

Abstract
The zymogen, factor XI, and the enzyme, factor XIa, interact specifically with functional receptors on the surface of activated platelets. The present studies were initiated to identify the molecular subdomain within factor XIa that binds to activated platelets. Both factor XIa (Ki ~1.4 nM) and a chimeric factor XIa containing the Apple 3 domain of prekallikrein (Ki ~2.7 nM) competed with 125I-factor XIa for binding sites on activated platelets suggesting that the factor XIa binding site for platelets is not located in the Apple 3 domain which mediates factor XI binding to platelets. The recombinant catalytic domain (Ile370-Val607) inhibited the binding of 125I-factor XIa to the platelets (Ki ~3.5 nM) whereas the recombinant factor XI heavy chain did not demonstrating that the platelet binding site is located in the light chain of factor XIa. A conformationally constrained cyclic peptide (Cys527-Cys542) containing a high-affinity (KD ~86 nM) heparin-binding site within the catalytic domain of factor XIa also displaced 125I-factor XIa from the surface of activated platelets (Ki ~5.8 nM), whereas a scrambled peptide of identical composition was without effect suggesting that the binding site in factor XIa that interacts with the platelet surface resides in the catalytic domain near the heparin binding site of factor XIa. These data support the conclusion that a conformational transition accompanies conversion of factor XI to factor XIa that conceals the Apple 3 domain factor XI (zymogen) platelet binding site and exposes the factor XIa (enzyme) platelet binding site within the catalytic domain possibly comprising residues Cys527-Cys542.
Keywords: Factor XIa, Platelet Receptors, Factor XI, Catalytic Domain Exosites, Apple Domains
Factor XI (FXI)1 is a homodimeric plasma coagulation protein (1–4), deficiency of which produces a mild hemostatic defect associated with trauma or surgery (5–8), in contrast to deficiencies of the “contact factors” (Factor XII [FXII], prekallikrein [PK], and high molecular weight kininogen [HK]), which lack a defined phenotype (9). This suggests that activation of FXI to the protease (FXIa) proceeds physiologically by a mechanism that is independent of FXII. This possibility was substantiated by the seminal observations of Naito and Fujikawa (10) and Gailani and Broze (11) who demonstrated that FXI could be activated by thrombin in the presence of dextran sulfate. Although the physiological surface that might potentiate FXI-activation by thrombin has not been definitively identified, it has been demonstrated that FXI binds to high-affinity (KD ~10 nM), saturable, specific receptors (Bmax ~1,500 sites/platelet) on activated platelets, in the presence of HK and ZnCl2 or prothrombin and CaCl2 (12–15). It is hypothesized that the formation of the FXI/HK or FXI/prothrombin complex leads to the exposure of residues within the Apple 3 (A3) domain that mediate FXI-binding (12, 15) to activated platelets where FXI can be cleaved to FXIa by FXIIa (16) and possibly also by thrombin (17). In contrast, neither resting nor thrombin-activated human umbilical vein endothelial cells have any capacity to bind FXI or promote its activation by thrombin (18). It has been demonstrated that the platelet membrane receptor for FXI consists of the glycoprotein Ib-IX-V complex (19).
FXIa recognizes its macromolecular substrate factor IX (FIX), which it cleaves at two scissile bonds, Arg145-Ala146 and Arg180-Val181, to generate factor IXa (FIXa) (2). The substrate-binding site on FXIa for FIX is within either the A2 or A3 domain (20–22). Similar rates for FIX activation by FXIa in the presence and absence of activated platelets have been demonstrated (23). Since FXIa binds to high-affinity (KD ~1.7 nM) receptors on the activated platelet surface (Bmax ~250 sites/platelet) (24, 25) as does FIX (KD ~2.5 nM; Bmax ~250 sites/platelet) (26) it is likely that the platelet surface provides a platform where FXIa and FIX can colocalize and FIX activation can efficiently occur. The regulation of soluble FXIa is mediated by the secretion from activated platelets of protease nexin 2 (PN2), a reversible, tight-binding (Ki ~500 pM) FXIa inhibitor, whereas platelet-bound FXIa is protected from inactivation by PN2 (25, 27, 28). These results are consistent with the view that the initiation of the consolidation phase of blood coagulation either by FXIIa (16) or by thrombin generated by the tissue factor pathway proceeds on the surface of activated platelets but not on the endothelium. Activated platelets provide a protective nidus for FIX activation by platelet-bound dimeric FXIa while secreting PN2 which potently inhibits unbound FXIa, thereby localizing blood coagulation to the hemostatic thrombus and preventing disseminated intravascular coagulation (28). The present study was initiated to identify the molecular domains within the enzyme, FXIa, that mediate its interaction with platelet receptors.
PK is a protein that is highly homologous to FXI, sharing 58% sequence identity and having similar domain structure. Two chimeras were generated, rFXI/PKA3 and rFXIa/PKA3, in which the A3 domain of FXI, the domain responsible for mediating the binding of FXI to the activated platelet surface (12, 14), was replaced with the A3 domain of PK in order to study the binding of FXIa to platelets. We have examined the ability of FXI and FXIa as well as rFXI/PKA3 and rFXIa/PKA3 chimeras to compete with 125I-FXIa for binding to the activated platelet surface. In addition, the isolated heavy chain (which contains the Apple domains) and the isolated catalytic domain of FXIa were examined to determine which domain was involved in FXIa binding to activate platelets. A number of peptides were designed for competition assays in order to further identify the particular residues involved in this interaction. Our data suggest that FXIa binds to the surface of activated platelets through the catalytic domain and that this domain is distinct from that which is involved in zymogen binding.
EXPERIMENTAL PROCEDURES
Reagents
The site-directed mutagenesis kit (Quikchange) was purchased from Strategene (La Jolla, CA). Lipofectamine 2000 Reagent was from Invitrogen (Carlsbad, CA). The chromogenic substrate S-2366 (L-pyroglutamyl-L-prolyl-L-arginyl-p-nitroaniline hydrochloride) was purchased from Chromogenix (Mölndal, Sweden). Glutamine, penicillin/streptomycin, bovine serum albumin, aprotinin, and cyanogen bromide-activated-sepharose 4B were purchased from Sigma Chemical Co. (St. Louis, MO). Dulbecco’s modified Eagle’s Medium (DMEM) was purchased from Mediatech (Herndon, VA). Geneticin (G-418) was purchased from Gibco (Grand Island, NY). Iodogen was obtained from Pierce (Rockford, IL). Na125I was obtained from GE Healthcare (Piscataway, NJ). All other reagents were of analytical grade and were the best quality commercially available.
Proteins
FXIIa, FXIa, and FXI purified from human plasma were purchased from Haematologic Technologies, Inc. (Essex Junction, VT). rFXI/PKA3 and rFXIa/PKA3 chimeric proteins were a kind gift from Dr. David Gailani (Vanderbilt University, Nashville, TN). Recombinant FXI with cysteine 362 and cysteine 482 mutated to serine (rFXI/C362S,C482S) was purified from stably transfected 293 human embryonic kidney cells (293-HEK). The monoclonal antibody 5F7 (directed against the A1 domain located within the heavy chain of FXI) was initially purified from the ascites fluids in a hybridoma cell line by Dr. Dipali Sinha (29) and now is commercially available from Green Mountain Antibodies (Burlington, VT). Corn trypsin inhibitor (coupled to Affi-Gel) columns were purchased from Enzyme Research Laboratories (South Bend, IN). The following peptides were synthesized at the Protein Chemistry Laboratory at the University of Pennsylvania (Dr. John Lambris, Director): the thrombin receptor activating peptide (TRAP, SFLLRN-amide) and 8 peptides that comprise the regions of greatest dissimilarity with PK, peptide 1 (383WQVTLHTTSPTQRHL397), peptide 2 (415FYGVESPKILRVYSG429), peptide 3 (433QSEIKEDTSFFGVQE447), peptide 4 (450IHDQYKMAESGYDIA446), peptide 5 (467KLETTVNYTDSQRPI481), peptide 6 (500GWGYRKLRDKIQNTL514), peptide 7 (527CQKRYRGHKITHKMIC542), which is the catalytic domain heparin binding loop, and peptide 8 (527CKQRYHMKGHIRTIKC542), which is a scrambled loop of identical amino acid composition as the heparin binding loop peptide (peptide 7).
Factor XI Mutant Construct
The cDNA for the full-length FXI sequence inserted in pJVCMV vector (a gift from Dr. David Gailani, Vanderbilt University, Nashville, TN) served as a template for the synthesis by PCR of the cDNA construct of rFXI/C362S,C482S. The appropriate mutagenic primers were used to incorporate the desired codon into the FXI cDNA sequence. The PCR products, containing the new mutations, were propagated in XL1-Blue bacteria. Each purified plasmid DNA was sequenced in the forward and reverse directions to verify that the appropriate mutation was incorporated.
Protein Expression in 293-Human Embryonic Kidney Cells (293-HEK)
293-HEK were transfected with 40 μg of the pJVCMV vector containing inserts of the cDNA sequence for rFXI/C362S,C482S and 2 μg of pRSVneo vector (containing the gene that confers resistance to neomycin and allows for the selection of positive clones) using Lipofectamine 2000. Positive clones were selected using Geneticin 418 (G-418) at a concentration of ~500 μg/ml and the expression levels were assessed by ELISA (described below). Cells were expanded in 2L roller bottles in DMEM containing 10% fetal bovine serum, penicillin/streptomycin, L-glutamine, and G-418 (~150 μg/ml final concentration) in a 5% CO2 incubator, 37°C. After the cells reached confluency in the roller bottles, the medium was replaced with serum free DMEM supplemented with penicillin/streptomycin, L-glutamine, G-418 (~150 μg/ml), insulin transferring selenium-A, 10 μg/ml of soya bean trypsin inhibitor, lima bean trypsin inhibitor, and aprotinin. Conditioned media were collected after 48–72 hrs, centrifuged and filtered through an acetate filter (0.45 μm pore size) to remove any cell debris, and made 5 mM EDTA and 5 mM in benzamidine to prevent any nonspecific protease cleavage of the protein and stored at −20°C until it was ready to be processed.
Enzyme Linked Immunosorbent Assay (ELISA)
A FXI-ELISA kit (Affinity Biologicals, Hamilton, Ontario, Canada) was used to determine the level of expression of various FXI mutants. The capture antibody, an affinity-purified polyclonal goat anti-human FXI IgG, was applied to the wells of a microtiter plate and incubated for 2 hours at 22°C. Each well was blocked with phosphate-buffered saline, 0.5% BSA for 2 hours. Wells were washed extensively with phosphate-buffered saline-Tween (0.1% Tween-20) before the addition of the detecting antibody, a peroxidase conjugated goat anti-FXI IgG. The wells were washed again with phosphate-buffered saline-Tween. O-Phenylenediamine substrate was added to each well and color was allowed to develop for 5–10 mins. 2.5 M H2SO4 was used to stop the color development reaction and the plate was read at 490 nm.
Preparation of the 5F7 Monoclonal Antibody Columns
The antibody linked resin was prepared by incubation with cyanogen bromide activated sepharose in 0.1 M NaHCO3 buffer containing 0.5 M NaCl, pH 8.4 (sodium bicarbonate coupling buffer). Briefly, the 5F7 antibody (8–10 mg) was diluted in coupling buffer and allowed to incubate overnight at 4°C under constant stirring with the cyanogen bromide activated resin (3.5 mL). The resin was washed with additional coupling buffer in order to remove any unreacted ligand. The resin was then blocked by incubation with 0.2 M glycine, pH 8.0 for 2 hours at 25°C under constant stirring. In order to remove all of the blocking solution, the resin was washed 4 times with alternating sodium acetate buffer (pH 4.0) and sodium bicarbonate buffer (pH 8.4). The washing was continued until absorbance at 280 nm was the same as that of the washing buffer. The 5F7-coupled resin was stored in 25 mM Tris-HCl, 100 mM NaCl, 5 mM benzamidine, pH 7.4 buffer containing 0.02% azide.
Purification of Factor XI Mutants
Expressed protein from cell supernatant was applied to the 5F7 monoclonal antibody affinity-column equilibrated in 25 mM Tris-HCl, 100 mM NaCl, and 5 mM benzamidine, pH 7.4. The column was washed with equilibration buffer until the A280 returned to base line. Adsorbed protein was eluted with 2 M potassium thiocyanate made in the equilibration buffer. The collected fractions were concentrated and dialyzed extensively against Tris-buffered saline, pH 7.4. The purity of the fractions was assessed by SDS-PAGE before being pooled and concentrated to 0.25 mL. Activation of FXI proteins was performed with the method described below.
Activation and Preparation of Factor XIa Light Chain
rFXI/C362S,C482S was incubated overnight with plasma FXIIa (10:1 molar ratio) at 37°C. FXIIa was removed from the activation mixture with corn trypsin inhibitor linked to Affi-gel for 1 hour at 25°C. The supernatant was run on an SDS-PAGE gel to verify complete activation and removal of FXIIa. Following activation, the rFXIa/C362S,C482S mutant was further purified using the 5F7 antibody coupled resin for 1 hour at 25°C in order to separate the heavy chain and catalytic domain. The supernatant containing the catalytic domain of FXIa was removed from the incubation mixture. The heavy chain was eluted using 2M potassium thiocyanate in 25 mM Tris-HCl, 100 mM NaCl, and 5 mM benzamidine, pH 7.4. The catalytic domain and the heavy chain were run on an SDS-PAGE gel to verify the purity of the isolated proteins.
Clotting Assay
Clotting time was determined by a modified version of the kaolin-activated partial thromboplastin time method. In brief, 25 μl of FXIa or FXIa mutant protein was added to 50 μl of FXI deficient plasma (George King Bio-Medical, Inc., Overland Park, KS). 25 μl of activated partial thromboplastin time reagent (Sigma Diagnostics, Inc., Saint Louis, MO) was added to the reaction mixture and allowed to incubate at 37°C for 2 min. Following this incubation, 50 mM CaCl2 (10 mM final concentration) was added to initiate clot formation and the time was recorded using an Amelung KC 4A microcoagulometer (Sigma Diagnostics, Inc., Saint Louis, MO). A standard curve was generated by titrating normal pooled plasma into congenitally FXI-deficient plasma. Results for all unknowns were quantified by comparison to the standard curve, which was generated on a logarithmic plot of clotting times versus concentration of normal pooled plasma.
Determination of the Michaelis-Menten Constant
To determine the Michaelis-Menten constant for pFXIa and rFXIa/C362S,C482S the hydrolysis of substrate 2366 (S-2366) was measured. Increasing concentrations of S-2366 (0–1.5 mM) were added to pFXIa or the rFXIa/C362S,C482S mutant (6.7 nM final concentration) and pNA generated was monitored by measuring absorbance at 405 nm in a Hewlett-Packard, model 8452A, diode array spectrophotometer and the data were then analyzed on Kaleidograph (Abelbeck Software, Reading, PA).
Radiolabeling with 125I
Plasma FXI and FXIa catalytic domain (rFXIac) were radiolabeled with minor modifications to the IODO-GEN method (30). In short, approximately 100 μg of protein was incubated with ~1 mCi of carrier-free Na125I for 20 minutes in a vial containing 15 μg/ml iodogen. The protein mixture was gel filtered through a 1 ml Sephadex G-50 column (blocked with 0.5–1% BSA) to separate the free iodine from the protein. Labeled proteins had specific activities of ~2 × 106 cpm/μg. The radiolabeled proteins retained >98% of their biological activity.
Measurement of Specific Radioactivity
Radiolabeled pFXI or rFXIac (1 μl) were added to 99 μl of 0.5 % BSA in Hepes buffered saline and 100 μl of 40 % trichloroacetic acid. The solution was vortexed and immediately placed on ice and incubated for 5 min. This mixture was then centrifuged for 5 min at 14,000 × g, and 100 μl was removed and put in a separate vial. Both vials, 100 μl of pellet (P) and 100 μl of supernatant (S), were placed in a gamma counter (Perkin Elmer, Inc., Wellesley, MA) and measured for γ-emission. To determine the percent radioactivity bound (Eq. 1) and specific radioactivity (SRA, Eq. 2):
| Eq. 1 |
| Eq. 2 |
Generation of 125I-Factor XIa
125I-FXIa was generated by activation of radiolabeled pFXI as described previously (25). Generally >98 % radioactivity was bound to the protein and the specific activity was ~2 × 106 cpm/μg. Radiolabeled FXI and rFXIac retained >90 % of the activity of non-labeled FXI.
Platelet Isolation
Platelets were prepared as described (31, 32). Blood from a normal donor was collected in 50 mL tubes containing 5 mL of ACD buffer (25 g/L of trisodium citrate, 15 g/L of citric acid and 20 g/L of glucose) in order to prevent clotting. The ACD-treated whole blood was centrifuged at 200g to obtain the platelet rich plasma. Platelet rich plasma was gel-filtered through a 50 ml Sepharose CL-2B column that was pre-equilibrated with HT buffer containing 2% BSA. Platelet eluates were counted electronically using a particle counter (Coulter Electronics, LOC, Hialeah, FL).
Platelet Activation
Platelets were activated using the thrombin receptor activation peptide (TRAP), with the sequence SFLLRN amide. TRAP (5 μM) was added to gel-filtered platelets 5 min prior to the addition of the radiolabeled protein (and competitor when described) reaction mixture.
Equilibrium Binding Assay
Increasing concentrations of 125I-rFXIac (0–50 nM) in Hepes Tyrode Buffer containing CaCl2 (2 mM) and ZnCl2 (25 μM) were incubated with TRAP (25 μM) activated platelets (1–2 × 108/ml) for 30 min at 37°C. Platelet bound radioactivity was separated from free proteins by centrifugation through silicone oil as previously described (31).
Competition Assay
125I-FXIa (2 nM) was incubated in Hepes Tyrode Buffer containing CaCl2 (2 mM) and ZnCl2 (25 μM) and TRAP (25 μM) activated platelets (1–2 × 108/ml) in the presence of increasing concentrations of non-radiolabeled FXI, FXIa, rFXI/PKA3, rFXIa/PKA3, or FXIa catalytic domain peptides for 30 min at 37°C. Platelet bound radioactivity was separated from free proteins by centrifugation through silicone oil and counted.
RESULTS
Expression and Purification of rFXI/C362S,C482S
Full length FXI was stably expressed in 293-HEK cells and then activated to FXIa with FXIIa by cleavage of the bond between Arg369 and Ile370. This cleavage reaction produced two separate chains, the heavy and the light chain. In order to generate these separate chains, two mutations were engineered at residues Cys362 and Cys482 (the cysteines were mutated to serines) so that the disulfide bond between the heavy and the light chain would not form. Thus cleavage of the scissile bond at Arg369-Ile370 generates heavy and light chains that are, unlike those in FXIa, free from each other. Purification via the 5F7 monoclonal antibody column directed against the A1 domain of the heavy chain allowed not only for the purification of full-length rFXI/C362S,C482S from the medium, but also for the subsequent purification of the light chain from the heavy chain. The expression level was approximately 0.4 μg/ml for rFXI/C362S,C482S and ~100 μg of FXIa light chain was obtained starting with ~400 μg of total protein.
rFXI/C362S,C482S migrated at an Mr ~160,000 Da on an SDS-PAGE gel (4–15 %) under non-reducing conditions indicating that the protein was secreted as a dimer (data not shown). Under reducing conditions (i.e. incubation with a reducing agent, β-mercaptoethanol) a band at ~80,000 Da was observed which is the appropriate size of the monomer (Figure 1). After activation, rFXIa/C362S,C482S was further purified to isolate the heavy chain from the catalytic domain and the products were run on an SDS-PAGE gel under reducing conditions (Figure 1). The heavy chain migrated at ~50,000 Da (lane 5) and the catalytic domain migrated at ~30,000 Da (lane 6).
Fig. 1. SDS-PAGE of rFXI/C362S,C482S.
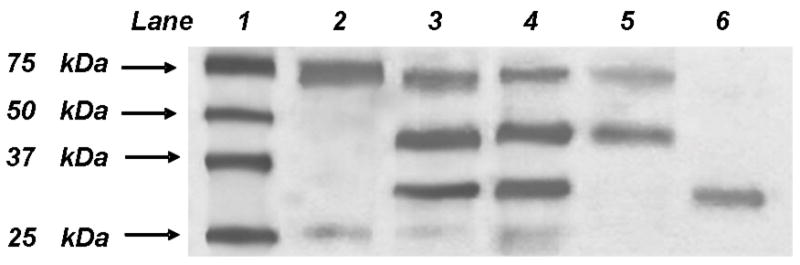
Plasma FXI (150 μg) or rFXI/C362S,C482S (150 μg) was incubated with FXIIa (~7.5 μg) for ~16 hrs at 37°C and fractionated on SDS-PAGE (4–15% Tris-HCl) under reducing conditions and stained with Coomassie Blue. Lane 1, size markers (sizes denoted in the figure). Lane 2, pFXI and lane 3, pFXIa. The upper band is unactivated FXI, the middle band is the 50kDa heavy chain, and the lower band is the 30kDa catalytic domain. Lane 4 is a sample of the rFXIa/C362S,C482S mutant after activation by FXIIa (FXIIa was removed by passing the activation mixture over a corn trypsin inhibitor column). Lane 5, the heavy chain and unactivated rFXI/C362S,C482S eluted from a 5F7 antibody column. Lane 6 is the isolated catalytic domain after purification with the 5F7 column.
Characterization of Factor XIa Catalytic Domain (rFXIac)
Functional characterization of the purified catalytic domain obtained from rFXIa/C362S,C482S was carried out by measuring the amidolytic activity, clotting activity, and protein concentration (determined by bicinchoninic acid protein assay). A standard curve generated using pFXIa was used to calculate a specific activity for FXIa of 416.6 moles of pNA generated sec−1 mol−1. By comparison the isolated catalytic domain (rFXIac) had a specific activity of 383.7 moles of pNA generated sec−1 mol−1 from which it can be concluded that the rFXIac retained >90 % of its functional active site concentration. Moreover when equimolar concentrations of FXIa and rFXIac were titrated with varying concentrations of S-2366, the Km for pFXIa (~339 μM) was very comparable to that for rFXIac (~389 μM) showing that the isolated catalytic domain and the full-length enzyme have similar active site architecture. In the activated partial thromboplastin time assay, however, the catalytic domain had less than 1% of the clotting activity of either normal pooled plasma or pFXIa at the same concentrations. This result is expected because the substrate binding site for FIX, the macromolecular substrate of FXIa, resides on the heavy chain of FXIa (20).
The Apple 3 Domain of Factor XIa does not Mediate the Binding of Factor XIa to Activated Platelets
FXI when complexed with HK in the presence of ZnCl2 or prothrombin in the presence of CaCl2 (13, 14) binds to the activated platelet surface through the A3domain (12, 15). It is hypothesized that the formation of the FXI/HK or FXI/FII complex leads to the exposure of residues within the A3 domain that mediate FXI-binding to platelets (14, 15). FXIa binds to high-affinity receptors on the activated platelet surface that are distinct from the receptors for FXI (24, 25). 125I-FXIa was shown to bind to saturable, specific, high-affinity (KD~1.7 nM) sites on TRAP (SFLLRN amide, 25 μM) activated platelets (n ~250 sites/platelet) in the presence of 25 μM ZnCl2 (25). FXIa binding to activated platelets required ZnCl2 but was not affected by the presence of HK (25). To determine whether FXIa utilizes its A3 domain, as does the zymogen, FXI, to bind to activated platelets, we initially carried out competition studies with wild-type FXI and FXIa and with chimeric proteins in which the A3 domain of PK replaced the A3 domain of FXI (rFXI/PKA3) or FXIa (rFXIa/PKA3). As shown in Figure 2A, neither FXI nor rFXI/PKA3 was able to compete with FXIa for binding sites on TRAP-activated platelets. In contrast, both FXIa (Ki ~1.4 nM) and a chimeric FXIa with the A3 domain of PK (rFXIa/PKA3, Ki ~2.7 nM) competed with 125I-FXIa for binding sites on activated platelets. These results suggest that the FXIa binding site for platelets is distinct from the binding site for FXI and that this site resides outside of the A3 domain of FXIa.
Fig. 2. Sites Within the Catalytic Domain but not the Heavy Chain Mediate the Binding of FXIa to Activated Platelets.
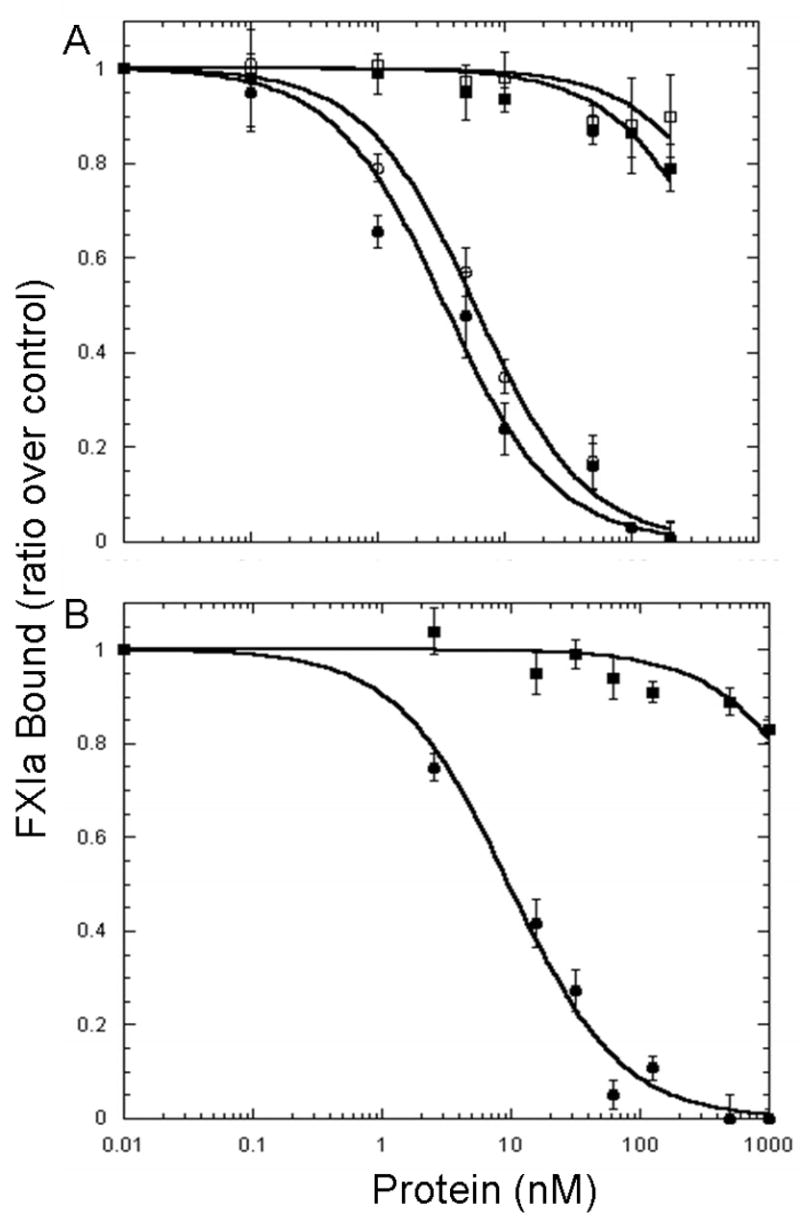
125I-FXIa (2 nM) was incubated with platelets (1 × 108platelets/ml), TRAP (25 μM) in the presence of CaCl2 (2 mM) and ZnCl2 (25 μM), and various concentrations of non-radiolabeled protein for 30 min prior to centrifugation through silicone oil. A, FXI (■) and rFXI/PKA3 (□) were unable to displace 125I-FXIa from the activated platelet surface whereas both FXIa (●) and rFXIa/PKA3 (○) were able to inhibit 125I-FXIa binding with Ki values of 1.4 ± 0.28 nM and 2.7 ± 0.32 nM respectively. B, The heavy chain (■) was unable to compete with 125I-FXIa for sites on the activated platelet whereas the catalytic domain (●) was effective in competing with 125I-FXIa for binding sites on the activated platelet surface with a Kiof 3.5 ± 0.42 nM. Values represent the mean ± standard deviation of three determinations each done in triplicate.
Factor XIa Binding to Activated Platelets is Mediated by the Catalytic Domain
To determine the location of molecular domains within FXIa that mediate its interaction with receptors exposed on the surface membrane of activated platelets, we prepared the heavy chain and the catalytic domain of FXI as recombinant proteins. They were utilized in competition studies examining the binding of 125I-FXIa to TRAP-activated platelets in the presence of ZnCl2(25 μM). These competition binding studies (Figure 2B) revealed that the heavy chain had no effect on the binding of 125I-FXIa to the platelet surface, whereas the recombinant catalytic domain (Ile370-Val607) displaced 125I-FXIa from binding sites on activated platelets with a Ki =3.5 ± 0.42 nM demonstrating that the FXIa platelet binding site is contained within the light chain or catalytic domain of FXIa.
Direct Binding Studies with the Catalytic Domain (rFXIac) of Factor XIa
To confirm the hypothesis that the FXIa platelet-binding site is contained within the catalytic domain of FXIa, the recombinant catalytic domain of FXIa (Ile370-Val607) was radiolabeled to high specific activity (~2 × 106 cpm/μg protein) with 125I and utilized in direct binding studies with TRAP-activated platelets in the presence of ZnCl2 (25 μM). 125I-rFXIac was shown to bind to high affinity sites (n = 270 ± 17 sites/platelet; KD = 8.1 ± 1.49 nM) in a saturable manner (Figure 3). These values are similar to those reported earlier for FXIa binding to platelets (n ~250 sites/platelets; KD ~2 nM) (24, 25). These results suggest that the totality of the platelet-binding energy of FXIa resides within the catalytic domain.
Fig 3. Direct Binding of rFXIac to the Activated Platelet Surface.
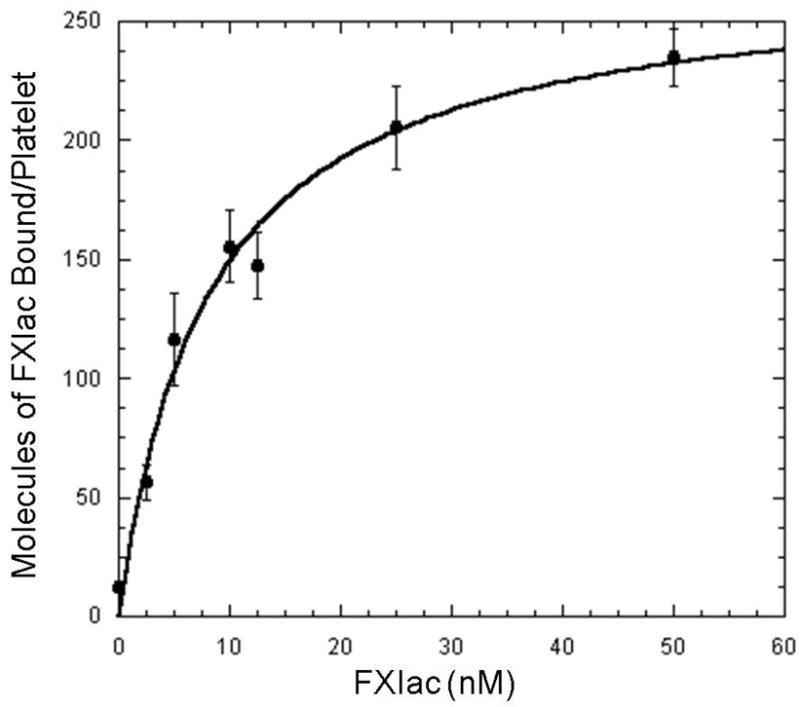
Increasing concentrations of 125I-rFXIac were incubated with TRAP (25 μM) activated platelets (1–2 × 108 platelets/ml) in the presence of CaCl2 (2 mM) and ZnCl2 (25 μM). Non-specific binding was measured and subtracted from the total binding. Shown here is the specific binding of rFXIac to activated platelets with a Bmax of 270 ± 17; KD of 8.1 ± 1.49 nM. Values represent the mean ± standard deviation of three determinations performed in triplicate.
Displacement of rFXIac by Factor XI and Factor XIa
FXI was unable to displace 125I-rFXIac from the surface of activated platelets whereas FXIa was able to displace 125I-rFXIac (Ki=1.3 nM) (Figure 4). This is similar to the KI value for rFXIac inhibition of FXIa binding to platelets (Ki = 3.5 nM, Figure 2B), further confirming that the catalytic domain is the only domain involved in the binding of the enzyme to the surface of activated platelets.
Fig 4. Displacement of rFXIac from the Activated Platelet Surface by FXI and FXIa.
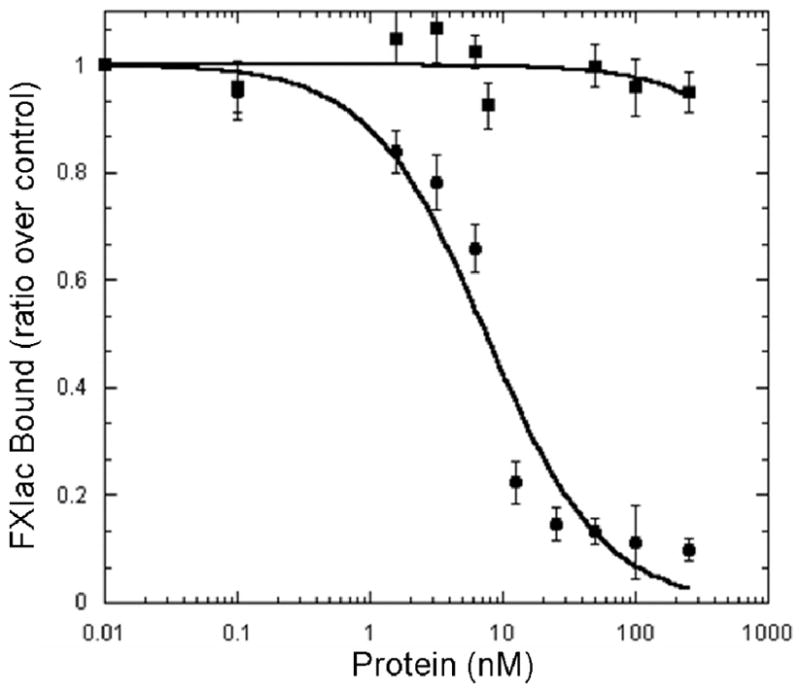
125I-rFXIac was incubated with non-radiolabeled FXI and FXIa and with platelets (1–2 × 108platelets/ml) and TRAP (25 μM) in the presence of CaCl2 (2 mM) and ZnCl2 (25 μM). FXIa (●) and not FXI (■) was able to displace rFXIac from the platelet surface with a Ki of 1.3 ± 0.16 nM, similar to that of rFXIac displacement of FXIa (Ki = 3.5 ± 0.42 nM). The values represent the mean ± standard deviation of three independent experiments each done in triplicate.
Localization of the Platelet-Binding Site within Factor XIa Catalytic Domain
The major approach in localizing the platelet-binding site within the catalytic domain was to compare the amino acid sequence of rFXIac with PK, another plasma protein that is unable to bind platelets but is 58% homologous in amino acid composition (Figure 5). Based on this comparison, the 7 subdomains with the greatest dissimilarities (boxed sequences in Figure 5) were identified, and 8 different peptides (Table 1; Figure 5) were synthesized (the 8th peptide being a scrambled peptide). An alternative approach to identifying FXIa residues that mediate binding to activated platelets comes from previous studies carried out in our laboratory (33). We previously have determined that the FXIa catalytic domain contains a cysteine-constrained α–helix–containing subdomain: 527CQKRYRGHKITHKMIC542, identified as a FXIa heparin-binding domain since a disulfide-constrained peptide comprising this sequence binds to heparin with a KD ~86 nM and competes with FXIa in binding to heparin, Ki ~240 nM (33). FXI binds to heparin (via Lys252 and Lys253) and to platelets (via residues Arg250, Lys255, Phe260, and Gln263) through overlapping amino acid sequences located in the A3 domain (12, 15, 34), and therefore it is possible that the region of FXIa that binds platelets is also located within the heparin binding regions (comprising the same sequence as Peptide 7) and/or a heparin binding consensus sequence (BXBBXBX, where B represents a basic residue and X is a hydrophilic residue); comprising the same sequence as Peptide 6). As shown in Figure 6A, 5 of the catalytic domain peptides (peptides 1, 2, 3, 5 and 6) were ineffective in displacing FXIa from the platelet surface. Peptide 4 was insoluble in deionized water or any of other buffers tested and therefore was not used in the competition study. As shown in Figure 6B, the conformationally constrained cyclic peptide (Cys527-Cys542; peptide 7, Table 1) containing a high-affinity (KD ~86 nM) heparin-binding site within the catalytic domain of FXIa also displaced 125I-FXIa from the surface of activated platelets (Ki ~5.8 nM), whereas a scrambled peptide (peptide 8) of identical composition was without effect suggesting that the binding site in FXIa that interacts with the platelet surface exists not in the A3 domain of FXIa but resides in the catalytic domain within a sequence of residues comprising the heparin binding site of FXIa (R7 in Figure 5).
Fig 5. Alignment of amino acid sequences of the catalytic domains of FXI and PK.
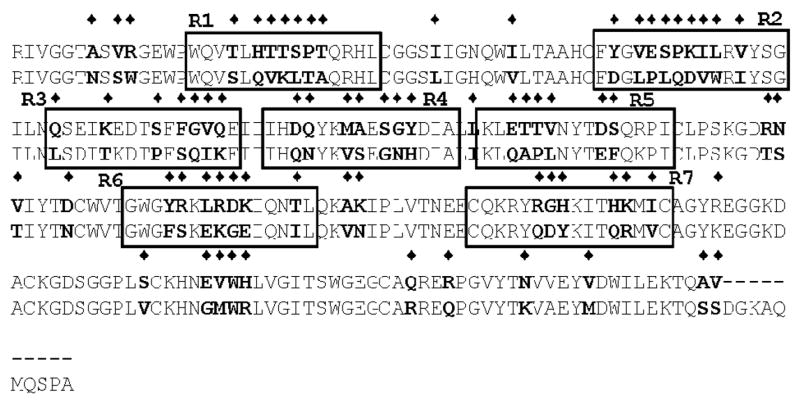
Shown is an alignment of the primary amino acid sequence of FXI and PK. The diamonds show the amino acids that are different. Based upon this alignment, synthetic peptides (labeled R1–R7) have been designed for the use in competition binding experiments to localize the region of FXIa light chain that mediates binding to the activated platelet surface.
Table 1.
Sequences of Factor XIa Synthetic Peptides
| Peptide | Sequence |
|---|---|
| TRP383-LEU397 (PEPTIDE 1) | 383WQVTLHTTSPTQRHL397 |
| PHE415-GLY429 (PEPTIDE 2) | 415FYGVESPKILRVYSG429 |
| GLN433-GLU447 (PEPTIDE 3) | 433QSEIKEDTSFFGVQE447 |
| ILE450-ALA446 (PEPTIDE 4) | 450IHDQYKMAESGYDIA446 |
| LYS467-ILE481 (PEPTIDE 5) | 467KLETTVNYTDSQRPI481 |
| GLY500-LEU514 (PEPTIDE 6) | 500GWGYRKLRDKIQNTL514 |
| CYS527-CYS542 (PEPTIDE 7)* | 527CQKRYRGHKITHKMIC542 |
| CYS527-CYS542 (PEPTIDE 8)* † SCR | 527CKQRYHMKGHIRTIKC542 |
Denotes a cyclized peptide
SCR denotes a scrambled peptide
Fig 6. Displacement of FXIa from the Activated Platelet Surface by Catalytic Domain Peptides.
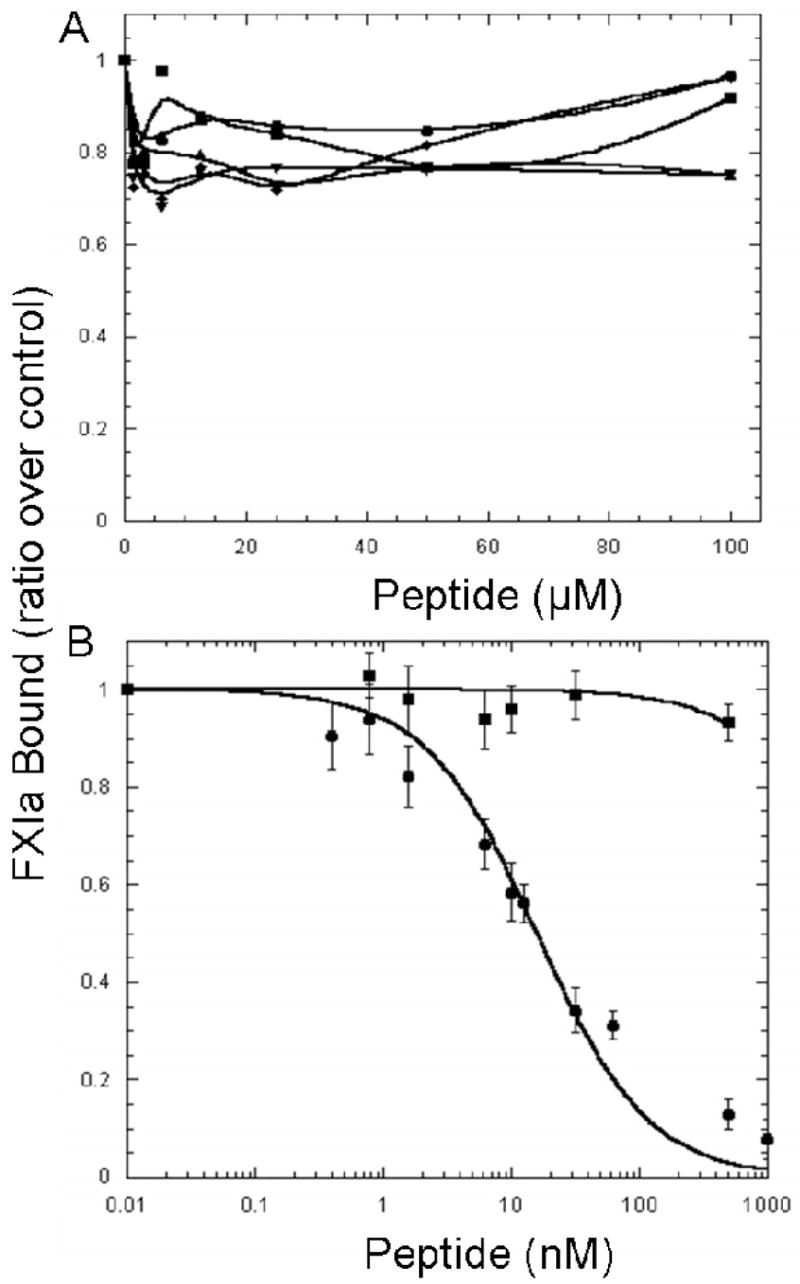
125I-FXIa (2 nM) was incubated with platelets (1–2 × 108 platelets/ml) and TRAP (25 μM) in the presence of CaCl2 (2 mM) and ZnCl2 (25 μM). Increasing concentrations of non-radiolabeled peptides were added to each reaction. The reaction mixture was incubated for 30 min. prior to centrifugation through silicone oil to separate the platelets with bound peptide from the free peptide. A, Peptide 1 (●), peptide 2 (■), peptide 3 (◆), peptide 5 (▲), and peptide 6 (▼) were unable to displace 125I-FXIa from the activated platelet surface. B, Peptide 7 (●), the cyclic heparin binding peptide was able to displace 125I-FXIa from the activated platelet surface (Ki =5.8 ± 0.78 nM), whereas peptide 8 (■), a scrambled peptide of identical composition was unable to displace 125I-FXIa. Values in 6B represent the mean ± standard deviation of three experiments repeated in triplicate.
DISCUSSION
Platelets circulate in the human vasculature in a dormant state until they encounter a blood vessel injury whereupon platelets bind to the exposed subendothelial collagen via von Willebrand factor. Platelets then become activated by a number of different agonists such as collagen, thrombin, ADP, and thromboxane A2. The activated platelets release the contents of their storage granules, such as ADP and fibrinogen, which promote platelet aggregation. Activated platelets, also participate in the assembly of coagulation complexes leading to the generation of sufficient quantities of thrombin to produce a hemostatic thrombus. This process requires the presence and normal activation of FXI, a protein that participates in the intrinsic phase of blood coagulation, as is evident from the fact that patients with FXI-deficiency have a bleeding diathesis whereas individuals with deficiencies in one of the contact factors (FXIIa, PK, and HK) do not. FXI can be proteolytically activated to FXIa by either FXIIa (1, 3, 4), FXIa or thrombin (10, 11). Reaction rates of FXI-activation by FXIIa are accelerated in the presence of activated platelets (16), and it has been suggested that feedback activation of FXI by thrombin is potentiated by activated platelets (17). However recent observations cast considerable doubt on the conclusion that activated platelets can promote the feedback activation of FXI by thrombin (35–37). Therefore, in a revised model of the interactions of FXI and FXIa with the activated platelet surface (Figure 7), dimeric FXI in complex with either HK or prothrombin is shown to expose a site within the A3 domain that binds to glycoprotein Ibα on the activated platelet surface (12–15, 19). Since this interaction has been demonstrated to be reversible (13), and since it has not been rigorously demonstrated whether it is the platelet-bound or soluble form of FXI that is converted to FXIa, the activation of FXI by FXIIa, FXIa or thrombin is shown in Figure 7 to occur in solution. PN2 is a potent FXIa inhibitor that is secreted from activated platelets (38–41). It has been shown that although soluble FXIa is rapidly inactivated by PN2, platelet-bound FXIa is protected from inhibition (25). This is potentially important physiologically since it could serve to localize the growing thrombus to the site of vessel injury and prevent propagation of thrombus growth in solution. FIX, which also binds to platelets (26), is believed to co-localize with FXIa thereby promoting efficient activation of FIX to FIXa, which is essential for the propagation of blood coagulation.
Fig 7. A Model for Factor IX Activation by Dimeric Factor XIa on the Platelet Surface.
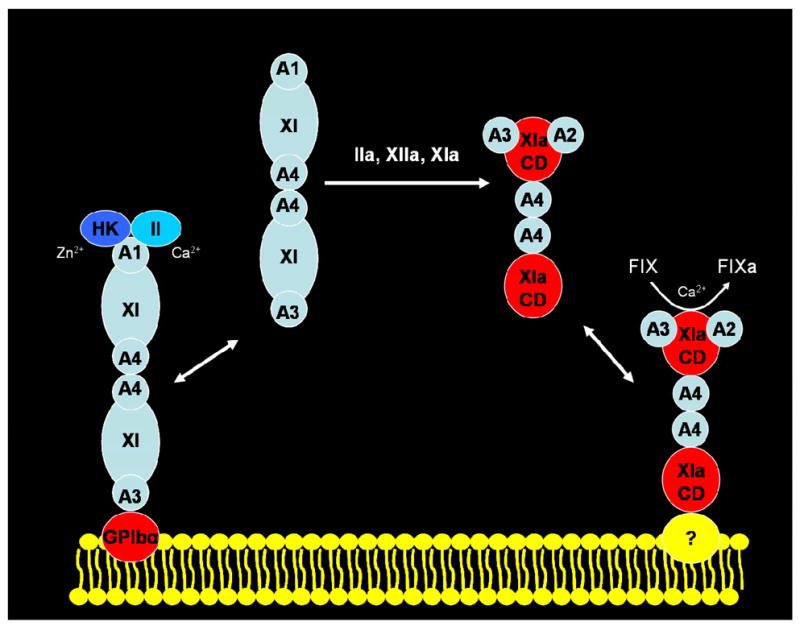
Dimeric FXI, consisting of two identical polypeptides, each containing four Apple domains (designated A1 through A4) and one catalytic domain (designated XI in the zymogen, light blue, or XIa in the enzyme, red), binds to ~1,500 sites (KD ~10 nM) on the platelet membrane (yellow bilayer) consisting of glycoprotein Ibα. Zymogen FXI can be activated by thrombin (IIa), FXIIa (XIIa) or FXIa to generate the enzyme which binds to an unknown (?) platelet receptor (~250 sites/platelet, KD ~1.7 nM) via the catalytic domain of one subunit, leaving the other subunit available to bind FIX (IX) via the substrate-binding site in the A2 and/or A3 domain and to utilize the active site within the FXIa catalytic domain (XIa, red) to activate zymogen FIX to the enzyme FIXa in the presence of calcium ions (Ca2+). Whereas platelet-bound FXIa is protected from inhibition by protease nexin-2, secreted by activated platelets, unbound FXIa is efficiently inhibited, thereby localizing FIX-activation to the platelet surface.
The focus of our present studies is to identify and characterize the molecular subdomain(s) within FXIa that interacts with activated platelets. We have previously shown that FXI binds to the activated platelet surface through the A3 domain (12). In the present study we show that FXIa binds to platelets utilizing residues that reside outside the A3 domain. FXI does not compete with FXIa for binding sites even though both proteins contain an identical amino acid sequence within the A3 domain (Figure 2A). PK, which contains Apple domains with 58% amino acid identity to FXI does not bind to activated platelets, and chimeric FXI proteins containing the PK A3 domain did not compete with FXIa for binding to the activated platelet surface whereas when the rFXI/PKA3 was activated with FXIIa it was able to compete with FXIa suggesting that the A3 domain is not involved in FXIa binding to platelets (Figure 2A). Competition studies with the heavy chain and the catalytic domain of FXIa (Figure 2B and 4) as well as direct binding studies (Figure 3) show that the subdomain of FXIa that binds to the activated platelet surface is located exclusively within the catalytic domain and not within any of the Apple domains. The modification of FXI that leads to enzymatic activity is proteolytic cleavage at Arg369-Ile370. This leads to a conformational change leading to active site availability for substrate cleavage, and also to changes in the quaternary structure of the protein involving the Apple domains and the catalytic domain. Our present studies indicate that the cysteine-constrained loop Cys527-Cys542 in the catalytic domain, which has been implicated in the binding of FXIa to heparin (33), and not the A3 domain which mediates the binding of FXI to both heparin and activated platelets (15) contains all of the binding energy required to mediate FXIa binding to the surface of activated platelets (Figure 6B).
Previously we have presented a model for FIX-activation by dimeric FXIa on activated platelets (42). These studies demonstrated that the dimeric structure of FXIa is required for normal rates of FIX-activation on the platelet surface. A monomeric form of FXIa, prepared by introducing the A4 domain of PK into chimeric FXI, was able to activate FIX in solution at rates comparable to those achieved by dimeric FXIa, whereas on the platelet surface only dimeric FXIa could activate FIX, and monomeric FXIa was inert. Our present studies provide the experimental basis for rationalizing these observations and revising our model for FIX activation on the platelet surface (Figure 7). Since FXIa binds to activated platelets through the catalytic domain of one monomer, access to the substrate, FIX, would be precluded by ligation to the platelet receptor whereas the catalytic domain of the other monomer, together with the substrate-binding site within the A2 and/or A3 domain (20–22), would be free to bind and catalyze the activation of FIX, either bound to the platelet surface or free in solution. The other inference to be drawn from this model explains our previous observation that platelet-bound FXIa is protected from inactivation by PN2, secreted at high concentration by activated platelets (25, 27,28), whereas unbound FXIa is potently inactivated by PN2. This monomeric FXIa would either be bound to platelets through its catalytic domain, thus preventing FIX-activation, or it would be efficiently inhibited in free solution by PN2. In contrast, dimeric FXIa would utilize the catalytic domain of one subunit to bind to platelets and the heavy and light chains of the other subunit to activate FIX in the vicinity of the platelet membrane where inactivation by PN2 is precluded.
Our present studies must be interpreted within the context of very recent structural information available for zymogen FXI (43), the catalytic domain of FXI in complex with the KPI domain of PN2 (44), and the A4 domain of FXI (45, 46). Shown in Figure 8 is the crystal structure of the catalytic domain of FXI with the regions of dissimilarity compared with PK (see Figure 5) highlighted in various colors and labeled R1 through R6. In addition, the platelet-binding domain (Cys527-Cys542) identified in the present study is also highlighted (in orange). We have also superimposed the catalytic domains of the zymogen FXI (PDB: 2F83) and the enzyme FXIa (PDB: 1ZJD) that demonstrates a close correspondence of the backbone structures of the zymogen and the enzyme (Figure 9). Therefore, it can be concluded that the conversion of the zymogen to the enzyme is not accompanied by any major discernible change in the secondary or tertiary structure of the disulfide-constrained loop structure (Cys527-Cys542) identified here as the platelet-binding site in FXIa. In contrast, we have recently reported that the solution structure of the isolated A4 domain exhibits a novel α-helix within the C-terminal strand connecting the A4 domain to the catalytic domain (45, 46). This striking conformational change in the structure of the A4 domain is accompanied by a major change in shape of FXI when it is converted to FXIa. We postulate that this conformational change accounts for the exposure of the catalytic domain platelet-binding site for the enzyme, FXIa, and obscures the A3 domain platelet-binding site for the zymogen, FXI.
Fig 8. X-Ray Crystal Structure of the Catalytic Domain of FXIa.

The isolated FXIa catalytic domain shown here is taken from the co-crystal structure of rFXIac complexed with PN2KPI (PDB:1ZJD) (44). The structure is displayed in the same orientation as that in which thrombin is conventionally shown. The residues comprising the catalytic triad (amino acids His413, Asp462, and Ser557 or His57, Asp189, Ser195, chymotrypsin numbering) are displayed in yellow, with the N-terminal isoleucine and the C-terminal valine shown in white. The six distinct regions of dissimilarity with PK are color-coded and marked in reference to those shown in Figure 5. Note the similarity of location and structure of region 3 (R3) to that of thrombin exosite I and of region 4 (R4) to that of exosite II of thrombin. Also the region identified as R6 by comparison with PK corresponds to the Autolysis Loop of FXIa, and the α-helix-containing subdomain (Cys527-Cys542, FXI numbering) contains the putative heparin- and platelet-binding residues proposed for mutational and mechanistic analysis. The catalytic domain numbering corresponds to Ile370-Val607 (mature plasma FXI) or Ile16-Val245 (chymotrypsin numbering).
Fig 9. Crystal Structure of the Protease Domains of FXI and FXIa.
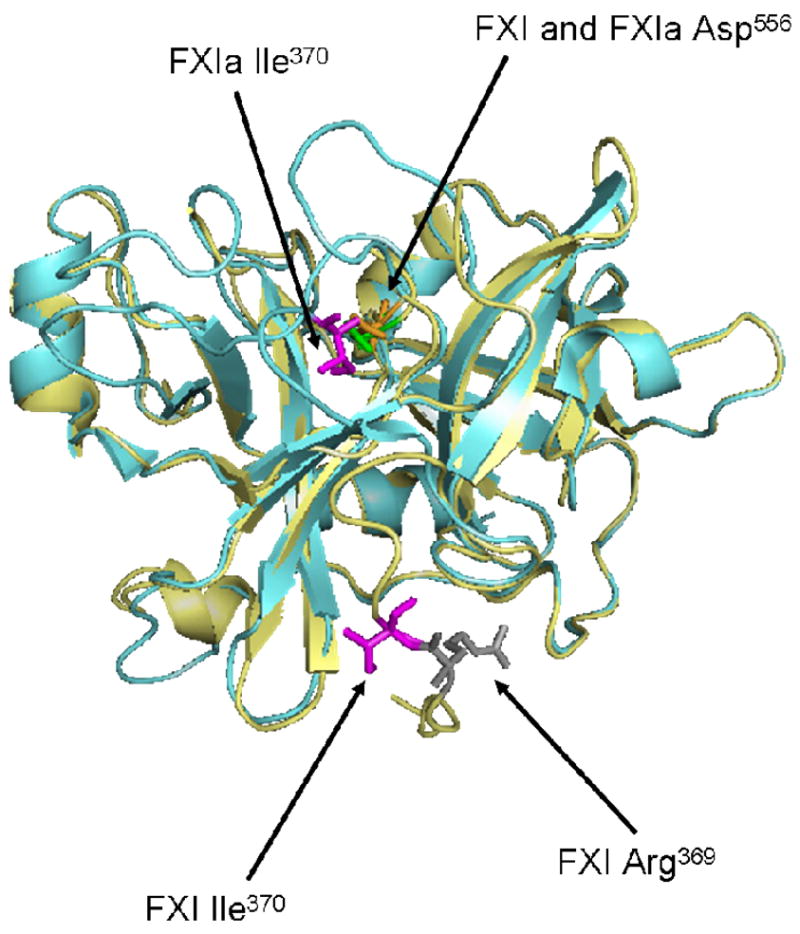
The protease domain of FXIa is shown here superimposed onto the protease domain of the zymogen, FXI. Ile370 (chymotrypsin Ile16) is shown in magenta for both molecules. For FXI, Arg369 is also shown in gray. Upon activation, the scissile bond between Arg369-Ile370 is cleaved and a new N-terminal sequence (Ile-Val-Gly-Gly) is formed. The new N-terminal Ile370 folds inward toward the catalytic triad and forms a salt bridge with Asp194. In the FXIa structure, the major conformational change is the positioning of Ile370, a movement of ~20Å from its placement in the zymogen structure. (PDB:2F83 for zymogen and PDB:1ZJD for enzyme were used to create this model using Pymol software (DeLano Scientific LLC, Palo Alto, CA)).
These studies support the conclusion that upon activation of FXI, sites within the A3 domain become concealed and a cryptic site within the catalytic domain becomes exposed that is able to mediate binding of FXIa to the activated platelet surface. Either HK with ZnCl2 (KD ~10 nM) or prothrombin with CaCl2 (KD ~250 nM) binds to the A1 domain of FXI. Upon binding, sites within the A3 domain become exposed which are then able to mediate FXI binding to the platelet surface (Bmax ~1500 sites/platelet; KD ~10 nM). FXI is then activated by FXIIa, thrombin, or autoactivation by cleavage of the scissile bond between Ile369 and Arg370. Cleavage at this site leads to a conformational change that conceals binding sites within the A3 domain and exposes sites within the catalytic domain that now are able to mediate binding of the enzyme to the platelet surface (Bmax ~250 sites/platelet; KD ~1.7 nM). We have previously presented data in support of the existence of an ordered, sequential mechanism for FIX and FIXa binding to platelet receptors in the assembly of the FX-activating complex (47), in which the reversible binding to platelets of the zymogen, FIX via its Gla domain, results in FIX-activation by FXIa to FIXa, which then assembles from solution via residues in the EGF-2 domain into the FX-activating complex on the platelet surface. Such an ordered, sequential mechanism may also exist for the interaction of the zymogen, FXI and the enzyme, FXIa, with the activated platelet surface, as displayed schematically in Figure 7.
TABLE 2.
Calculated Ki values for Proteins and Peptides that Compete for Factor XIa Binding to the Activated Platelet Surface
| Competitor | Ki (10−9 M) |
|---|---|
| FXIa | 1.4 ± 0.28 |
| FXI | NE |
| rFXIa/PKA3 | 2.7 ± 0.32 |
| rFXI/PKA3 | NE |
| TRP383-LEU397 (peptide 1) | NE |
| PHE415-GLY429 (peptide 2) | NE |
| GLN433-GLU447 (peptide 3) | NE |
| ILE450-ALA446 (peptide 4) | ND |
| LYS467-ILE481 (peptide 5) | NE |
| GLY500-LEU514 (peptide 6) | NE |
| CYS527-CYS542 (peptide 7)* | 5.8 ± 0.78 |
| CYS527-CYS542 (peptide 8)*† scr | NE |
| Heavy Chain | NE |
| Catalytic Domain | 3.5 ± 0.42 |
Denotes a cyclized peptide
scr denotes scrambled peptide
NE denotes no effect of the ligand at concentrations of up to 200 nM for FXI and rFXI/PKA3, up to 1 μM for the heavy chain, 100 μM for peptides 1–3, 5–6, and 1 μM for peptide 8.
ND denotes a value that was not determined.
Values represent the mean ± standard deviation for three independently performed experiments each carried out in triplicate.
Footnotes
This work is supported by research grants from the National Institute of Health: (HL74124 and HL46213 to PNW and from the American Heart Association (99100069U to TRB).
Abbrevations used: FXI, factor XI; FXIa, factor XIa; FXII, factor XII; FXIIa, factor XIIa; PK, prekallikrein; HK, high molecular weight kininogen; A3, Apple 3; FIX, factor IX; FIXa, factor IXa; PN2, protease nexin 2; pFXIa, plasma FXIa; rFXI/PKA3, recombinant FXI with the A3 domain of FXI replaced with the A3 of PK; rFXIa/C362S,C482S, recombinant FXI with cysteine 362 and cysteine 482 mutated to serines; rFXIac, factor XIa catalytic domain; TRAP, thrombin receptor activation peptide.
References
- 1.Bouma BN, Griffin JH. Human blood coagulation factor XI. Purification, properties, and mechanism of activation by activated factor XII. J Biol Chem. 1977;252:6432–6437. [PubMed] [Google Scholar]
- 2.Fujikawa K, Legaz ME, Kato H, Davie EW. The mechanism of activation of bovine factor IX (Christmas factor) by bovine factor XIa (activated plasma thromboplastin antecedent) Biochemistry. 1974;13:4508–4516. doi: 10.1021/bi00719a006. [DOI] [PubMed] [Google Scholar]
- 3.Koide T, Kato H, Davie EW. Isolation and characterization of bovine factor XI (plasma thromboplastin antecedent) Biochemistry. 1977;16:2279–2286. doi: 10.1021/bi00629a037. [DOI] [PubMed] [Google Scholar]
- 4.McMullen BA, Fujikawa K, Davie EW. Location of the disulfide bonds in human coagulation factor XI: the presence of tandem apple domains. Biochemistry. 1991;30:2056–2060. doi: 10.1021/bi00222a008. [DOI] [PubMed] [Google Scholar]
- 5.Leiba H, Ramot B, Many A. Hereditary and coagulation studies in ten families with factor XI (plasma thromboplastin antecedent) deficiency. Br J Haematol. 1965;11:654–665. doi: 10.1111/j.1365-2141.1965.tb00114.x. [DOI] [PubMed] [Google Scholar]
- 6.Ragni MV, Sinha D, Seaman F, Lewis JH, Spero JA, Walsh PN. Comparison of bleeding tendency, factor XI coagulant activity, and factor XI antigen in 25 factor XI-deficient kindreds. Blood. 1985;65:719–724. [PubMed] [Google Scholar]
- 7.Rapaport SI, Proctor RR, Patch MJ, Yettra M. The mode of inheritance of PTA deficiency: evidence for the existence of a major PTA deficiency and a minor PTA deficiency. Blood. 1961;18:149–155. [PubMed] [Google Scholar]
- 8.Rosenthal RL, Dreskin OH, Rosenthal N. New hemophilia-like disease caused by deficiency of a third plasma thromboplastin factor. Proc Soc Exp Biol Med. 1953;82:171–174. doi: 10.3181/00379727-82-20057. [DOI] [PubMed] [Google Scholar]
- 9.Colman RW. Contact activation pathway: inflammatory, fibrinolytic, anticoagulant, antiadhesive and antiangiogenic activities. In: Colman RW, Hirsh J, Marder VJ, Clowes AW, George JN, editors. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 103–122. [Google Scholar]
- 10.Naito K, Fujikawa K. Activation of human blood coagulation factor XI independent of factor XII. Factor XI is activated by thrombin and factor XIa in the presence of negatively charged surfaces. J Biol Chem. 1991;266:7353–7358. [PubMed] [Google Scholar]
- 11.Gailani D, Broze GJ., Jr Factor XI activation in a revised model of blood coagulation. Science. 1991;253:909–912. doi: 10.1126/science.1652157. [DOI] [PubMed] [Google Scholar]
- 12.Baglia FA, Jameson BA, Walsh PN. Identification and characterization of a binding site for platelets in the Apple 3 domain of coagulation factor XI. J Biol Chem. 1995;270:6734–6740. doi: 10.1074/jbc.270.12.6734. [DOI] [PubMed] [Google Scholar]
- 13.Greengard JS, Heeb MJ, Ersdal E, Walsh PN, Griffin JH. Binding of coagulation factor XI to washed human platelets. Biochemistry. 1986;25:3884–3890. doi: 10.1021/bi00361a022. [DOI] [PubMed] [Google Scholar]
- 14.Ho DH, Badellino K, Baglia FA, Sun MF, Zhao MM, Gailani D, Walsh PN. The role of high molecular weight kininogen and prothrombin as cofactors in the binding of factor XI A3 domain to the platelet surface. J Biol Chem. 2000;275:25139–25145. doi: 10.1074/jbc.M001890200. [DOI] [PubMed] [Google Scholar]
- 15.Ho DH, Baglia FA, Walsh PN. Factor XI binding to activated platelets is mediated by residues R(250), K(255), F(260), and Q(263) within the Apple 3 domain. Biochemistry. 2000;39:316–323. doi: 10.1021/bi991851q. [DOI] [PubMed] [Google Scholar]
- 16.Walsh PN, Griffin JH. Contributions of human platelets to the proteolytic activation of blood coagulation factors XII and XI. Blood. 1981;57:106–118. [PubMed] [Google Scholar]
- 17.Oliver JA, Monroe DM, Roberts HR, Hoffman M. Thrombin activates factor XI on activated platelets in the absence of factor XII. Arterioscler Thromb Vasc Biol. 1999;19:170–177. doi: 10.1161/01.atv.19.1.170. [DOI] [PubMed] [Google Scholar]
- 18.Baird TR, Walsh PN. Activated platelets but not endothelial cells participate in the initiation of the consolidation phase of blood coagulation. J Biol Chem. 2002;277:28498–28503. doi: 10.1074/jbc.M203427200. [DOI] [PubMed] [Google Scholar]
- 19.Baglia FA, Shrimpton CN, Emsley J, Kitagawa K, Ruggeri ZM, Lopez JA, Walsh PN. Factor XI interacts with the leucine-rich repeats of glycoprotein Ibalpha on the activated platelet. J Biol Chem. 2004;279:49323–49329. doi: 10.1074/jbc.M407889200. [DOI] [PubMed] [Google Scholar]
- 20.Sinha D, Seaman FS, Walsh PN. Role of calcium ions and the heavy chain of factor XIa in the activation of human coagulation factor IX. Biochemistry. 1987;26:3768–3775. doi: 10.1021/bi00387a005. [DOI] [PubMed] [Google Scholar]
- 21.Baglia FA, Jameson BA, Walsh PN. Identification and chemical synthesis of a substrate-binding site for factor IX on coagulation factor XIa. J Biol Chem. 1991;266:24190–24197. [PubMed] [Google Scholar]
- 22.Sun Y, Gailani D. Identification of a factor IX binding site on the third apple domain of activated factor XI. J Biol Chem. 1996;271:29023–29028. doi: 10.1074/jbc.271.46.29023. [DOI] [PubMed] [Google Scholar]
- 23.Sinha D, Marcinkiewicz M, Lear JD, Walsh PN. Factor XIa dimer in the activation of factor IX. Biochemistry. 2005;44:10416–10422. doi: 10.1021/bi050361x. [DOI] [PubMed] [Google Scholar]
- 24.Sinha D, Seaman FS, Koshy A, Knight LC, Walsh PN. Blood coagulation factor XIa binds specifically to a site on activated human platelets distinct from that for factor XI. J Clin Invest. 1984;73:1550–1556. doi: 10.1172/JCI111361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baird TR, Walsh PN. The interaction of factor XIa with activated platelets but not endothelial cells promotes the activation of factor IX in the consolidation phase of blood coagulation. J Biol Chem. 2002;277:38462–38467. doi: 10.1074/jbc.M205902200. [DOI] [PubMed] [Google Scholar]
- 26.Ahmad SS, Rawala-Sheikh R, Walsh PN. Comparative interactions of factor IX and factor IXa with human platelets. J Biol Chem. 1989;264:3244–3251. [PubMed] [Google Scholar]
- 27.Badellino KO, Walsh PN. Protease nexin II interactions with coagulation factor XIa are contained within the Kunitz protease inhibitor domain of protease nexin II and the factor XIa catalytic domain. Biochemistry. 2000;39:4769–4777. doi: 10.1021/bi9925468. [DOI] [PubMed] [Google Scholar]
- 28.Scandura JM, Zhang Y, Van Nostrand WE, Walsh PN. Progress curve analysis of the kinetics with which blood coagulation factor XIa is inhibited by protease nexin-2. Biochemistry. 1997;36:412–420. doi: 10.1021/bi9612576. [DOI] [PubMed] [Google Scholar]
- 29.Sinha D, Koshy A, Seaman FS, Walsh PN. Functional characterization of human blood coagulation factor XIa using hybridoma antibodies. J Biol Chem. 1985;260:10714–10719. [PubMed] [Google Scholar]
- 30.Tuszynski GP, Knight L, Piperno JR, Walsh PN. A rapid method for removal of [125I]iodide following iodination of protein solutions. Anal Biochem. 1980;106:118–122. doi: 10.1016/0003-2697(80)90126-8. [DOI] [PubMed] [Google Scholar]
- 31.Scandura JM, Ahmad SS, Walsh PN. A binding site expressed on the surface of activated human platelets is shared by factor X and prothrombin. Biochemistry. 1996;35:8890–8902. doi: 10.1021/bi9525029. [DOI] [PubMed] [Google Scholar]
- 32.Walsh PN, Mills DC, White JG. Metabolism and function of human platelets washed by albumin density gradient separation. Brit J Haematol. 1977;36:287–296. doi: 10.1111/j.1365-2141.1977.tb00649.x. [DOI] [PubMed] [Google Scholar]
- 33.Badellino KO, Walsh PN. Localization of a heparin binding site in the catalytic domain of factor XIa. Biochemistry. 2001;40:7569–7580. doi: 10.1021/bi0027433. [DOI] [PubMed] [Google Scholar]
- 34.Ho DH, Badellino K, Baglia FA, Walsh PN. A binding site for heparin in the apple 3 domain of factor XI. J Biol Chem. 1998;273:16382–16390. doi: 10.1074/jbc.273.26.16382. [DOI] [PubMed] [Google Scholar]
- 35.Walsh PN. Retraction to “Prothrombin is a cofactor for the binding of factor XI to the platelet surface and for platelet-mediated factor-XI activation by thrombin, Biochemistry 37:2271–2281, 1998”. Biochemistry bi-2007-0150k. 2007 doi: 10.1021/bi972113+. In Press. [DOI] [PubMed] [Google Scholar]
- 36.Walsh PN. Retraction to “Baglia, F. A. and Walsh, P. N. Thrombin-mediated feedback activation of factor XI on the activated platelet surface is preferred over contact activation by factor XIIa or factor XIa. J Biol Chem 275:20514–20519, 2000”. J Biol Chem. 2007;282:29067-a. doi: 10.1074/jbc.M000464200. [DOI] [PubMed] [Google Scholar]
- 37.Pedicord DL, Seiffert D, Blat Y. Feedback activation of factor XI by thrombin does not occur in plasma. Proc Natl Acad Sci U S A. 2007;104:12855–12860. doi: 10.1073/pnas.0705566104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bush AI, Martins RN, Rumble B, Moir R, Fuller S, Milward E, Currie J, Ames D, Weidemann A, Fischer P, Multhaup G, Beyreuther K, Masters CL. The amyloid precursor protein of Alzheimer’s disease is released by human platelets. J Biol Chem. 1990;265:15977–15983. [PubMed] [Google Scholar]
- 39.Smith RP, Higuchi DA, Broze GJ., Jr Platelet coagulation factor XIa-inhibitor, a form of Alzheimer amyloid precursor protein. Science. 1990;248:1126–1128. doi: 10.1126/science.2111585. [DOI] [PubMed] [Google Scholar]
- 40.Van Nostrand WE, Schmaier AH, Farrow JS, Cunningham DD. Protease nexin-II (amyloid beta-protein precursor): a platelet alpha-granule protein. Science. 1990;248:745–748. doi: 10.1126/science.2110384. [DOI] [PubMed] [Google Scholar]
- 41.Smith RP, Broze GJ., Jr Characterization of platelet-releasable forms of beta-amyloid precursor proteins: the effect of thrombin. Blood. 1992;80:2252–2260. [PubMed] [Google Scholar]
- 42.Gailani D, Ho D, Sun MF, Cheng Q, Walsh PN. Model for a factor IX activation complex on blood platelets: dimeric conformation of factor XIa is essential. Blood. 2001;97:3117–3122. doi: 10.1182/blood.v97.10.3117. [DOI] [PubMed] [Google Scholar]
- 43.Papagrigoriou E, McEwan PA, Walsh PN, Emsley J. Crystal structure of the factor XI zymogen reveals a pathway for transactivation. Nat Struct Mol Biol. 2006;13:557–558. doi: 10.1038/nsmb1095. [DOI] [PubMed] [Google Scholar]
- 44.Navaneetham D, Jin L, Pandey P, Strickler JE, Babine RE, Abdel-Meguid SS, Walsh PN. Structural and Mutational Analyses of the Molecular Interactions between the Catalytic Domain of Factor XIa and the Kunitz Protease Inhibitor Domain of Protease Nexin 2. J Biol Chem. 2005;280:36165–36175. doi: 10.1074/jbc.M504990200. [DOI] [PubMed] [Google Scholar]
- 45.Samuel D, Cheng H, Riley PW, Canutescu AA, Nagaswami C, Weisel JW, Bu Z, Walsh PN, Roder H. Solution structure of the A4 domain sheds light on the mechanism of zymogen activation. PNAS. 2007 doi: 10.1073/pnas.0703080104. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Riley PW, Cheng H, Samuel D, Roder H, Walsh PN. Dimer Dissociation and Unfolding Mechanism of Coagulation Factor XI Apple 4 Domain: Spectroscopic and Mutational Analysis. J Mol Biol. 2007;367:558–573. doi: 10.1016/j.jmb.2006.12.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang X, Walsh PN. An ordered sequential mechanism for Factor IX and Factor IXa binding to platelet receptors in the assembly of the Factor X-activating complex. Biochem J. 2005;390:157–167. doi: 10.1042/BJ20050029. [DOI] [PMC free article] [PubMed] [Google Scholar]