm.34 Small-angle neutron scattering experiments on Igepal reverse micelles prepared using the same method employed here give the following relationship for the size of the water pool, dIG = (0.38w0 + 1.40) nm.36 While Igepal reverse micelles were not studied for sizes smaller than w0 = 10 the data demonstrate that reverse micelles somewhat smaller than w0 = 10 will be stable and their sizes will follow the same linear relationship.36 In addition, MD simulations on AOT and Igepal type surfactants show similar ellipticity characteristics for reverse micelles made from both surfactants with sizes smaller than w0 = 10.37,40 We have prepared Igepal reverse micelles with w0 = 7, and smaller. For w0 = 7, the equation predicts an Igepal reverse micelle nanoscopic water pool with the same size as the AOT w0 = 10 reverse micelle, that is, d = 4 nm. A diameter of 4 nm was chosen because in previous studies of nanoscopic water in AOT, the w0 = 10 reverse micelle was shown to have water dynamics significantly different from bulk water.14–17 AOT reverse micelles have water dynamics that rapidly approach those of bulk water as the size is increased beyond 4 nm.14 Therefore, significantly larger sizes are not useful.
Smaller sizes are problematic for two important reasons. First, the size of smaller Igepal reverse micelles becomes increasingly uncertain as the size is decreased. Second, in the experiments described below, the OD stretch of dilute HOD in H2O provides a probe of the hydrogen bond dynamics of water, free from vibrational excitation transfer contributions41–43 that occur in pure water. Excitation transfer causes a decay of the measured anisotropy, and therefore, prevents determination of orientational relaxation. The hydroxyl group of Igepal can exchange its hydrogen with the water in the reverse micelle water pool. The deuterium from an HOD molecule will exchange with the Igepal hydroxyl group so that instead of studying only water, the experiment also has a contribution from the hydroxyl group of Igepal. This is a real effect, but it will only be significant for very small reverse micelles, in which the number of surfactant molecules is comparable to the number of water molecules. For the w0 = 7 reverse micelle studied here, there are seven water molecules per surfactant molecule which means there are fourteen water hydroxyl groups for each Igepal hydroxyl group. Statistically, the contribution to the signal from a water OD stretch will outweigh the Igepal hydroxyl OD by a factor of 14. So, the contribution from the Igepal hydroxyl group should be ~7% of the signal for the w0 = 7 reverse micelle.
Therefore, comparing 4 nm AOT and Igepal reverse micelle water nanopools to understand the effects of interfacial differences on the dynamics of the confined water is appropriate and essentially the only size that can provide well defined results to answer the question of whether or not interfacial charges dictate the dynamics of confined water. A single counter example is sufficient to demonstrate that interfacial charges are not solely responsible for confinement effects on water dynamics.
A. IR Spectroscopy
The IR spectrum of water is very sensitive to the local environment of the water molecules, particularly the strength and type of the hydrogen bonds and the local electric fields.44,45 Figure 1 shows the background subtracted IR spectra of the OD stretch of HOD in the w0 = 7 Igepal reverse micelle, the w0 = 10 AOT reverse micelle, and bulk water for comparison. The spectrum of the OD stretch in Igepal reverse micelles (maximum at 2522 cm−1) lies between that of bulk water (maximum at 2509 cm−1) and the AOT reverse micelle spectrum (maximum at 2537 cm−1). Both reverse micelle water spectra are blue shifted compared to bulk water, but the AOT spectrum is shifted to even higher frequency than the Igepal spectrum. In addition, both reverse micelles have water spectra that are ~8% broader than that of bulk water.
Figure 1.

Linear IR spectrum of water in the w0 = 10 AOT reverse micelle (dashed line), the w0 = 7 Igepal reverse micelle (dash-dot line), and bulk water (solid line).
Piletic et al. recently showed that the IR spectra of the OD stretch in the water nanopool of various sizes of AOT reverse micelles can be reproduced with great accuracy (peak position, linewidth, and shape) using a weighted sum of the spectra of bulk water and the smallest reverse micelle studied, w0 = 2, in which essentially all of the water molecules are associated with the sulfonate head groups.14 The experiments and analysis demonstrated that for any size, the spectrum is composed of a core spectrum (bulk water spectrum) and a shell region (spectrum of w0 = 2, water associated with head groups). Vibrational population relaxation was observed to be biexponential. For any size reverse micelle, the biexponential population decay was found to be the weighted sum of the core lifetime (bulk water, 1.7 ps) and the shell lifetime (w0 = 2 lifetime, 5.2 ps). However, the same study established that dynamics involving hydrogen bond structural evolution, such as orientational relaxation and spectral diffusion, could not be described in terms of two distinct subensembles.14 The dynamics of the core and shell regions are coupled through the hydrogen bond network rendering their dynamics dependent on the properties of both regions. The important results from the study of AOT showed that spectra and vibrational lifetimes are local observables that depend on the distinct environments associated with the core and shell, while observables that depend on hydrogen bond dynamics are not separable by region. Dokter et al., studying HOD in D2O in AOT reverse micelles found evidence through a frequency dependent study that water molecules associated with the head groups reoriented more slowly than those in the core.19 The difference in these results from those of Piletic et al was attributed to the greater inhomogeneous broadening of OH compared to OD which allowed greater spectral isolation of water molecules interacting with the head groups.
The Igepal reverse micelles have not been subjected to a similar spectral analysis as the size and shape of the smallest reverse micelles are not characterized, and the spectrum of very small reverse micelles would be dominated by the head group hydroxyls. However, it is likely that the same considerations are responsible for the spectral shift and increased linewidth of the IR spectrum of water in the Igepal reverse micelle that have been shown to describe the spectra of water in AOT reverse micelles. One of the key observables in AOT was the broadening of the spectrum for sizes in between the extremes of pure water and the smallest AOT reverse micelle. Both water and AOT w0 = 2 spectra are narrower than the w0 = 10 spectrum, and the w0 = 10 peak falls in between the peak positions of the two other spectra. The broadening occurs because the w0 = 10 spectrum is the sum of the two other spectra, one at higher frequency and one at lower frequency. The Igepal w0 = 7 spectrum has the same characteristics; it is shifted from and broader than the bulk water spectrum. However, as seen in figure 1, the shift of the water in the Igepal spectrum is smaller than for AOT although the two reverse micelles have the same size water nanopool.
The presence of ionic sulfonate groups in AOT may explain the greater blue spectral shift of water in the AOT reverse micelle compared to water in the Igepal reverse micelle. Many anions including sulfonate groups are known to perturb the hydrogen bonding network of water in their first solvation shells causing a blue shift in the absorption frequency.10,46–50 Some cations, including Mg2+ can induce a red shift in the absorption frequency, but Omta et al. have shown that sodium cations have very little effect on the spectrum and dynamics of water.51 Bergström and coworker found that when the O-D group of an HOD molecule formed a hydrogen bond with a sulfonate group its distribution of absorption frequencies was centered at 2605 cm−1. In comparison, water in an AOT w0=2 reverse micelle absorbs at ~2575 cm−1. However, this value includes both the O-D groups which are hydrogen bonding to sulfonate groups and the O-D groups hydrogen bonded to other water molecules.
In terms of the core-shell model, the sulfonate groups in AOT can play a role in the increased blue shift of the water spectrum relative to Igepal because the absorption spectrum of water molecules hydrating the sulfonate groups (shell) is blue shifted. In the core-shell model, the shift of the water spectrum arises because the spectrum is the sum of the spectra of water molecules associated with the head groups (the shell) and water molecules away from the head groups (the core). In AOT reverse micelles, the spectrum of the shell waters is shifted substantially to the blue of the core water spectrum so the combined spectrum is blue shifted. The spectrum of the Igepal shell waters is unknown and may include contributions from water hydrogen bonding to both the ether moieties and the alcohol group of the Igepal head group. However, the smaller blue shift of the Igepal spectrum is consistent with different interactions between the water molecules and the head groups in Igepal as compared with AOT.
B. Vibrational Lifetime
Like the IR spectrum, the vibrational lifetime is sensitive to the local environment of the excited OD stretch.12–16 Vibrational relaxation occurs because of fluctuating forces acting on the oscillator,52–54 and the rate of vibrational relaxation depends strongly on the availability of lower energy inter- and intramolecular modes to dissipate the vibrational energy.54 Different hydroxyl stretch frequencies will require different amounts of energy to be dissipated in a combination of internal and intermolecular degrees of freedom.
The vibrational population relaxation and the HOD orientational relaxation were measured using ultrafast IR pump-probe experiments. The pump-probe signal decays were measured with the probe polarization parallel to the pump polarization ( I||) and perpendicular to the pump polarization ( I⊥). With an accurate measurement of I|| and I⊥, vibrational population relaxation, P(t) (lifetime) is obtained using
| (1) |
and the orientational relaxation, r(t) (anisotropy) is given by
| (2) |
where C2 (t) is the second Legendre polynomial orientational correlation function. Vibrational relaxation in water leads to a deposition of heat in the sample which causes a shift in the absorption spectrum and a long lived signal. This effect has been well documented in both bulk water and reverse micelles and is corrected for here using the standard procedure described in the references.14,16,55,56
For the OD stretch of HOD in bulk water, the vibrational lifetime is virtually wavelength independent because of extremely fast spectral diffusion that allows water molecules to sample almost all possible hydrogen bonding configurations on a timescale fast compared to the vibrational lifetime. 57–60 In a heterogeneous system like a reverse micelle, distinct environments (shell and core) interconvert slowly. The slow interconversion makes it possible to observe distinct vibrational lifetimes associated with the multiple environments.14 This slow interconversion between different environments in AOT reverse micelles has been observed in MD simulations20. The MD simulation found that the residence time for water at the interface of an AOT w0=10 reverse micelle is between 18 and 34 ps depending on the proximity to the sulfonate head groups. For AOT reverse micelles, the core-shell model, with its two subensembles, was sufficient to fit the biexponential vibrational population decay for every size reverse micelle studied. The fast component was fixed at the vibrational lifetime of the OD stretch in bulk water (1.7 ps) and the slow component was fixed at the vibrational lifetime of the OD stretch of HOD in the w0 = 2 reverse micelle (5.2 ps).14 The fact that the AOT population decays can be fit with these two numbers demonstrates that exchange of water molecules between the shell and core regions is much slower than 5 ps because fast exchange would produce averaging of the two decay times. Figure 2 is a semilog plot of the vibrational population relation, P(t), of the OD stretch in the two types of reverse micelles. The solid lines are the best fit biexponential decays. The fitting parameters are listed in Table 1.
Figure 2.

Semilog plot of the vibrational lifetime of water in the w0 = 10 AOT reverse micelle and the w0 = 7 Igepal reverse micelle. The solid lines are the biexponential fits to the experimental data.
Table 1.
Vibrational Population Relaxation Parameters
| Sample | A1 | t1 (ps) | A2 | t2 (ps) |
|---|---|---|---|---|
| Igepal | 0.55 ± 0.01 | 1.2 ± 0.1 | 0.51 ± 0.01 | 3.2 ± 0.1 |
| AOT | 0.80 ± 0.01 | 1.7 ± 0.1 | 0.26 ± 0.01 | 5.2 ± 0.1 |
| Water | 1.0 | 1.7 ± 0.1 |
The first important observation is that the vibrational population decay of the OD stretch in the Igepal water nanopools is also a biexponential. However, the vibrational lifetime of water in the w0 = 7 Igepal reverse micelle is faster on both time scales than water in the AOT reverse micelle. A biexponential fit gives time constants of 1.2 ps and 3.2 ps. While the numbers are somewhat different, the biexponential vibrational population relaxation observed in Igepal has the same basic nature as in AOT, which is distinct from the single exponential observed in bulk water.
The behavior of water in Igepal reverse micelles is consistent with the core-shell model used to describe the population relaxation in AOT.14 The data presented in figure 2 was taken near the peak of the Igepal spectrum. As the detection wavelength is tuned to the blue, the amplitude of the slow component becomes greater relative to the fast component. (The wavelength dependence of the data will be discussed in detail in a subsequent publication.) In the core-shell model, a bluer detection wavelength will reflect more of the shell vibrational relaxation. In AOT, the slower component of the biexponential decay was shown to be the shell component.
The fast component of the Igepal population decay is actually faster than that of bulk water. In the population relaxation of the OD stretch of HOD in AOT, the core vibrational lifetime was taken to be that of bulk water. However, the lifetime is sensitive to the local environment experienced by the OD. If the core-shell model applies to the water in Igepal reverse micelles, the results suggest that the core water has relaxation pathways available to it which are not available in the core of AOT or bulk water. Vibrational relaxation is very sensitive to the coupling of the relaxing mode to other discrete modes and to the low frequency continuum of states associated with the medium.52–54,61 For example, a shortening of the vibrational lifetime of bulk water was observed upon lowering the temperature toward the freezing point.62
The IR spectra and the vibrational lifetimes provide information on the local, essentially static core and shell structures of water in reverse micelles. As discussed in the next section, the hydrogen bond network dynamics of the water nanopools is not the same as that of bulk water in either AOT14 or Igepal.
C. Orientational Relaxation
The dynamics of the hydrogen bond network of water molecules can be probed using the IR pump-probe anisotropy decay of the OD stretch, which measures the orientational relaxation dynamics of the water.14,63 The anisotropy decay is directly related to the second Legendre polynomial correlation function, C2(t) (see equation 2). The anisotropy decay of water in a reverse micelle is sensitive to the dynamics of the hydrogen bond network because the hydrogen bonds place significant restrictions on the orientational freedom of water molecules. Both the short time and long time scale orientational motions of water in small reverse micelles deviate from those observed in bulk water. Following an ultrafast inertial component, the decay of orientational anisotropy in bulk water is single exponential while it is biexponential for small AOT reverse micelles.14,15,17
For a molecule to undergo full orientational randomization a global restructuring of the hydrogen bond network is required, that is, hydrogen bonds must break and reform with a new geometry. Theoretical models can be applied to provide a physical picture for the anisotropy decay of water in confined environments. The wobbling-in-a-cone model, followed by complete orientational relaxation can describe the dynamics observed in AOT reverse micelles.14,15,64 Wobbling-in-a-cone describes the short time angular relaxation that is limited by the intact hydrogen bond network. On a longer time scale, hydrogen bond network restructuring leads to complete orientational randomization. Recently the wobbling-in-a-cone model followed by jump reorientation65,66 for the long time decay of the anisotropy was used to analyze the orientational relaxation of water molecules confined in Nafion membranes,12 and in concentrated salt solutions.50 MD simulations have provided evidence at a molecular level for these reorientation mechanisms.65,67 The purpose of this paper is to compare the orientational motions of water molecules in two types of reverse micelles. Detailed discussions of the anisotropy decay of HOD in AOT reverse micelles of various sizes have been published previously.14–16 A discussion of the anisotropy decay for various sizes of Igepal reverse micelles will follow in a later publication.
Figure 3a shows the anisotropy decays of the OD stretch of HOD in the water nanopools (d = 4 nm) in AOT w0=10 and Igepal w0=7 reverse micelles at the same wavelength, 2539 cm−1, close to the peak of both spectra. The orientational relaxation dynamics in these two reverse micelles are very similar. The decays in both samples are biexponential with a fast component of ~0.6 ps and a slow component of ~10 ps (see Table 2 for fit parameters). (There is also an ultrafast inertial component that decays completely in <200 fs and is hidden beneath the non-resonant solvent signal (not shown).15) In contrast, bulk water’s anisotropy decays as a single exponential of 2.6 ps (following the ultrafast inertial component).14,63 Figure 3b shows the anisotropy decays of the two reverse micelles in comparison to the anisotropy decays of bulk water and neat Igepal. It is clear that the water in the reverse micelles have similar dynamics, both of which are much slower than bulk water. A comparison with neat Igepal provides evidence that the reorientational dynamics measured for the Igepal reverse micelle are not dominated by the Igepal hydroxyl groups. The key point here is that the orientational relaxation in Igepal water nanopools is very different from that of bulk water and very similar to AOT water nanopools even though Igepal has a non-ionic head group.
Figure 3.
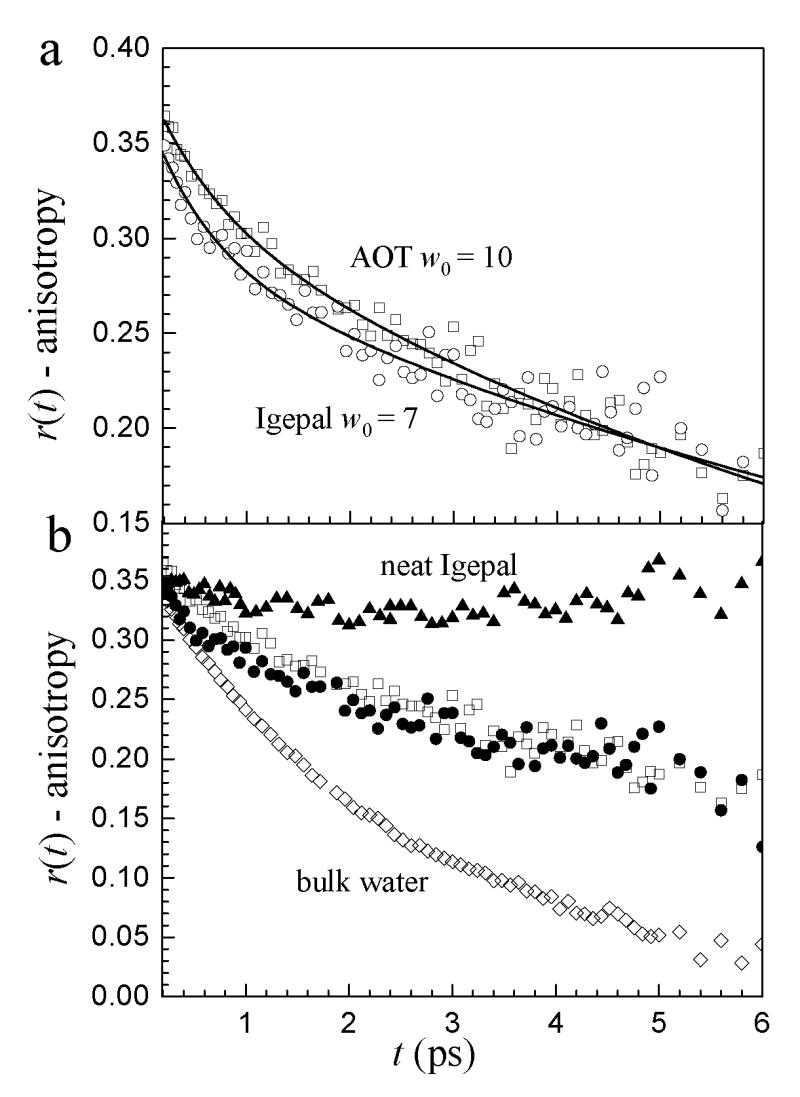
a. Orientational relaxation (anisotropy decay) of water in the w0 = 10 AOT reverse micelle (open squares), and the w0 = 7 Igepal reverse micelle (open circles). Solid lines are biexponential fits to the data. b. Orientational relaxation of the two reverse micelles (AOT - open squares, Igepal - closed circles) with bulk water (open diamonds) and neat Igepal (closed triangles) for comparison. Note the different vertical scales for the two graphs.
Table 2.
Orientational Relaxation Parameters
| sample | a1 | τ1(ps) | a2 | τ2 (ps) |
|---|---|---|---|---|
| Igepal | 0.08 ± 0.01 | 0.6 ± 0.2 | 0.29 ± 0.01 | 11.6 ± 1 |
| AOT | 0.07 ± 0.01 | 0.6 ± 0.2 | 0.32 ± 0.01 | 9.6 ± 0.8 |
| Water | 0.36 ± 0.01 | 2.6 ± 0.1 |
In the reverse micelles, the short time component of the biexponential has been attributed to wobbling-in-a-cone, that is, a sampling of a limited range of angles (the cone).14 The angular range is initially restricted by the hydrogen bond network. At short times, orientational diffusion is limited by the constraints of the initial hydrogen bond network connectivity. A water molecule is only able to sample the angular space allowed within its initial local hydrogen bond network geometry. Full orientational randomization can only occur through relaxation of the angular restrictions by hydrogen bond breaking and reformation in a different configuration. The complete orientational relaxation probably arises from jump reorientation65,68 rather than Gaussian diffusion, which is the usual description of orientational relaxation in simpler liquids. The rate of complete orientational relaxation by jump reorientation depends on the jump angle and the jump rate, both of which are determined by the nature of the hydrogen bond network. Basically, there are orientational motions that are restricted by the hydrogen bond network and occurring on a time scale faster than hydrogen bond breaking. For orientational excursions that are greater than the angular cone allowed by the initial local hydrogen bond network geometry to occur, hydrogen bonds must break and reform. The reformed network must have a different configuration with the OD bond vector pointing in a new direction. This shift in direction is one of many angular jumps that randomize the orientation.
The fact that orientational relaxation in the ~4 nm diameter nanopools of water in AOT and Igepal reverse micelles is very similar in the two nanoscopic systems and is very different from that of bulk water demonstrates that the hydrogen bond dynamics of nanoscopic water reflects the impact of confinement by an interface rather than the presence or absence of charges at the interface. Both the sulfonate head groups of AOT and the alcohol head groups of Igepal form hydrogen bonds with water. In both cases, the restricted orientational geometry and dynamics at the interface propagate into the core of the reverse micelle because of the connectivity of the hydrogen bond network. The result is a significant alteration of the hydrogen bond network dynamics as observed by the change in orientational relaxation compared to bulk water whether or not there are interfacial charges. The influence on the dynamics in ionic reverse micelle systems is, therefore, not predominantly an electric field effect produced by charged groups.
Several MD simulations have produced results similar to the experimental data for models of several types of reverse micelles, as well as hydrophobic cavities.15,20,21,69 Based on the slowing of the orientational relaxation even when the walls of the cavity were hydrophobic, Senapati et al. concluded that both hydrogen bonding to the interface and a confined geometry play a major role in slowing the water dynamics.21 It is important to note that the uncharged hydrophobic interfaces that have been simulated are quite different from the non-ionic reverse micelles studied experimentally here.
While the spectra and the vibrational population relaxation can be described in terms of a core-shell model, in which the observables are determined by the sum of the observables for water molecules in the interfacial boundary layer and in the core of the reverse micelles, the dynamics of the hydrogen bond network are not separable in the same manner.14 The core and shell regions, while physically distinct, are intimately connected through the hydrogen bond network. Although the detailed structure of water associated with the head groups is unknown, it is safe to say that the arrangement of water molecules in a shell of waters at the interface will differ substantially from bulk water. Because core waters are hydrogen bonded to interfacial waters, the rearrangements responsible for orientational relaxation in small reverse micelles require motions that depend on both regions. The results shown in figure 3 demonstrate that the perturbation of the hydrogen bond network, which is responsible for the distinct dynamics of nanoscopic water, is not strongly dependent on the presence of charges at the interface. Rather the differences between nanoscopic water and bulk water are determined by the fact that the water is influenced by the interface on a nanometer length scale, and therefore, small confined systems display water dynamics that are distinct from the bulk. This should also be true for water within a few nanometers of an interface, such as a cell membrane, regardless of whether it is charged or neutral.
The dynamics shown in figure 3 are very similar, but they are not identical. The results at very short time suggest that there is a somewhat larger inertial decay (the vertical intercept) for Igepal. The rest of the dynamics for the two systems are quite similar (see Table 2). Some differences would be expected because the interfaces are not identical; hence there will be differences in the interfacial water structures that will propagate through the hydrogen bonding network. However, the results show that the interfacial differences do not play a central role in determining the hydrogen bond dynamics of the confined water. At the interface, the water geometry will not be that of bulk water. This difference will propagate into the core, influencing hydrogen bond network dynamics.
Several groups have found that the polyether groups in Igepal reverse micelles are somewhat hydrated.70,71 Molecular dynamics simulations70, as well as electron spin and fluorescence probes71 show that the polyether chains interact with water although the dominant portion of the water is segregated into the central water pool. The presence of a water nanopool containing most of the water in small Igepal reverse micelles has been confirmed by SANS data.36 The partial hydration of the polyether chains may influence the structure of the Igepal reverse micelle interface making it structurally distinct from the closely packed arrangement of the AOT head groups. The result is that the nature of the combined core-shell hydrogen bond network is not the same in the two types of reverse micelles, giving rise to somewhat different hydrogen bond dynamics, and, therefore, mildly different orientational relaxation dynamics.
It is possible to observe some differences between water in AOT and Igepal reverse micelles that will be discussed in detail in a subsequent publication. As the detection wavelength is shifted substantially to higher frequency than the center of the Igepal spectrum, the anisotropy develops a very slow, apparently constant component. Dokter and coworkers have also observed a slow component in the high frequency regions of HOD in D2O confined in AOT reverse micelles.19 They attribute this slow component to an ensemble of water molecules which is strongly hydrogen bonded to the sulfonate head groups. In Igepal, the nature of the interface is different from AOT and the slow component may come from several sources. One source is the hydroxyl group of the Igepal surfactant. Figure 3b shows that after a fast inertial decay, virtually no reorientation occurs for the hydroxyl group of neat Igepal. Physically this observation makes sense since the hydroxyl group is bound to a long surfactant molecule which will reorient very slowly. While the hydroxyl group of Igepal may play a large role in the anisotropy decay for small reverse micelles, as the number of water molecules in the reverse micelle increases the contribution to the signal from the Igepal hydroxyl groups becomes small. This is the case for the Igepal reverse micelles studied here. Another likely source contributing to the slow anisotropy decay detected in larger reverse micelles at bluer wavelengths is water hydrating the polyether groups of Igepal. Experiments have shown that these groups are somewhat hydrated and isolated water molecules may become trapped between surfactant molecules making it difficult for reorientation to occur on the experimental time scale. While the degree of hydration of the head group’s polyether moiety as the size of the reverse micelle increases is not well known, preliminary orientational relaxation experiments show that for Igepal reverse micelles up to w0 = 25 there appears to be a persistent long lived component in the anisotropy decay. The sizable long lived component is consistent with significant hydration of the polyether groups for large Igepal reverse micelles.
IV. Concluding Remarks
The question of the influence of nanoscopic confinement on the water hydrogen bond dynamics has been addressed by comparing spherical nanopools of water of the same size (4 nm) in two types of reverse micelles, AOT and Igepal. It is well documented that the dynamics of water in confined environments, near interfaces, and in the solvation shell of ions differ from the dynamics of bulk water. A persistent question surrounding the study of water in confined environments or near interfaces has been, ‘is it the presence of or the nature of the confining interface that has the largest effect on the dynamics of the water molecules?’ AOT has an ionic head group while Igepal’s head group is non-ionic. Therefore, the nature of the water interface is substantially different in the two types of reverse micelles. By studying the properties of water in the same size nanopools with and without ions, the issue of confinement vs. the nature of the interface was examined.
The infrared absorption spectrum of the hydroxyl stretch and the vibrational population relaxation in both types of reverse micelles differ from bulk water. The general nature of the differences for the two reverse micelles is the same, the spectra are blue shifted relative to bulk water, and the vibrational population relaxation is biexponential rather than the single exponential decay found for bulk water. While the nature of the differences from bulk water are the same, AOT has a larger spectral blue shift, and its biexponential population decay has longer components of 1.7 ps and 5.2 ps compared to Igepal’s 1.2 ps and 3.2 ps. These two observables were discussed using the core-shell model.14 In terms of the core-shell model, these observables are local and might be expected to be influenced more by the particular details of the interfacial properties.
In contrast, orientational relaxation of water in the nanopools depends on the global dynamics and structure of the hydrogen bond network. The orientational relaxation of water in the nanopools of both types of reverse micelles is distinct from that of bulk water, biexponential decays in contrast to the single exponential decay for bulk water. Figure 3 shows that in spite of the differences in their head group interfaces, orientational relaxation in the AOT and Igepal reverse micelles is very similar, but very different from either bulk water or neat Igepal. Does the interface matter? Yes. Does the interface need to be ionic to matter? No. The experimental results demonstrate that nanoscopic confinement has a major impact on water hydrogen bond network dynamics regardless of the nature of the interface.
Acknowledgments
This work was supported by the Department of Energy (DE-FG03-84ER13251) and the National Institutes of Health (2 R01 GM-061137-05). D.E.M. thanks the NDSEG for a graduate research fellowship. NEL thanks the Colorado State University for support of a research leave.
References
- 1.Liu L, Faraone A, Mou CY, Yen CW, Chen SH. J Phys: Condens Matter. 2004;16:S5403–S5436. [Google Scholar]
- 2.Ramsay JDF, Poinsignon C. Langmuir. 1987;3:320–326. [Google Scholar]
- 3.Floquet N, Coulomb JP, Dufau N, Andre G, Kahn R. Physica B. 2004;350:265–269. [Google Scholar]
- 4.Mitra S, Mukhopadhyay R, Tsukushi I, Ikeda S. J Phys: Condens Matter. 2001;13:8455–8465. [Google Scholar]
- 5.Bellisent-Funel MC, Chen SH, Zanotti JM. Phys Rev E. 1995;51:4558–4569. doi: 10.1103/physreve.51.4558. [DOI] [PubMed] [Google Scholar]
- 6.Zhu Y, Granick S. Phys Rev Lett. 2001;87:096104. doi: 10.1103/PhysRevLett.87.096104. [DOI] [PubMed] [Google Scholar]
- 7.Raviv U, Perkin S, Laurat P, Klein J. Langmuir. 2004;20:5322–5332. doi: 10.1021/la030419d. [DOI] [PubMed] [Google Scholar]
- 8.Endom L, Hertz HG, Thul B, Zeidler MD. Berichte der Bunsengesellschaft. 1967;71:1008. [Google Scholar]
- 9.Lisitza N, Bryant RG. J Chem Phys. 2007;126:101102. doi: 10.1063/1.2714942. [DOI] [PubMed] [Google Scholar]
- 10.Kropman MF, Bakker HJ. J Chem Phys. 2001;115:8942–8948. [Google Scholar]
- 11.Halle B. Phil Trans R Soc London, B. 2004;359:1207–1224. doi: 10.1098/rstb.2004.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moilanen DE, Piletic IR, Fayer MD. J Phys Chem C. 2007;111:8884–8891. doi: 10.1021/jp067460k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moilanen DE, Piletic IR, Fayer MD. J Phys Chem A. 2006;110:9084–9088. doi: 10.1021/jp0623084. [DOI] [PubMed] [Google Scholar]
- 14.Piletic IR, Moilanen DE, Spry DB, Levinger NE, Fayer MD. J Phys Chem A. 2006;110:4985–4999. doi: 10.1021/jp061065c. [DOI] [PubMed] [Google Scholar]
- 15.Tan HS, Piletic IR, Fayer MD. J Chem Phys. 2005;122:174501–174509. doi: 10.1063/1.1883605. [DOI] [PubMed] [Google Scholar]
- 16.Piletic IR, Tan HS, Fayer MD. J Phys Chem B. 2005;109:21273–21284. doi: 10.1021/jp051837p. [DOI] [PubMed] [Google Scholar]
- 17.Tan HS, Piletic IR, Riter RE, Levinger NE, Fayer MD. Phys Rev Lett. 2004;94 doi: 10.1103/PhysRevLett.94.057405. 057405(057404) [DOI] [PubMed] [Google Scholar]
- 18.Cringus D, Lindner J, Milder MTW, Pshenichnikov MS, Vohringer P, Wiersma DA. Chem Phys Lett. 2005;408:162–168. [Google Scholar]
- 19.Dokter A, Woutersen S, Bakker HJ. Proc Natl Acad Sci. 2006;103:15355–15358. doi: 10.1073/pnas.0603239103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faeder J, Ladanyi BM. J Phys Chem B. 2000;104:1033–1046. [Google Scholar]
- 21.Senapati S, Berkowitz ML. J Chem Phys. 2003;118:1937–1944. [Google Scholar]
- 22.Brubach JB, Mermet A, Filabozzi A, Colavita P, Gerschel A, Roy P. Journal de Physique IV France. 2000;10:Pr7–215. [Google Scholar]
- 23.Pal SK, Peon J, Zewail AH. Proc Natl Acad Sci. 2002;99:1763 – 1768. doi: 10.1073/pnas.042697899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levinger NE. Science. 2002;298:1722–1723. doi: 10.1126/science.1079322. [DOI] [PubMed] [Google Scholar]
- 25.Sando G, Dahl K, Owrutsky JC. J Phys Chem B. 2005;109:4084–4095. doi: 10.1021/jp045287r. [DOI] [PubMed] [Google Scholar]
- 26.Sando GM, Dahl K, Owrutsky JC. J Phys Chem A. 2004;108:11209–11217. [Google Scholar]
- 27.Riter RE, Undiks EP, Levinger NE. J Am Chem Soc. 1998;120:6062–6067. [Google Scholar]
- 28.Riter RE, Willard DM, Levinger NE. J Phys Chem B. 1998;102:2705–2714. [Google Scholar]
- 29.Deak JC, Pang Y, Sechler TD, Wang Z, Dlott DD. Science. 2004;306:473 – 476. doi: 10.1126/science.1102074. [DOI] [PubMed] [Google Scholar]
- 30.Patzlaff T, Janich M, Seifert G, Graener H. Chem Phys. 2000;261:381–389. [Google Scholar]
- 31.Seifert G, Patzlaff T, Graener H. Phys Rev Lett. 2002;88:147402. doi: 10.1103/PhysRevLett.88.147402. [DOI] [PubMed] [Google Scholar]
- 32.Giustini M, Palazzo G, Colafemmina G, Della Monica M, Giomini M, Ceglie A. J Phys Chem. 1996;100:3190–3198. [Google Scholar]
- 33.Zhong Q, Baronavski AP, Owrutsky JC. J Chem Phys. 2003;119:9171–9177. [Google Scholar]
- 34.Kinugasa T, Kondo A, Nishimura S, Miyauchi Y, Nishii Y, Watanabe K, Takeuchi H. Colloid Surface A. 2002;204:193–199. [Google Scholar]
- 35.Zulauf M, Eicke HF. J Phys Chem. 1979;83:480–486. [Google Scholar]
- 36.Lipgens S, Schubel D, Schlicht L, Spilgies JH, Ilgenfritz G, Eastoe J, Heenan RK. Langmuir. 1998;14:1041–1049. [Google Scholar]
- 37.Abel S, Waks M, Marchi M, Urbach W. Langmuir. 2006;22:9112–9120. doi: 10.1021/la060978v. [DOI] [PubMed] [Google Scholar]
- 38.Tan HS, Piletic IR, Fayer MD. J Op Soc Am B. 2005;22:2009–2017. [Google Scholar]
- 39.Hansen RS. Appl Optics. 2003;42:4819–4826. doi: 10.1364/ao.42.004819. [DOI] [PubMed] [Google Scholar]
- 40.Abel S, Sterpone F, Bandyopadhyay S, Marchi M. J Phys Chem B. 2004;108:19458–19466. [Google Scholar]
- 41.Woutersen S, Bakker HJ. Nature. 1999;402:507–509. [Google Scholar]
- 42.Gaffney KJ, Piletic IR, Fayer MD. J Chem Phys. 2003;118:2270–2278. [Google Scholar]
- 43.Gochanour CR, Fayer MD. J Phys Chem. 1981;85:1989–1994. [Google Scholar]
- 44.Corcelli SA, Skinner JL. J Phys Chem A. 2005;109:6154–6165. doi: 10.1021/jp0506540. [DOI] [PubMed] [Google Scholar]
- 45.Glew DN, Rath NS. Can J Chem. 1971;49:837–856. [Google Scholar]
- 46.Kropman MF, Bakker HJ. Science. 2001;291:2118–2120. doi: 10.1126/science.1058190. [DOI] [PubMed] [Google Scholar]
- 47.Bergstrom PA, Lindgren J. J Phys Chem. 1991;95:8575–8580. [Google Scholar]
- 48.Bergstrom PA, Lindgren J. J Mol Struct. 1990;239:103–111. [Google Scholar]
- 49.Park S, Kwak K, Fayer MD. Laser Phys Lett. 2007;4:704–718. [Google Scholar]
- 50.Park S, Fayer MD. Proc Nat Acad Sci. 2007 accepted. [Google Scholar]
- 51.Omta AW, Kropman MF, Woutersen S, Bakker HJ. J Chem Phys. 2003;119:12457–12461. [Google Scholar]
- 52.Oxtoby DW. Annu Rev Phys Chem. 1981;32:77. doi: 10.1146/annurev.pc.46.100195.002421. [DOI] [PubMed] [Google Scholar]
- 53.Oxtoby DW. Adv Chem Phys. 1981;47:487–519. [Google Scholar]
- 54.Kenkre VM, Tokmakoff A, Fayer MD. J Chem Phys. 1994;101:10618. [Google Scholar]
- 55.Steinel T, Asbury JB, Fayer MD. J Phys Chem A. 2004;108:10957–10964. doi: 10.1021/jp046711r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rezus YLA, Bakker HJ. J Chem Phys. 2005;123 doi: 10.1063/1.2009729. 114502-114501-114507. [DOI] [PubMed] [Google Scholar]
- 57.Eaves JD, Loparo JJ, Fecko CJ, Roberts ST, Tokmakoff A, Geissler PL. Proc Natl Acad Sci. 2005;102:13019–13022. doi: 10.1073/pnas.0505125102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fecko CJ, Eaves JD, Loparo JJ, Tokmakoff A, Geissler PL. Science. 2003;301:1698–1702. doi: 10.1126/science.1087251. [DOI] [PubMed] [Google Scholar]
- 59.Asbury JB, Steinel T, Kwak K, Corcelli S, Lawrence CP, Skinner JL, Fayer MD. J Chem Phys. 2004;121:12431–12446. doi: 10.1063/1.1818107. [DOI] [PubMed] [Google Scholar]
- 60.Asbury JB, Steinel T, Stromberg C, Corcelli SA, Lawrence CP, Skinner JL, Fayer MD. J Phys Chem A. 2004;108:1107–1119. doi: 10.1063/1.1803532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moore P, Tokmakoff A, Keyes T, Fayer MD. J Chem Phys. 1995;103:3325. [Google Scholar]
- 62.Woutersen S, Emmerichs U, Nienhuys HK, Bakker HJ. Phys Rev Lett. 1998;81:1106. [Google Scholar]
- 63.Rezus YLA, Bakker HJ. J Chem Phys. 2006;125 doi: 10.1063/1.2353831. 144512-144511-144519. [DOI] [PubMed] [Google Scholar]
- 64.Lipari G, Szabo A. J Am Chem Soc. 1982;104:4546–4559. [Google Scholar]
- 65.Laage D, Hynes JT. Science. 2006;311:832–835. doi: 10.1126/science.1122154. [DOI] [PubMed] [Google Scholar]
- 66.Laage D, Hynes JT. Proc Natl Acad Sci. 2007;104:11167–11172. doi: 10.1073/pnas.0701699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ohmine I, Saito S. Acc Chem Res. 1999;32:741–749. [Google Scholar]
- 68.Ivanov EN. Soviet Physics JETP. 1964;18:1041–1045. [Google Scholar]
- 69.Senapati S, Berkowitz ML. J Phys Chem A. 2004;108:9768–9776. [Google Scholar]
- 70.Allen R, Bandyopadhyay S, Klein ML. Langmuir. 2000;16:10547–10552. [Google Scholar]
- 71.Caldararu H, Caragheorgheopol A, Vasilescu M, Dragutan I, Lemmetyinen H. J Phys Chem. 1994;98:5320–5331. [Google Scholar]