

Abstract
Because α-synuclein (Snca) has a role in brain lipid metabolism, we determined the impact that the loss of α-synuclein had on brain arachidonic acid (20:4n-6) metabolism in vivo using Snca-/- mice. We measured [1-14C]20:4n-6 incorporation and turnover kinetics in brain phospholipids using an established steady-state kinetic model. Liver was used as a negative control and no changes were observed between groups. In Snca-/- brains, there was a marked reduction in 20:4n-6-CoA mass and in microsomal acyl-CoA synthetases (Acsl) activity toward 20:4n-6. Microsomal Acsl activity was completely restored after the addition of exogenous wt mouse or human α-synuclein, but not by A30P, E46K, and A53T forms of α-synuclein. Acsl and acyl-CoA hydrolase expression was not different between groups. The incorporation and turnover of 20:4n-6 into brain phospholipid pools was markedly reduced. The dilution coefficient lambda, which indicates 20:4n-6 recycling between the acyl-CoA pool and brain phospholipids, was increased 3.3-fold, indicating more 20:4n-6 was entering the 20:4n-6-CoA pool from the plasma relative to that being recycled from the phospholipids. This is consistent with the reduction in Acsl activity observed in the Snca-/- mice. Using titration microcalorimetry, we determined that α-synuclein bound free 20:4n-6 (Kd of 3.7 μM), but did not bind 20:4n-6-CoA. These data suggest α-synuclein is involved in substrate presentation to Acsl rather than product removal. In summary, our data demonstrate that α-synuclein has a major role in brain 20:4n-6 metabolism through its modulation of endoplasmic reticulum localized acyl-CoA synthetase activity, although mutants forms of α-synuclein fail to restore this activity.
Keywords: alpha-synuclein, arachidonic acid, kinetics, brain, phospholipid, acyl-CoA synthetase
α-Synuclein is a 140 amino acid soluble protein that is highly expressed in the central nervous system (1, 2) and is abundant in presynaptic terminals of neurons (1, 3-5). α-Synuclein is also found in other regions of neurons, in astrocytes, and in oligodendroglia (6-11). Overexpression of and mutations in α-synuclein are associated with early onset Parkinson’s disease (12-15) and other neurodegenerative diseases (16-20). Despite this association with neurodegenerative diseases, the physiological function of this protein remains unclear.
Several lines of evidence suggest that α-synuclein can influence brain lipid metabolism. It has structural similarities to class A2 apolipoproteins (21, 22) and to fatty acid binding proteins (23), suggesting that α-synuclein may alter intracellular lipid trafficking, the regulation of lipid metabolism, and may act to stabilize lipid membranes. α-Synuclein binds to small phospholipid vesicles (22, 24, 25) and to brain vesicles (26). Consistent with this binding, the lack of α-synuclein decreases the resting/reserve pool of synaptic vesicles (27, 28). Although the direct binding of fatty acids is controversial (23, 29), recent studies indicate a strong potential for an important role of α-synuclein in fatty acid uptake and metabolism (11, 29, 30). In astrocytes lacking α-synuclein palmitic (16:0) and arachidonic (20:4n-6) acid uptake and metabolism is depressed through an unknown mechanism (11). Lack of α-synuclein in brain depresses 16:0 uptake and significantly alters its metabolism (29), although the impact on brain 20:4n-6 uptake and metabolism is unknown.
In addition, α-synuclein modulates phospholipase C and D activities in vitro (22, 31-33), suggesting a role in lipid-mediated signal transduction. Studies in vivo also demonstrate that α-synuclein affects enzymes involved in lipid metabolism, because α-synuclein overexpression leads to phospholipase D inhibition in yeast (34) and down regulates expression of phospholipase A2 and of long chain fatty acid CoA synthetase in Drosophila (35). These additional findings suggest that α-synuclein impacts lipid-mediated signal transduction, including 20:4n-6 release. Release of 20:4n-6 during signal transduction is crucial for proper CNS function (36-41) and pathophysiological responses (42-45).
To address the potential role for α-synuclein in brain 20:4n-6 uptake and metabolism, we used steady-state kinetic modeling of [1-14C]20:4n-6 metabolism in vivo coupled with studies using enzyme assays and mRNA expression of key fatty acid metabolic enzymes. These data show, for the first time, a mechanistic explanation for the impact of α-synuclein deficiency on brain lipid metabolism. Herein, we demonstrate that, in the absence of α-synuclein, brain microsomal acyl-CoA synthetase activity was decreased, which in turn reduced 20:4n-6 uptake and turnover in brain phospholipid pools. . More importantly, addition of wt human or mouse α-synuclein completely restored acyl-CoA synthetase activity in these microsomes, while the A30P, E46K, and A53T mutant forms of α-synuclein failed to restore activity.
MATERIALS and METHODS
Mice
This study was conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals (NIH publication 80-23) and under an animal protocol approved by the IACUC at the University of North Dakota (Protocol #0110-1). α-Synuclein gene-ablated mice (Snca-/-) were generated from 129/SvEv strain by gene targeted deletion (28). Male mice (25-30 g) were maintained on standard laboratory chow diet and water ad libitum. The ages of the mice used in this study were between 9-11 months in both groups.
Mouse surgery and tracer infusion
The mouse surgery and tracer infusion was performed as previously described (29, 46, 47). Briefly, [1-14C]20:4n-6 (Moravek Biochemical, Brea, CA) was solubilized in 5 mM HEPES (pH 7.4) buffer containing “essentially fatty acid free” bovine serum albumin (50 mg/ml, Sigma Chemical Co., St. Louis, MO). Awake (3-4 hours post-operative) fasted, male mice (Snca+/+ or Snca-/-) were infused with 170 μCi/kg of [1-14C]20:4n-6 into the femoral vein over 10 min at a constant rate of 30 μL/min. Prior to and during the infusion, arterial blood samples (∼20 μL) were taken to determine plasma radioactivity and confirm steady-state plasma radioactivity. Following infusion, each mouse was killed using pentobarbital (100 mg/kg, i.v.) and immediately subjected to focused microwave irradiation to heat denature enzymes in situ. Brain and liver were rapidly removed, frozen in liquid nitrogen and pulverized under liquid nitrogen temperatures to a fine, homogeneous powder.
Lipid extraction
Lipids from the tissue powder, plasma, and blood samples were extracted using a two-phase extraction procedure (48). The radioactivity in the aqueous and organic fractions was determined by liquid scintillation counting. The extracts were concentrated under a stream of N2 at 40°C and dissolved in n-hexane : 2-propanol : water (56.7 : 37.8 : 5.5 by vol.).
Thin layer chromatography
Tissue phospholipids (PL) were separated by thin-layer chromatography (TLC) on heat-activated Whatman silica gel-60 plates (20 × 20 cm, 250 μm) and developed in chloroform : methanol : acetic acid : water (55 : 37.5 : 3 : 2 by vol.) (49). Brain, liver, and plasma neutral lipids (NL) were separated by TLC on heat-activated Whatman silica gel-60 plates (20 × 20 cm, 250 μm) and developed in petroleum ether : diethyl ether : acetic acid (75 : 25 : 1.3 by vol.) (50). Once separated, individual PL and NL were isolated and used to quantify individual PL and NL class fatty acid mass and radioactivity.
Gas liquid chromatography
The 20:4n-6 mass in phosphatidylinositol (PtdIns), phosphatidylserine (PtdSer), choline glycerophospholipids (ChoGpl), and ethanolamine glycerophospholipids (EtnGpl) was determined following base-catalyzed transesterification to form the fatty acid methyl esters (51).
Sphingomyelin (CerPCho) individual fatty acid mass, as well as mass of free fatty acids in plasma, brain, and liver, were measured by gas liquid chromatography (GLC) after acid-catalyzed esterification (52). The gas liquid chromatograph (Trace GC, ThermoElectron, Austin, TX) was equipped with a capillary column (SP 2330; 30 m × 0.32 mm i.d., Supelco, Bellefonte, PA) and a flame ionization detector. Fatty acids were quantified using a standard curve from commercially purchased standards (NuChek Prep, Elysian, MN) and 17:0 was the internal standard (29).
Acyl-CoA extraction, separation and quantification
Acyl-CoA from brain and liver were extracted and purified using a solid-phase extraction procedure and separated by HPLC on C-18(2) column (Luna, Phenomenex, Torrance, CA) (53). The system was controlled by Beckman 127 solvent module (Fullerton, CA, USA). The eluent was monitored at 260 nm using a Beckman 166 UV/Vis detector. (53). Mass of 20:4n-6-CoA was determined using a standard curve from commercially purchased standards (Sigma Chemical Co., St. Louis, MO) and 17:0-CoA was the internal standard. Radioactivity of 20:4n-6-CoA was determined by liquid scintillation counting.
Liquid scintillation counting
Samples were placed into 20-ml liquid scintillation vials, and 0.5 ml of H2O was added, followed by 10 ml of Scintiverse BD (Fisher). After mixing, the samples were quantified by liquid scintillation counting using a Beckman LS5000 CE liquid scintillation counter (Beckman Instruments, Fullerton, CA) at least 1 h after the addition of the liquid scintillation mixture.
Kinetic analysis
Our study was performed at steady-state conditions using a quantitative kinetic model as previously described (29, 36, 54-56). The main aspect of this model is that it accounts for fatty acid recycling between phospholipid pools and acyl-CoA pool. The dilution coefficient, λ, represents this recycling and is defined as the steady-state specific activity of brain 20:4n-6-CoA relative to specific activity of plasma 20:4n-6.
Titration microcalorimetry
Titration microcalorimetry was carried out as previously described (29, 57). Briefly, recombinant α-synuclein (50 μM) was titrated with 20:4n-6 at 27°C in 20 mM potassium phosphate buffer containing 50 mM KCl (pH 7.2). The sample was titrated with 25-30 aliquots of the fatty acid containing solution (1 mM) added at 3 min intervals. Raw data were integrated using the supplied ORIGIN software (MicroCal, Inc.) and isotherms were analyzed as previously described (58, 59).
Northern blot analysis
Northern blot analysis was carried out as previously described (60). Briefly, 20 μg of isolated mRNA from Snca-/- and Snca+/+ mouse brains was resolved on a 1.2% agarose-formaldehyde gel and transferred to a Biodyne B membrane. Probes were chosen for each Acsl that were for specific regions of the mRNA that differed significantly from each other. Probes were radioactively labelled by random priming using [α-32P]dCTP and High Prime solution (Roche). A murine cDNA (position 9-969; accession no. BC011106) of the ubiquitously expressed acidic ribosomal phosphoprotein P0 (36B4) was used as a loading control. To detect murine Acsl3, Acsl4, Acsl6, and Bach mRNA, the following probes were used: mouse Acsl3 fragment of 342 bp (nucleotides 1060-1402 of accession no. NM_028817); mouse Acsl4 cDNA fragment of 329 bp (nucleotides 878-1206 of accession no. NM_019477); mouse Acsl6 fragment of 398 bp (nucleotides 877-1274 of accession no. NM_144823); and mouse Bach cDNA fragment of 386 bp (position 559-944 of accession no. NM_133348). The blot was hybridized using ExpressHyb solution (BD Biosciences, Franklin Lakes, NJ) at 68°C and washed to high stringency according to manufacturer’s instructions.
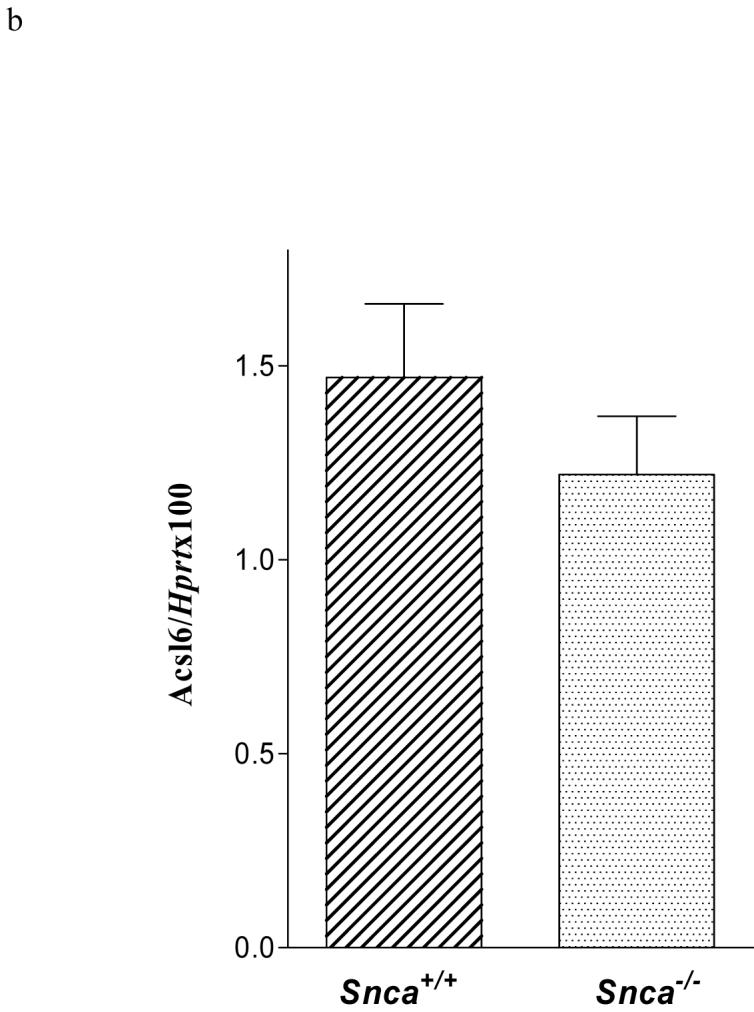
Changes in Acsl6 expression were confirmed using quantitative real time PCR. For each assay, 4 ng cDNA (generated by GeneAmp RNA PCR Kit, Applied Biosystems) was analyzed using the iCycler iQ real-time PCR detection system (Bio-Rad). Mouse Acsl6 expression was quantified using the primers nt877 5′- atagaggactgtggccgaga -3′ (forward) and nt979 5′- ctttggggttccctgttgta -3′ (reverse) (Acc. No. NM_144823) with Sybr-Green. As a control, hydroxy-phosphoribosyl transferase (Hprt) mRNA levels were quantified using the primers nt503, 5′- ctggtaaaacaatgcaaac -3′ (forward), nt622, 5′- caaagtctggcctgtatc -3′ (reverse), nt569, 5′-FAM-caagcttgctggtgaaaagga -DABCYL-3′ (TaqMan probe). Standard curves were generated by serial dilution of plasmids containing Acsl6 or Hprt cDNA (kindly provided by Dr. D.W. Melton, University of Edinburgh, U.K). The thermocycler was programmed: 95°C for 10 min followed by 50 cycles at 95°C for 20 s and 58°C for 50 s for Hprt and 95°C for 10 min followed by 35 cycles at 95°C for 10 s and 60°C for 50 s for Acsl6. At the end of the Acsl6 Sybr-Green-detection, a melt curve was performed for each well to ensure the absence of unspecific PCR products.
Brain homogenate and brain microsomes associated acyl-CoA synthetase activity
Brain microsomal fractions, from wt and snca-/- mice, containing acyl-CoA synthetases were prepared by centrifugation and long chain fatty acyl-CoA synthetase activity was measured (61, 62). The protein content in microsomal fraction and whole brain homogenate was measured using a dye-binding assay (63). Acsl activity was measured in three different groups: 1). microsomes from wt mice; 2). microsomes from snca-/- mice; and 3). microsomes from snca-/- mice incubated with 1.0% wt mouse α-synuclein for 30 min. In all groups, Acsl activity was measured in whole brain homogenate or in microsomal homogenate by adding 20 μg of protein to the assay cocktail, containing 670 μM Triton X-100, having a final [1-14C]20:4n-6 concentration between 3.6 and 190 μM. The reactions were terminated after incubation at 37°C for 4 min. After extraction of non-esterified 20:4n-6, the radioactivity of 20:4n-6-CoA was determined by liquid scintillation counting. The initial rates of reaction were calculated based on the specific radioactivity of the reaction substrate. The Vmax and Km of Acsl activity with 20:4n-6 as a substrate was calculated using standard Michaelis-Menton kinetic analysis.
Rescue of acyl-CoA synthetase activity
Purified, recombinant human and mouse wt α-synuclein or its mutant forms (A30P, E46K, and A53T) were purified (64) and used to determine if these different forms of α-synuclein could rescue microsomal Acsl activity. To determine which concentration of wt or mutant α-synuclein was the ideal concentration to restore Acsl activity, we incubated microsomes with 0.01 to 10% α-synuclein relative to total microsomal protein. This corresponds to a final concentration for α-synuclein of 0.6-634 μM in the assay. The α-synuclein, in Tris buffer (pH 7.4), was added 30 min prior to incubating the Snca-/- brain microsomal fractions (20 μg of protein) with [1-14C]20:4n-6 (190 μM). Acsl activity was determined as stated above. To assess potential endogenous acyl-CoA synthetase activity of α-synuclein, recombinant α-synuclein (1.2 μg) was added to 50 μl of solubilization buffer, which was then added to the assay cocktail.
Western blot analysis of α-synuclein
Brain microsomal protein (40 μg) from Snca+/+ mice (n=8) was resolved on a 15% polyacrylamide gel and transferred to PVDF membrane (11, 65). The positive control was the recombinant α-synuclein and the antibody used to detect α-synuclein was a monoclonal anti-synuclein antibody 4D6 (1:1,000 Signet, Dedham, MA. Blots were developed using chemiluminiscence super-signal (Amersham, Piscataway, NJ).
Statistics
Statistical significance was assessed using an unpaired, two-way, Student’s t-test, with a p < 0.05 considered to be statistically significant. For microsomal Acsl assays, statistical significance was assessed using a one-way ANOVA and a Tukey-Kramer post-hoc test, with a p<0.05 considered to be significant. The number of mice used per experiment is indicated in the figure and table legends.
RESULTS
α-Synuclein binds 20:4n-6 but not 20:4n-6-CoA
We have shown that α-synuclein does not bind palmitate or oleate using titration microcalorimetry (29), although others have shown oleate binding using the classical Lipidex assay (23). To assess if α-synuclein bound monomeric 20:4n-6, we again used titration microcalorimetry. We determined that α-synuclein bound 20:4n-6 with a Kd of 3.7 ± 1.7 μM (n=3) (Figure 1a), but did not bind 20:4n-6-CoA (Figure 1b). The binding affinity for 20:4n-6 is 1 to 2 orders of magnitude less than that of a classical FABP (66).
Figure 1.


a). α-Synuclein binding of 20:4n-6 as determined using titration microcalorimetry, n=3.
b). Lack of α-synuclein binding of 20:4n-6-CoA as determined using titration microcalorimetry, n=3.
Absence of α-synuclein reduced brain 20:4n-6-CoA mass
We noted that the mass of the brain 20:4n-6-CoA pool was reduced 58% in the Snca-/-mice, while the mass in liver 20:4n-6-CoA was unchanged (Figure 2a). Because liver tissue does not contain α-synuclein (29), we used it as a negative control in these experiments. This reduction in brain 20:4n-6-CoA mass was not accounted for by reduced incorporation of 20:4n-6 from plasma into the brain 20:4n-6-CoA pool (k*), as these values were equivalent between groups (data not shown). This indicates that the reduced 20:4n-6-CoA pool was not the result of a reduction in plasma derived 20:4n-6, but rather from a potential reduction in 20:4n-6 recycling from brain phospholipid pools. This was confirmed by an increase in the dilution coefficient, λ, in the Snca-/- brains, indicating an increase in the amount of fatty acid entering the 20:4n-6-CoA pool via the plasma relative to the amount coming from the recycling of 20:4n-6 from endogenous phospholipid pools (Figure 2b). From these results, we would predict that the markedly reduced 20:4n-6-CoA pool would cause a reduction in the incorporation of 20:4n-6-CoA into Snca-/- brain phospholipid pools.
Figure 2.


a). Mass of 20:4n-6-CoA in brain and liver isolated from snca-/- and snca+/+ mice. Values represent mean ± SD, n=8-10. The * indicates statistical significance from snca+/+ mice, p<0.05.
c). Dilution coefficient λ for snca-/- and snca+/+ mice. Values represent mean ± SD, n=8-10. The * indicates statistical significance from snca+/+ mice, p<0.05.
Absence of α-synuclein decreased brain 20:4n-6-CoA incorporation into phospholipids
We next examined the impact of α-synuclein deficiency on changes in the kinetics of 20:4n-6 incorporation and turnover in brain phospholipid pools. In brain tissue, we found the incorporation coefficient k* for 20:4n-6 entering PtdIns, PtdSer, ChoGpl, and CerPCho was decreased 25%, 34%, 22%, and 50%, respectively, in Snca-/- compared to Snca+/+ mice (Table 1). The incorporation rate (JFA,i) for 20:4n-6-CoA into EtnGpl, PtdIns, PtdSer, ChoGpl, and CerPCho was also decreased 48%, 46%, 41%, 47%, and 66%, respectively, in Snca-/- compared to Snca+/+ mice. As a result, the half-life (T1/2) for 20:4n-6 in brain EtnGpl, PtdIns, PtdSer, ChoGpl, and CerPCho was increased 2.8-fold, 3.3-fold, 2.4-fold, 2.9-fold, and 3.7-fold, respectively, Snca-/- compared to Snca+/+ mice. Importantly, there were no significant differences in liver individual phospholipid kinetics between groups (Table 2), indicating that these changes were brain specific.
Table 1.
Effects of α-synuclein ablation on the unilateral incorporation coefficient (k*), rate of incorporation into individual brain phospholipid pools, and turnover in these individual phospholipid pools
| ki* ×10-5 sec-1 | JFA,i nmol/hour | T1/2,i hour | Cbr,i nmol/g ww | |||||
|---|---|---|---|---|---|---|---|---|
| Snca+/+ | Snca-/- | Snca+/+ | Snca-/- | Snca+/+ | Snca-/- | Snca+/+ | Snca-/- | |
| EtnGpl | 2.7±.7 | 2.5±0.6 | 102±37 | 53±28* | 11.2±3.7 | 31.6±12.1* | 1802±619 | 1892±266 |
| PtdIns | 6.8±1.8 | 5.2±1.3* | 252±110 | 135±72* | 1.1±0.3 | 3.6±0.9* | 357±72 | 348±80 |
| PtdSer | 1.2±.4 | 0.8±0.2* | 39±16 | 23±9* | 2.7±1.2 | 6.6±2.1* | 110±21 | 135±33 |
| ChoGpl | 9.3±2.3 | 7.3±1.1* | 350±160 | 186±93* | 1.4±0.3 | 4.0±1.4* | 783±163 | 901±118 |
| CerPCho | 0.8±0.6 | 0.4±0.2* | 32±10 | 11±4* | 0.9±0.3 | 3.3±1.6* | 42±6 | 38±9 |
Values represent mean ± SD, n=7-9. The * indicates statistical significance from control mice, p<0.05. Abbreviations are EtnGpl - ethanolamine glycerophospholipids; PtdIns - phosphatidylinositol; PtdSer - phosphatidylserine; ChoGpl - choline glycerophospholipids; CerPCho - sphingomyelin; ki* - incorporation coefficient of 20:4n-6 from plasma into individual phospholipid; JFA,i - net rate of incorporation from 20:4n-6-CoA into individual phospholipid; T1/2,i - half life of 20:4n-6 in individual phospholipid; Cbr,i - mass of 20:4n-6 in individual phospholipid.
Table 2.
Effects of α-synuclein ablation on the unilateral incorporation coefficient (k*), rate of incorporation into individual liver phospholipid pools, and turnover in these individual phospholipid pools
| ki* ×10-5 sec-1 | JFA,i nmol/hour | T1/2,i hour | Cliv,i nmol/g ww | |||||
|---|---|---|---|---|---|---|---|---|
| Snca+/+ | Snca-/- | Snca+/+ | Snca-/- | Snca+/+ | Snca-/- | Snca+/+ | Snca-/- | |
| EtnGpl | 42.6±5.8 | 51.9±14.6 | 551±207 | 562±164 | 1.3±0.5 | 1.2±0.3 | 989±148 | 1045±118 |
| PtdIns | 24.2±2.9 | 23.2±5.4 | 261±46 | 233±68 | 1.7±0.6 | 1.6±0.5 | 694±116 | 627±77 |
| PtdSer | 10.0±1.8 | 9.6±2.9 | 127±28 | 107±28 | 1.1±0.3 | 1.4±0.5 | 189±30 | 180±30 |
| ChoGpl | 191.7±15.1 | 208.3±59.8 | 2443±817 | 2271±692 | 0.6±0.2 | 0.6±0.2 | 1935±155 | 1990±82 |
| CerPCho | 4.6±1.0 | 4.5±1.4 | 57±18 | 47±12 | 1.9±0.5 | 2.2±0.6 | 166±14 | 155±24 |
Values represent mean ± SD, n=7-9. Abbreviations are as found in Table 1.
While this reduction in 20:4n-6-CoA affected the kinetics of incorporation of 20:4n-6 into brain phospholipid pools, it did not cause a reduction in the absolute mass of 20:4n-6 in individual brain phospholipids (Table 1). These results are consistent with a reduction in the 20:4n-6-CoA available as substrate for phospholipid fatty acid remodeling. We then turned our attention to examining whether this effect was mediated by decreased synthesis of 20:4n-6-CoA and 20:4n-6 uptake.
Absence of α-synuclein reduced brain 20:4n-6 uptake
Previously, we demonstrated that α-synuclein deficiency decreased brain 16:0 uptake in vivo (29) and reduced 16:0 and 20:4n-6 uptake in cultured astrocytes (11). We assessed the effect of α-synuclein on brain 20:4n-6 uptake in vivo by infusing Snca+/+ and Snca-/- mice with [1-14C]20:4n-6 (i. v.). There were no differences in input function (integrated plasma radioactivity) for Snca+/+ (1394 ± 445 nCi × ml-1 × min-1) and Snca-/- (1391 ± 204 nCi × ml-1 × min-1) mice, indicating that there were no overt differences in 20:4n-6 metabolism between groups. In brain extracts, the total and organic (lipid containing) phase incorporation coefficient for Snca-/- mice was modestly decreased 15% and 12%, respectively, (Figure 3). There was no significant difference in tracer entering the brain aqueous compartment (Figure 3), which represents products of β-oxidation (46, 67-69). The lack of difference in β-oxidation adds additional evidence supporting the idea that the changes observed herein are the result of altered ER based Acsl expression or activity, rather than changes in acyl-CoA pools associated with mitochondrial β-oxidation. However, the lack of a large reduction in brain 20:4n-6 uptake suggests 20:4n-6-CoA formation may account for the reduction observed in 20:4n-6-CoA mass.
Figure 3.

Uptake of [1-14C] 20:4n-6 into brain from snca-/- and snca+/+ mice expressed as total uptake or uptake into the aqueous and organic brain fractions. Values were normalized to the plasma curve radioactivity and expressed as the unidirectional incorporation coefficient k*×10-5 (s-1). Values were corrected for radioactivity associated with the residual blood left in brain and represent mean ± SD, n=7-9. The * indicates statistical significance from snca+/+ mice, p<0.05.
Acyl-CoA synthetase and hydrolase expression is unaffected by the lack of α-synuclein
One explanation for the reduction in 20:4n-6-CoA mass would be a reduction in brain acyl-CoA synthetase expression or an increase in brain acyl-CoA hydrolase expression in the Snca-/- mice. Because Acsl3, 4, and 6 are expressed in brain (70-75), we measured the mRNA levels of these three synthetases using Northern blot hybridization and real time PCR analyses. All three synthetases were equally expressed in Snca+/+ and Snca-/- mice (Figure 4a). Expression of Acsl6 was also assessed by real time PCR relative to a housekeeping mRNA for a housekeeping enzyme, Hprt, and was found to be unchanged in Snca-/- mice (Figure 4b). We also measured expression of type II Acyl-CoA thioesterase (Bach), which is highly expressed in brain (76, 77) and hydrolyzes 20:4n-6-CoA very efficiently (78). There were no differences in Bach expression (Figure 4a). Although there are other thioesterases (Type-I), which hydrolyze 20:4n-6-CoA, they do so with much less efficiency and it seems unlikely that these could account for the observed reduction in brain 20:4n-6-CoA levels. Hence, while acyl-CoA synthetase expression was not altered by α-synuclein deficiency, such measurements do not assess a reduction in enzymic activity.
Figure 4.
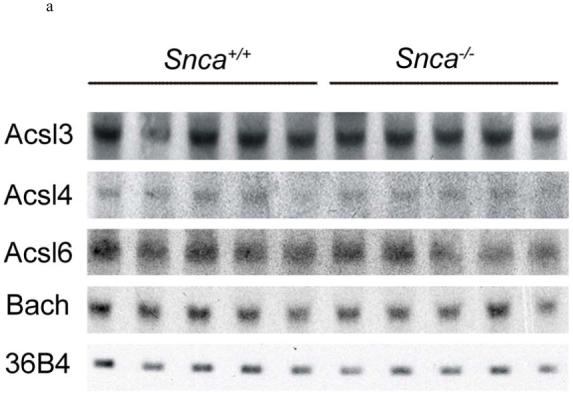
a) Northern blot analysis of brain acyl-CoA synthetase and acyl-CoA hydrolase mRNA expression in snca-/- and snca+/+ mice. Abbreviations are: Acsl - acyl-CoA synthetase mRNA; Bach - acyl-CoA hydrolase mRNA; 36B4 - acidic ribosomal phosphoprotein P0 (loading control).
b). Quantitative real-time RT-PCR of Acsl6 mRNA isolated from snca-/- and snca+/+ mice.
Absence of α-synuclein decreased brain microsomal acyl-CoA synthetase activity
Because α-synuclein deficiency reduced brain 20:4n-6-CoA mass without altering acyl-CoA synthetase and hydrolase expression, we measured acyl-CoA synthetase activities, using 20:4n-6 as a substrate, in whole brain homogenates and in brain microsomal fractions. No differences in acyl-CoA synthetase activity were found in whole brain homogenate (Snca+/+ 1.6 ± 0.1 and Snca-/- 1.5 ± 0.1 pmol × min-1 × mg-1 (n=8)), consistent with the similar incorporation coefficients from plasma into the 20:4n-6-CoA pools. These data suggested an alteration in the activity of endoplasmic reticulum (microsomal) expressed acyl-CoA synthetases which was confirmed using 20:4n-6 as a substrate (Figure 5). Addition of wt mouse α-synuclein to brain microsomes isolated from snca-/- mice clearly demonstrates a complete restoration of activity at all but the lowest concentration of 20:4n-6. Indeed, α-synuclein deficiency reduced Vmax and Km for microsomal acyl-CoA synthetase activity, using 20:4n-6 as a substrate, 32% and 16%, respectively, but these values were completely restored by addition of wt mouse α-synuclein (Table 3). Calculating the ratio of Vmax to Km, a situation that may more accurately reflect activity under physiological conditions (75), we demonstrate a significant 19% reduction in this ratio in the microsomes from snca-/- mice as compared to control mice. Because ER localized acyl-CoA synthetase activity is crucial for 20:4n-6 recycling from phospholipid pools into the 20:4n-6-CoA pool, this reduction in enzymic activity accounts for the observed increase in the dilution coefficient (λ) (Fig. 2b) and the reduction in 20:4n-6 uptake (Figure 3).
Figure 5.

Acsl activity was measured in brain microsomes as described in the Experimental Procedures using 20:4n-6 as a substrate. Values represent mean ± SD, n=8. Samples were from individually prepared microsomes isolated from 8 snca-/- and snca+/+ mice. Inset is a representative double reciprocal plot of these data.
Table 3.
| Vmax (pmol × min-1 × mg-1) | Km (μM) | Vmax/Km × 10-6 | |
|---|---|---|---|
| Snca+/+ | 15.8±1.8 | 33.7±3.4 | 0.47±0.05 |
| Snca-/- | 10.8±0.8* | 28.4±1.5* | 0.38±0.05* |
| Snca-/- + WT Snca | 17.7±2.1 | 35.9±4.0 | 0.49±0.05 |
The Vmax and Km of Acsl activity with 20:4n-6 as a substrate was calculated using standard Michaelis-Menton kinetic analysis. The Vmax/Km ratio was also calculated for individual groups. All values represent mean ± SD, n=8. The * indicates statistical significance from snca+/+ and snca-/- + wt snca.
Wt but not mutant α-synuclein restores acyl-CoA synthetase activity
To further establish a modulatory role for >wt α-synuclein on acyl-CoA synthetase activity in the endoplasmic reticulum, we performed an add back experiment with wt mouse and human α-synuclein and the A30P, E46K, and A53T forms of α-synuclein. Others have shown that α-synuclein is enriched in microsomes (23) and total brain protein is comprised of between 0.1% to 1.0% α-synuclein (79, 80). We confirmed the presence of α-synuclein in our wt brain microsomes using Western blot analysis (Figure 6). Using a dose-response curve, we established that 0.1% to 10% wt mouse and human α-synuclein elicited the same effect on Acsl activity with 20:4n-6 as the substrate (Figure 7a). (Note that this corresponds to a final assay concentration for α-synuclein of 6.3 to 634 μM) However, only wt mouse and human α-synuclein increased Acsl activity in snca-/- microsomes, while E46K and A53T α-synuclein had no effect on activity (Figure 7b). Surprisingly, A30P α-synuclein depressed enzyme activity. In addition, note that 0.05% α-synuclein had significantly less activity than 0.1% and significantly more activity than 0.01% α-synuclein, indicating that the lower concentration of α-synuclein needed to modulate Acsl activity is quite narrow. Importantly, the mutant forms of α-synuclein did not restore activity, demonstrating an important physiological impact of these mutations on brain 20:4n-6 metabolism. The presence of α-synuclein in microsomes isolated from Snca+/+ brain and the restoration of acyl-CoA synthetase activity by exogenously supplied α-synuclein, demonstrate a clear mechanistic role for α-synuclein in brain 20:4n-6 metabolism. Whether α-synuclein interacts with the enzyme directly or is involved in substrate presentation remains to be determined.
Figure 6.
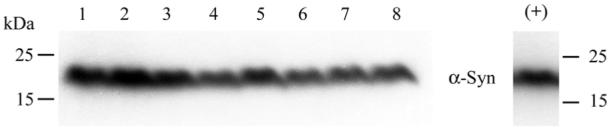
Western blot analysis confirmed the presence α-synuclein in brain microsomes from snca+/+ mice used in 5 . Microsomes were isolated from mouse brains as described in the Experimental Procedures. Lanes 1-8 contain 50 μg of total protein from snca+/+ brain microsomes. The positive control (+) is 0.3 μg of total protein from α-synuclein expressing HEK-293 cells;
Figure 7.


a). Dose-response curve for microsomal Acsl activity in brain microsomes isolated from snca-/- mice following incubation for 30 min with 0.01% to 10% of wt (mouse and human), A30P, E46K, and A53T α-synuclein. This corresponds to a final assay concentration of 0.6 to 634 μM for α-synuclein. Values represent activity in pmol × min-1 × mg protein-1, n=8 for each group. The * indicates statistical significance from microsomes deficient in α-synuclein.
b).Restoration of Acsl activity was determined by measuring Acsl activity in snca-/- brain microsomes following the addition of 1.0% of of wt (mouse and human), A30P, E46K, and A53T α-synuclein. This corresponds to a final assay concentration of 63 μM for the α-synuclein. Microsomes were isolated from 8 snca-/- mouse brains as described in the Experimental Procedures. The substrate was 20:4n-6 (190 μM). All experimental groups were assayed in the same experiment. Values represent mean ± SD, n=8. The * indicates statistical significance from snca+/+ mice, p<0.05, and the ** indicates significance from snca-/- mice, p<0.05.
DISCUSSION
There are two major driving forces for cellular fatty acid uptake: binding to proteins (46, 47, 81-83) and metabolic utilization, specifically acyl-CoA formation (84-86). For instance, expression of Acsl6 in PC-12 cells substantially increases fatty acid uptake (84, 86), whereas expression of FABP increases fatty acid uptake into cells (81-83) and the loss of FABP expression reduces tissue fatty acid uptake in vivo (46, 47). We have previously shown in our studies of 16:0 incorporation and turnover in brain phospholipid pools in Snca-/- mice that α-synuclein does not bind 16:0, yet Snca-/-mice have decreased 16:0 incorporation and turnover in brain without changing 16:0-CoA mass (29), suggesting that altered 16:0 metabolism was the driving force for this reduction in uptake. Others have shown that α-synuclein binds oleate (18:1n-9) using a classical Lipidex competition assay (23), although we demonstrated no 18:1n-9 binding using titration microcalorimetry (29). Although α-synuclein binds to 20:4n-6, it does so with an affinity considerably lower than for a classical FABP (66, 87), suggesting that the demonstrated effect of α-synuclein on brain fatty acid uptake is more likely linked to acyl-CoA formation rather than fatty acid binding.
Absence of α-synuclein depressed brain 20:4n-6 uptake and altered fatty acid targeting to the brain organic fraction, although targeting to the aqueous fraction was unaltered (Figure 3). These results are consistent with those observed for uptake of 16:0 into brains lacking α-synuclein (29) and with its affect on 20:4n-6 and 16:0 uptake and metabolism in primary astrocytes (11). Brain cytosolic, unesterified polyunsaturated fatty acid mass is reduced when α-synuclein is absent (88), suggesting a connection between α-synuclein expression and polyunsaturated fatty acids. The observed reduction in targeting to the organic fraction is accounted by reduced incorporation coefficients for 20:4n-6 into individual brain phospholipids in Snca-/-mice. Thus, there is a modest reduction in 20:4n-6 uptake in Snca-/-mice that is related to reduced incorporation into brain phospholipids pools.
We also observed a marked reduction in 20:4n-6-CoA mass in Snca-/- mice. This reduction was associated with depressed kinetics of 20:4n-6 incorporation into brain phospholipid pools, as evidenced by the elevation in lambda and prolonged half-life for 20:4n-6 in phospholipid pools. The net rate of 20:4n-6 incorporation from 20:4n-6-CoA into individual phospholipids was decreased 40-65% and brain individual phospholipid half-lives were increased 2.4-fold to 3.7-fold in α-synuclein deficient conditions (Table 1). It is important to note that absolute levels of 20:4n-6 in phospholipids pools was not altered, but rather the kinetics of 20:4n-6 incorporation and the duration that 20:4n-6 remained on the phospholipid was increased. The dilution coefficient, λ, was increased 3.3-fold in the absence of α-synuclein, indicating that there was significantly less fatty acid recycling from the endogenous pools of 20:4n-6 found in brain phospholipids and more fatty acid entered 20:4n-6-CoA pool from plasma (Figure 8). These data are consistent with the interpretation that α-synuclein modulates specific brain acyl-CoA synthetases that target fatty acids to phospholipids.
Figure 8.

A model for the role of α-synuclein in brain 20:4n-6 metabolism is presented. Abbreviations are: 20:4n-6 - arachidonic acid; k* 20:4-CoA - incorporation coefficient of 20:4 from plasma into 20:4-CoA pool; FATP - fatty acid binding protein; [20:4-CoA] - mass of 20:4-CoA in tissue; Lamda - dilution coefficient; and Incorp. Rate 20:4-CoA - net rate of 20:4 incorporation from 20:4-CoA into individual phospholipids.
Several mechanisms could lead to decreased 20:4n-6-CoA mass in the absence of α-synuclein. Because α-synuclein overexpression alters expression of acyl-CoA synthetase in Drosophila (35), we determined if α-synuclein deficiency has an effect on the expression of key enzymes involved in acyl-CoA metabolism (Figure 4a,b). Our data indicate that neither acyl-CoA synthetase expression nor acyl-CoA hydrolase expression was altered, hence this possibility does not account for the reduction in observed 20:4n-6-CoA levels. On the other hand, we demonstrated a 32% reduction in brain microsomal acyl-CoA synthetase activity (Vmax) in preparations from α-synuclein deficient mouse brains (Figure 5 and Table 3). In addition, the Vmax/Km ratio was also significantly depressed in the gene-ablated mice (Table 3), demonstrating that under physiological conditions (75) there would be an effect by α-synuclein on Acsl activity. While we demonstrated the presence of α-synuclen in snca+/+- microsomes (Figure 6), others have also noted that α-synuclein is particularly enriched in microsomal fractions containing primarily ER (23) as well as colocalized with ER in synaptosomal preparations (21, 80, 89), but not found associated with synaptic vesicles (80).
More importantly, addition of a physiologically relevant amount of recombinant wt mouse or human α-synuclein completely restored acyl-CoA synthetase activity towards 20:4n-6 (Figure 7a,b). These results indicate that α-synuclein can modulate acyl-CoA synthetase activity, while its mutant (A53T and A30P) forms failed to do so. In fact, A30P significantly reduced acyl-CoA synthetase activity when added to α-synuclein deficient microsomes. Although wt mouse α-synuclein has a natural A53T mutation, it has a number of significant differences in the carboxy tail region as compared to human wt α-synuclein or α-synuclein bearing the A53T mutation. We speculate that these differences may account for the ability of the wt mouse α-synuclein but not the human A53T form of α-synuclein to restore Acsl activity in microsomes isolated from snca-/- mice. These results indicate, for the first time, a significant impact of the mutant forms of α-synuclein on a physiological activity, implying a potentially important reduction in brain 20:4n-6 metabolism in subjects expressing these forms.
To address if α-synuclein may be involved in substrate presentation or product removal, we determined the ability of α-synuclein to bind 20:4n-6 and 20:4n-6-CoA. The lack of 20:4n-6-CoA binding by α-synuclein does not support modulation of acyl-CoA synthetase activity via product removal, while the binding of free 20:4n-6 supports modulation via substrate presentation, but does not rule out a direct protein-to-protein interaction. These results clearly indicate that α-synuclein interacts with the enzyme in a manner that modulates activity, either by a direct protein-protein interaction or through substrate presentation.
Although microsomal Acsl activity was significantly reduced in Snca-/- brains, total brain acyl-CoA synthetase activity toward 20:4n-6 was not altered. This lack of altered activity for total brain acyl-CoA formation is consistent with the similar incorporation coefficients (k*) from plasma into the 20:4n-6-CoA pools. Taken together, these data indicate that α-synuclein modulates specific acyl-CoA synthetase pools. Acyl-CoA pools are spatially arranged in organelles, with pools ranging from the mitochondrial pool to those in the endoplasmic reticulum (29). The lack of differences in the amount of tracer entering brain aqueous fraction, which represents products of β-oxidation (46, 67-69) (Figure 3), indicates that α-synuclein does not impact the mitochondrial pool relative to the endoplasmic reticulum pool, similar to the effect observed using 16:0 (29). Moreover, fatty acid transport protein (FATP) is associated with plasma membrane and exhibits both fatty acid transport and acyl-CoA synthetase activities (90-92) and FATP-4 is expressed in the brain (93). The potential involvement of this enzyme in acyl-CoA formation may account for the lack of differences between the groups in whole brain homogenate acyl-CoA activity and the similar incorporation coefficients for 20:4n-6 from the plasma into the total brain acyl-CoA pool. This suggests that α-synuclein modulates specific acyl-CoA synthetases localized in the ER, consistent with our data presented above. Taking into account that brain Acsl6 targets fatty acids to phospholipids and neutral lipids (84), but that Ascl1 targets fatty acids only to triglycerides (75), we speculate that α-synuclein selectively affects one or more specific acyl-CoA synthetases (Acsl 3, 4, or 6) associated with phospholipid synthesis. It is important to note that in α-synuclein deficient astrocytes, there is significantly more fatty acid targeted for incorporation into neutral lipids, e.g. triacylglycerols and cholesteryl esters as well as a substantial increase in neutral lipid mass (11). In addition, brain neutral lipid mass was also increased in the Snca-/- brains (unpublished data), suggesting a similar situation occurs in vivo.
Collectively, our results demonstrate that α-synuclein is critical for maintenance of brain 20:4n-6-CoA levels that are absolutely essential for incorporation of 20:4n-6 into brain phospholipid pools (Figure 6). This effect on metabolism accounts for the small, but significant reduction in 20:4n-6 uptake. There was a large reduction in 20:4n-6-CoA mass in Snca-/- brains, resulting from a reduction in endoplasmic reticulum (microsomal) acyl-CoA synthetase activity. This reduction in activity decreased the net 20:4n-6-CoA pool size, which caused a substrate limiting reduction in 20:4n-6-CoA resulting in decreased incorporation into individual phospholipids. As a result, the half-life of individual phospholipids was increased, indicating less recycling of 20:4n-6 from endogenous phospholipid stores. These results are important because they are the first demonstration that α-synuclein impacts brain 20:4n-6 metabolism through modulation of an ER based acyl-CoA synthetase rather than through the influence on fatty acid uptake via the direct binding of 20:4n-6 by α-synuclein. Because of the fundamental importance of 20:4n-6 metabolism in brain function, we demonstrate herein a key means of regulating 20:4n-6 metabolism through modulation of brain 20:4n-6-CoA pools; thereby providing a means to channel 20:4n-6 to phospholipids. This newly described physiological function for α-synuclein provides a broader view of how it regulates brain lipid metabolism and how changes in this protein may impact brain function, contributing to the pathophysiology associated with neurodegenerative disease. In addition, we demonstrate the inability of the mutant forms of α-synuclein to restore brain microsomal 20:4n-6-CoA production via microsomal Acsl, suggesting a potentially critical reduction in 20:4n-6 metabolism in subjects expressing these mutant forms of α-synuclein.
ACKNOWLEDGEMENTS
We thank Dr. Carole Haselton for her excellent surgical and technical work. The anti α-synuclein antibodies were kindly provided by Signet Laboratories, Inc. We thank Cindy Murphy for typed preparation of the manuscript.
This work was supported by NIH grants NS043697-01A to EJM and, in part, by a project (EJM) on NIH grant 1P20 RR17699-01 and the Intramural Research Program of the National Genome Research Institute, NIH.
ABBREVIATIONS
- Snca
alpha-synuclein
- Acsl
acyl-CoA synthetase
- Bach
acyl-CoA hydrolase mRNA
- 36B4
acidic ribosomal phosphoprotein P0 (italics gene and non-italics protein)
- EtnGpl
ethanolamine glycerophospholipids
- PtdIns
phosphatidylinositol
- PtdSer
phosphatidylserine
- ChoGpl
choline glycerophospholipids
- CerPCho
sphingomyelin
- 20:4n-6
arachidonic acid
- ki*
incorporation coefficient of 20:4n-6 from plasma into individual phospholipid
- JFA,i
net rate of incorporation from 20:4n-6-CoA into individual phospholipid
- T1/2,i
half life of 20:4n-6 in individual phospholipid
- k* 20:4-CoA
incorporation coefficient of 20:4 from plasma into 20:4-CoA pool
- [20:4-CoA]
mass of 20:4-CoA in tissue
- Lambda
dilution coefficient
- Incorp. Rate 20:4-CoA
net rate of 20:4 incorporation from 20:4-CoA into individual phospholipids
- FATP
fatty acid binding protein
REFERENCES
- 1.Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994;345:27–32. doi: 10.1016/0014-5793(94)00395-5. [DOI] [PubMed] [Google Scholar]
- 2.Lavedan C. The synuclein family. Genome Research. 1998;8:871–880. doi: 10.1101/gr.8.9.871. [DOI] [PubMed] [Google Scholar]
- 3.Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized in the nucleus and presynaptic nerve terminal. J. Neurosci. 1988;8:2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jo E, McLaurin J, Yip CM, St.George-Hyslop P, Fraser PE. α-synuclein membrane interactions and lipid specificity. J. Biol. Chem. 2000;275:34328–34334. doi: 10.1074/jbc.M004345200. [DOI] [PubMed] [Google Scholar]
- 5.McLean PJ, Ribich S, Hyman BT. Subcellular localization of alpha-synuclein in primary neuronal cultures: effect of missense mutations. J. Neural. Transm. Suppl. 2000;(58):53–63. doi: 10.1007/978-3-7091-6284-2_5. [DOI] [PubMed] [Google Scholar]
- 6.Richter-Landsberg C, Gorath M, Trojanowski JQ, Lee VM. Alpha-synuclein is developmentally expressed in cultured rat brain oligodendrocytes. J. Neurosci. Res. 2000;62:9–14. doi: 10.1002/1097-4547(20001001)62:1<9::AID-JNR2>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 7.Mori F, Tanji K, Yoshimoto M, Takahashi H, Wakabayashi K. Demonstration of α-synuclein immunoreactivity in neuronal and glial cytoplasm in normal human brain tissue using proteinkinase K and formic acid pretreatment. Exp. Neurol. 2002;176:98–104. doi: 10.1006/exnr.2002.7929. [DOI] [PubMed] [Google Scholar]
- 8.Cheng SY, Trombetta LD. The induction of amyloid precursor protein and α-synuclein in rat hippocampal astrocytes by diethyldithiocarbamate and copper with or without glutathione. Toxicology Letters. 2004;146:139–149. doi: 10.1016/j.toxlet.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 9.Sung JY, Park SM, Lee CH, Um JW, Lee HJ, Kim J, Oh YJ, Lee ST, Paik SR, Chung KC. Proteolytic cleavage of extracellular secreted α-synuclein via matrix metalloproteinases. J. Biol. Chem. 2005;280:25216–25224. doi: 10.1074/jbc.M503341200. [DOI] [PubMed] [Google Scholar]
- 10.Ziolkowska B, Gieryk A, Bilecki W, Wawrzczak-Bargiela A, Wedzony K, Chocyk A, Danielson PE, Thomas EA, Hilbush BS, Sutcliff JG, Przewlocki R. Regulation of α-synuclein expression in limbic and motor brain regions of morphine-treated mice. J. Neurosci. 2005;25:4996–5003. doi: 10.1523/JNEUROSCI.4376-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castagnet PI, Golovko MY, Barceló-Coblijn GC, Nussbaum RL, Murphy EJ. Fatty acid incorporation is decreased in astrocytes cultured from α-synuclein gene-ablated mice. J. Neurochem. 2005;94:839–849. doi: 10.1111/j.1471-4159.2005.03247.x. [DOI] [PubMed] [Google Scholar]
- 12.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, DiIorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 13.Kruger R, Kuhn W, muller T, Woitalla D, Graeber S, Kosel S, Przuntek H, Epplen JT, Schols L, Reiss O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nature Genetics. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 14.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. α-synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 15.Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Gomez Tortosa E, del Ser T, Muñoz DG, de Yebenes JG. The new mutation, E46K, of α-synuclein causes Parkinson and Lewy Body dementia. Ann. Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 16.Trojanowski JQ, Goedert M, Iwatsubo T, Lee VM. Fatal attractions: abnormal protein aggregation and neuron death in Parkinson’s disease and Lewy body dementia. Cell Death Differentiation. 1998;5:832–837. doi: 10.1038/sj.cdd.4400432. [DOI] [PubMed] [Google Scholar]
- 17.Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci. Lett. 1998;251:205–208. doi: 10.1016/s0304-3940(98)00504-7. [DOI] [PubMed] [Google Scholar]
- 18.Takeda A, Hashimoto M, Mallory M, Sundsumo M, Hansen L, Sisk A, Masliah E. Abnormal distribution of the non-Abeta component of Alzheimer’s disease amyloid precursor/alpha-synuclein in Lewy body disease as revealed by proteinase K and formic acid pretreatment. Lab. Invest. 1998;78:1169–1177. [PubMed] [Google Scholar]
- 19.Lippa CF, Schmidt ML, Lee VM, Trojanowski JQ. Antibodies to alpha-synuclein detect Lewy bodies in many Down’s syndrome brains with Alzheimer’s disease. Ann. Neurol. 1999;45:353–357. doi: 10.1002/1531-8249(199903)45:3<353::aid-ana11>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 20.Iseki E, Marui W, Kosaka K, Akiyama H, Ueda K, Iwatsubo T. Degenerative terminals of the perforant pathway are human alpha-synuclein-immunoreactive in the hippocampus of patients with diffuse Lewy body disease. Neurosci. Lett. 1998;258:81–84. doi: 10.1016/s0304-3940(98)00856-8. [DOI] [PubMed] [Google Scholar]
- 21.George JM, Jin H, Woods WS, Clayton DF. Characterization of a novel protein regulated during the critical period for song learning in the Zebra Finch. Neuron. 1995;15:361–372. doi: 10.1016/0896-6273(95)90040-3. [DOI] [PubMed] [Google Scholar]
- 22.Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 23.Sharon R, Goldberg MS, Bar-Josef I, Betensky RA, Shen J, Selkoe DJ. α-Synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc. Natl. Acad. Sci. USA. 2001;98:9110–9115. doi: 10.1073/pnas.171300598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Narayanan V, Scarlata S. Membrane binding and self-association of α-synucleins. Biochemistry. 2001;40:9927–9934. doi: 10.1021/bi002952n. [DOI] [PubMed] [Google Scholar]
- 25.Perrin RJ, Woods WS, Clayton DF, George JM. Exposure to long-chain polyunsaturated fatty acids triggers rapid multimerization of synucleins. J. Biol. Chem. 2001;276:41958–41962. doi: 10.1074/jbc.M105022200. [DOI] [PubMed] [Google Scholar]
- 26.Jensen PH, Nielsen MS, Jakes R, Dotti CG, Goedert M. Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J. Biol. Chem. 1998;273:26292–26294. doi: 10.1074/jbc.273.41.26292. [DOI] [PubMed] [Google Scholar]
- 27.Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 2000;20:3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cabin DE, Shimazu K, Murphy D, Cole NB, Gottschalk W, McIlwain KC, Orrison B, Chen A, Ellis CE, Paylor R, Lu B, Nussbaum RL. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking α-synuclein. J. Neurosci. 2002;22:8797–8807. doi: 10.1523/JNEUROSCI.22-20-08797.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Golovko MY, Faergeman NJ, Cole NB, Castagnet PI, Nussbaum RL, Murphy EJ. Alpha-synuclein gene deletion decreases brain palmitate uptake and alters the palmitate metabolism in the absence of alpha-synuclein palmitate binding. Biochemistry. 2005;44:8251–8259. doi: 10.1021/bi0502137. [DOI] [PubMed] [Google Scholar]
- 30.Ellis CE, Murphy EJ, Mitchell DC, Golovko MY, Scaglia F, Barcelo-Coblijn G, Nussbaum RL. Mitochondrial lipids abnormality and electron transport chain impairment in mice lacking α-synuclein. Molecular and Cellular Biology. 2005;25:10190–10201. doi: 10.1128/MCB.25.22.10190-10201.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jenco JM, Rawlingson A, Daniels B, Morris AJ. Regulation of phospholipase D2: selective inhibition of mammalian phospholipase D isoenzymes by alpha- and beta-synucleins. Biochemistry. 1998;37:4901–4909. doi: 10.1021/bi972776r. [DOI] [PubMed] [Google Scholar]
- 32.Payton JE, Perrin RJ, Woods WS, George JM. Structural determinants of PLD2 inhibition by alpha-synuclein. J. Mol. Biol. 2004;337:1001–1009. doi: 10.1016/j.jmb.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 33.Narayanan V, Guo Y, Scarlata S. Fluorescence studies suggest a role for α-synuclein in the phosphatidylinositol lipid signaling pathway. Biochemistry. 2005;44:462–470. doi: 10.1021/bi0487140. [DOI] [PubMed] [Google Scholar]
- 34.Outeiro TF, Lindquist S. Yeast cells provide insight into α-synuclein biology and pathobiology. Science. 2003;302:1772–1775. doi: 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scherzer CR, Jensen RV, Gullans SR, Feany MB. Gene expression changes presage neurodegeneration in a Drosophila model of Parkinson’s disease. Hum. Mol. Genet. 2003;12:2457–2466. doi: 10.1093/hmg/ddg265. [DOI] [PubMed] [Google Scholar]
- 36.Rosenberger TA, Villacreses NE, Contreras MA, Bonventre JV, Rapoport SI. Brain lipid metabolism in the cPLA2 knockout mouse. J. Lipid Res. 2003;44:109–117. doi: 10.1194/jlr.m200298-jlr200. [DOI] [PubMed] [Google Scholar]
- 37.Lesa GM, Palfreyman M, Hall DH, Clandinin MT, Rudolph C, Jorgensen EM, Schiavo G. Long chain polyunsaturated fatty acids are required for efficient neurotransmission in C. elegans. Journal of Cell Science. 2003;116:4965–4975. doi: 10.1242/jcs.00918. [DOI] [PubMed] [Google Scholar]
- 38.Bazan NG. Synaptic lipid signaling: significance of polyunsaturated fatty acis and platelet-activating factor. J. Lipid Res. 2003;44:2221–2233. doi: 10.1194/jlr.R300013-JLR200. [DOI] [PubMed] [Google Scholar]
- 39.Williams JH, Errington ML, Lynch MA, Bliss TVP. Arachidonic acid induces a long-term activity-dependent enhancement of synaptic transmission in the hippocampus. Nature. 1989;341:739–742. doi: 10.1038/341739a0. [DOI] [PubMed] [Google Scholar]
- 40.Wolf MJ, Izumi Y, Zorumski CF, Gross RW. Long-term potentiation requires activation of calcium-independent phospholipase A2. FEBS Lett. 1995;377:358–362. doi: 10.1016/0014-5793(95)01371-7. [DOI] [PubMed] [Google Scholar]
- 41.Massicotte G, Vanderklish P, Lynch G, Baudry M. Modulation of a DL-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid/quisqualate receptors by phospholipase A2: a necessary step in long-term potentiation. Proc. Natl. Acad. Sci. USA. 1991;88:1893–1897. doi: 10.1073/pnas.88.5.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee H, Villacreses NE, Rapoport SI, Rosenberger TA. In vivo imaging detects a transient increase in brain arachidonic acid metabolism: a potential marker of neuroinflammation. J. Neurochem. 2004;91:936–945. doi: 10.1111/j.1471-4159.2004.02786.x. [DOI] [PubMed] [Google Scholar]
- 43.Bazan NG. Changes in free fatty acids of brain by drug-induced convulsions, electroshock and anesthesia. J. Neurochem. 1971;18:1379–1385. doi: 10.1111/j.1471-4159.1971.tb00002.x. [DOI] [PubMed] [Google Scholar]
- 44.Rosenberger TA, Villacreses NE, Hovda JT, Boestti F, Weerasinghe G, Wine RN, Harry GJ, Rapoport SI. Rat brain arachidonic acid metabolism is increased by a 6-day intracerebral ventricular infusion of bacterial lipopolysaccharide. J. Neurochem. 2004;88:1168–1178. doi: 10.1046/j.1471-4159.2003.02246.x. [DOI] [PubMed] [Google Scholar]
- 45.Arai K, Ikegaya Y, Nakatani Y, Kudo I, Nishiyama N, Matsuki N. Phospholipase A2 mediates ischemic injury in the hippocampus: a regional difference of neuronal vulnerability. Eur. J. Neurosci. 2001;13:2319–2323. doi: 10.1046/j.0953-816x.2001.01623.x. [DOI] [PubMed] [Google Scholar]
- 46.Murphy EJ, Barcelo-Coblijn G, Binas B, Glatz JF. Heart fatty acid uptake is decreased in heart-fatty acid binding protein gene-ablated mice. J. Biol. Chem. 2004;279:34481–34488. doi: 10.1074/jbc.M314263200. [DOI] [PubMed] [Google Scholar]
- 47.Murphy EJ, Owada Y, Kitanaka N, Konda H, Glatz JFC. Brain arachidonic acid incorporation is decreased in heart-fatty acid binding protein gene-ablated mice. Biochemistry. 2005;44:6350–6360. doi: 10.1021/bi047292r. [DOI] [PubMed] [Google Scholar]
- 48.Folch J, Lees M, Sloan Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 49.Jolly CA, Hubbell T, Behnke WD, Schroeder F. Fatty acid binding protein: stimulation of microsomal phosphatidic acid formation. Arch. Biochem. Biophys. 1997;341:112–121. doi: 10.1006/abbi.1997.9957. [DOI] [PubMed] [Google Scholar]
- 50.Marcheselli VL, Scott BL, Reddy TS, Bazan NG. Quantitative analysis of acyl group composition of brain phospholipids, neutral lipids, and free fatty acids. In: Boulton AA, Baker GB, Horrocks LA, editors. Neuromethods 7 Lipids and Related Compounds. Humana Press; Clifton, NJ: 1988. pp. 83–110. [Google Scholar]
- 51.Brockerhoff H. Determination of the positional distribution of fatty acids in glycerolipids. Methods Enzymol. 1975;35:315–325. doi: 10.1016/0076-6879(75)35171-9. [DOI] [PubMed] [Google Scholar]
- 52.Akesson B, Elovsson J, Arvidsson G. Initial incorporation into rat liver glycerolipids of intraportally injected [3H] glycerol. Biochim. Biophys. Acta. 1970;210:15–27. doi: 10.1016/0005-2760(70)90057-3. [DOI] [PubMed] [Google Scholar]
- 53.Golovko MY, Murphy EJ. An improved method for tissue long chain acyl-CoA extraction and analysis. J. Lipid Res. 2004;45:1777–1782. doi: 10.1194/jlr.D400004-JLR200. [DOI] [PubMed] [Google Scholar]
- 54.Robinson PJ, Noronha J, DeGeorge JJ, Freed LM, Nariai T, Rapoport SI. A quantitative method for measuring regional in vivo fatty acid incorporation into and turnover within brain phospholipids: review and critical analysis. Brain Res. Rev. 1992;17:187–214. doi: 10.1016/0165-0173(92)90016-f. [DOI] [PubMed] [Google Scholar]
- 55.Rapoport SI, Chang MCJ, Spector AA. Delivery and turnover of plasma-derived essential PUFAs in mammalian brain. J. Lipid Res. 2001;42:678–685. [PubMed] [Google Scholar]
- 56.Rosenberger TA, Oki J, Purdon AD, Rapoport SI, Murphy EJ. Rapid synthesis and turnover of brain microsomal ether phospholipids in the adult rat. J. Lipid Res. 2002;43:59–68. [PubMed] [Google Scholar]
- 57.Rolf B, Oudenampsen-Krüger E, Börchers T, Færgeman NJ, Knudsen J, Lezius A, Spener F. Analysis of the ligand binding properties of recombinant bovine liver-type fatty acid binding protein. Biochim. Biophys. Acta. 1995;1259:245–253. doi: 10.1016/0005-2760(95)00170-0. [DOI] [PubMed] [Google Scholar]
- 58.Sigurskjold BW, Altman E, Bundle DR. Sensitive titration microcalorimetric study of the binding of Salmonella O-antigenic oligosaccharides by a monoclonal antibody. Eur. J. Biochem. 1991;197:239–246. doi: 10.1111/j.1432-1033.1991.tb15904.x. [DOI] [PubMed] [Google Scholar]
- 59.Faergeman NJ, Sigurskjold BW, Kragelund BB, Andersen KV, Knudsen J. Thermodynamics of ligand binding to acyl-coenzyme A binding protein studied by titration calorimetry. Biochemistry. 1996;35:14118–14126. doi: 10.1021/bi960545z. [DOI] [PubMed] [Google Scholar]
- 60.Fraisl P, Forss-Petter S, Zigman M, Berger J. Murine bubblegum orthologue is a microsomal very long-chain acyl-CoA synthetase. Biochem. J. 2004;377:85–93. doi: 10.1042/BJ20031062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wilson DB, Prescott SM, Majerus PW. Discovery of an arachidonoyl coenzyme A synthetase in human platelets. J. Biol. Chem. 1982;257:3510–3515. [PubMed] [Google Scholar]
- 62.Saunders C, Voigt JM, Weis MT. Evidence for a single non-arachidonic acid-specific fatty acyl-CoA synthetase in heart which is regulated by Mg2+ Biochem. J. 1996;313:849–853. doi: 10.1042/bj3130849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 64.Cole NB, Murphy DD, Lebowitz J, DiNoto L, Levine RL, Nussbaum RL. Metal-catalyzed oxidation of α-synuclein: helping to define the relationship between oligomers, protofilaments and filaments. J. Biol. Chem. 2005;280:9678–9690. doi: 10.1074/jbc.M409946200. [DOI] [PubMed] [Google Scholar]
- 65.Cole NB, Murphy DD, Grider T, Rueter S, Brasaemle D, Nussbaum RL. Lipid droplet binding and oligomerization properties of the Parkinson’s disease protein α-synuclein. J. Biol. Chem. 2002;277:6344–6352. doi: 10.1074/jbc.M108414200. [DOI] [PubMed] [Google Scholar]
- 66.Richieri GV, Ogata RT, Kleinfeld AM. Equilibrium constants for the binding of fatty acids with fatty acid-binding proteins from adipocyte, intestine, heart, and liver measured with the fluorescent probe ADIFAB. J. Biol. Chem. 1994;269:23918–23930. [PubMed] [Google Scholar]
- 67.Miller JC, Gnaedinger JM, Rapoport SI. Utilization of plasma fatty acid in rat brain: distribution of [14C]palmitate between oxidative and synthetic pathways. J. Neurochem. 1987;49:1507–1514. doi: 10.1111/j.1471-4159.1987.tb01021.x. [DOI] [PubMed] [Google Scholar]
- 68.Gnaedinger JM, Miller JC, Latker CH, Rapoport SI. Cerebral metabolism of plasma [14-C]palmitate in awake adult rat: subcellular localization. Neurochem. Res. 1988;13:21–29. doi: 10.1007/BF00971850. [DOI] [PubMed] [Google Scholar]
- 69.Murphy EJ, Rosenberger TA, Patrick CB, Rapoport SI. Intravenously injected [1-14 C]arachidonic acid targets phospholipids, and [1-14C]palmitic acid targets neutral lipids in hearts of awake rats. Lipids. 2000;35:891–898. doi: 10.1007/s11745-000-0598-7. [DOI] [PubMed] [Google Scholar]
- 70.Iijima H, Fujino T, Minekura H, Suzuki H, Kang MJ, Yamamoto T. Biochemical studies of two rat acyl-CoA synthetases, ACS-1 and ACS-2. Eur. J. Biochem. 1996;242:196–190. doi: 10.1111/j.1432-1033.1996.0186r.x. [DOI] [PubMed] [Google Scholar]
- 71.Oikawa E, Iijima H, Suzuki T, Sasano H, Sato H, Kamataki A, Nagura H, Kang MJ, Fujino T, Suzuki H, Yamamoto TT. A novel acyl-CoA synthetase, ACS5, expressed in intestinal epithelial cells and proliferating preadipocytes. J. Biochem. (Tokyo) 1998;124:679–685. doi: 10.1093/oxfordjournals.jbchem.a022165. [DOI] [PubMed] [Google Scholar]
- 72.Kang M-J, Fujino T, Sasano H, Minekura H, Yabuki N, Nagura H, Iijima H, Yamamoto TT. A novel arachidonate-preferring acyl-CoA synthetase is present in steroidogenic cells of the rat adrenal, ovary, and testis. Proc. Natl. Acad. Sci. USA. 1997;94:2880–2884. doi: 10.1073/pnas.94.7.2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fugino T, Kang M-J, Suzuki H, Iijima H, Yamamoto T. Molecular characterization and expression of rat acyl-CoA synthetase 3. J. Biol. Chem. 1996;271:16748–16752. doi: 10.1074/jbc.271.28.16748. [DOI] [PubMed] [Google Scholar]
- 74.Lee EJ, Kim HC, Cho YY, Byun SJ, Lim JM, Ryoo ZY. Alternative promotion of the mouse acyl-CoA synthetase 6 (mAcsl6) gene mediates the expression of multiple transcripts with 5′-end heterogeneity: genetic organization of mAcsl6 variants. Biochem. Biophys. Res. Commun. 2005;327:84–93. doi: 10.1016/j.bbrc.2004.11.141. [DOI] [PubMed] [Google Scholar]
- 75.Van Horn CG, Caviglia JM, Lei OL, Wang S, Granger DA, Coleman RA. Characterization of recombinant long-chain rat acyl-CoA synthetase isoforms 3 and 6: identification of a novel variant of isoform 6. Biochemistry. 2005;44:1635–1642. doi: 10.1021/bi047721l. [DOI] [PubMed] [Google Scholar]
- 76.Kuramochi Y, Takagi-Sakuma M, Kitahara M, Emori R, Asaba Y, Sakaguchi R, Watanabe T, Kuroda J, Hiratsuka K, Nagae Y, Suga T, Yamada J. Characterization of mouse homolog of brain acyl-CoA hydrolase: molecular cloning and neuronal localization. Brain Res. Mol. Brain Res. 2002;98:81–92. doi: 10.1016/s0169-328x(01)00323-0. [DOI] [PubMed] [Google Scholar]
- 77.Yamada J, Kuramochi Y, Takagi M, Watanabe T, Suga T. Human brain acyl-CoA hydrolase isoforms encoded by a single gene. Biochem. Biophys. Res. Commun. 2002;299:49–56. doi: 10.1016/s0006-291x(02)02587-1. [DOI] [PubMed] [Google Scholar]
- 78.Broustas CG, Hajra AK. Purification, properties, and specificity of rat brain cytosolic fatty acyl coenzyme A hydrolase. J. Neurochem. 1995;64:2345–2353. doi: 10.1046/j.1471-4159.1995.64052345.x. [DOI] [PubMed] [Google Scholar]
- 79.Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- 80.Shibayama-Imazu T, Okahashi I, Omata K, Nakajo S, Ochiai H, Nakai Y, Hama T, Nakamura Y, Nakaya K. Cell and tissue distribution and developmental change of neuron specific 14 kDa protein (phoshoneuroprotein 14) Brain Res. 1993;622:17–25. doi: 10.1016/0006-8993(93)90796-p. [DOI] [PubMed] [Google Scholar]
- 81.Murphy EJ, Prows DR, Jefferson JR, Schroeder F. Liver fatty acid binding protein expression in transfected fibroblasts stimulates fatty acid uptake and metabolism. Biochim. Biophys. Acta. 1996;1301:191–196. doi: 10.1016/0005-2760(96)00024-0. [DOI] [PubMed] [Google Scholar]
- 82.Prows DR, Murphy EJ, Schroeder F. Intestinal and liver fatty acid binding proteins differentially affect fatty acid uptake and esterification in L-cells. Lipids. 1995;30:907–910. doi: 10.1007/BF02537481. [DOI] [PubMed] [Google Scholar]
- 83.Murphy EJ. L-FABP and L-FABP expression increases NBD-stearate uptake and cytoplasmic diffusion in L cells. Am. J. Physiol. 1998;275:G244–G249. doi: 10.1152/ajpgi.1998.275.2.G244. [DOI] [PubMed] [Google Scholar]
- 84.Marszalek JR, Kitidis C, DiRusso CC, Lodish HF. Long-chain acyl-CoA synthetase 6 preferentially promotes DHA metabolism. J. Biol. Chem. 2005;280:10817–10826. doi: 10.1074/jbc.M411750200. [DOI] [PubMed] [Google Scholar]
- 85.Muoio DM, Lewin TM, Wiedmer P, Coleman RA. Acyl-CoAs are functionally channeled in liver: potential role of acyl-CoA synthetase. Am. J. Physiol. Endocrinol. Metab. 2000;279:E1366–E1373. doi: 10.1152/ajpendo.2000.279.6.E1366. [DOI] [PubMed] [Google Scholar]
- 86.Marszalek JR, Kitidis C, Dararutana A, Lodish HF. Acyl-CoA synthetase 2 overexpression enhances fatty acid internalization and neurite outgrowth. J. Biol. Chem. 2004;279:23882–23891. doi: 10.1074/jbc.M313460200. [DOI] [PubMed] [Google Scholar]
- 87.Richieri GV, Ogata RT, Zimmerman AW, Veerkamp JH, Kleinfeld AM. Fatty acid binding proteins from different tissues show distinct patterns of fatty acid interactions. Biochemistry. 2000;39:7197–7204. doi: 10.1021/bi000314z. [DOI] [PubMed] [Google Scholar]
- 88.Sharon R, Bar-Joseph I, Mirick GE, Serhan CN, Selkoe DJ. Altered fatty acid composition of dopaminergic neurons expressing α-synuclein and human brains with α-synucleinopathies. J. Biol. Chem. 2003;278:49874–49881. doi: 10.1074/jbc.M309127200. [DOI] [PubMed] [Google Scholar]
- 89.Kahle PJ, Neumann M, Ozman L, Müller V, Jacobsen H, Schindzielorz A, Okochi M, Leimer U, van der Putten H, Probst A, Kremmer E, Kretzschmar HA, Haass C. Subcellular localization of wild-type and Parkinson’s disease-associated mutant α-synuclein in human and transgenic mouse brain. J. Neurosci. 2000;20:6365–6373. doi: 10.1523/JNEUROSCI.20-17-06365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hall AM, Smith AJ, Bernlohr DA. Characterization of the Acyl-CoA synthetase activity of purified murine fatty acid transport protein 1. J. Biol. Chem. 2003;278:43008–43013. doi: 10.1074/jbc.M306575200. [DOI] [PubMed] [Google Scholar]
- 91.Hall AM, Wiczer BM, Herrmann T, Stremmel W, Bernlohr DA. Enzymatic properties of purified murine fatty acid transport protein 4 and analysis of acyl-CoA synthetase activities in tissues from FATP4 null mice. J. Biol. Chem. 2005;280:11948–11954. doi: 10.1074/jbc.M412629200. [DOI] [PubMed] [Google Scholar]
- 92.DiRusso CC, Li H, Darwis D, Watkins PA, Berger J, Black PN. Comparative biochemical studies of the murine fatty acid transport proteins (FATP) expressed in yeast. J. Biol. Chem. 2005;280:16829–16837. doi: 10.1074/jbc.M409598200. [DOI] [PubMed] [Google Scholar]
- 93.Herrmann T, Buchkremer F, Gosch I, Hall AM, Bernlohr DA, Stremmel W. Mouse fatty acid transport protein 4 (FATP4): characterization of the gene and functional assessment as a very long chain acyl-CoA synthetase. Gene. 2001;270:31–40. doi: 10.1016/s0378-1119(01)00489-9. [DOI] [PubMed] [Google Scholar]