

Abstract
A total synthesis of 22-hydroxyacuminatine, a cytotoxic alkaloid isolated from Camptotheca acuminata, is reported. The key step in the synthesis involves the reaction of 2,3-dihydro-1H-pyrrolo[3,4-b]quinoline with a brominated phthalide to generate a substituted pentacyclic 12H-5,11a-diazadibenzo[b,h]fluoren-11-one intermediate. Despite its structural resemblance to camptothecin and luotonin A, a biological evaluation of 22-hydroxyacuminatine in a topoisomerase I-deficient cell line P388/CPT45 has confirmed that the observed cytotoxicity is not due to topoisomerase I inhibition. This result is consistent with the hypothesis that π-π stacking is more important than hydrogen bonding interactions in determining topoisomerase I inhibitor binding in the ternary cleavage complex.
Introduction
Camptothecin (CPT, 1) is a potent topoisomerase I (Top1) inhibitor with significant cytotoxicity against a variety of different cancer cell lines.1 Top1 is a nuclear enzyme that relaxes the DNA supercoils generated during DNA replication and transcription. The enzyme operates by carrying out a transesterification reaction involving a nucleophilic attack of the Tyr723 hydroxyl group on a DNA phosphodiester bond, thus creating a transient nick that is rapidly resealed by the reverse transesterification reaction.2 CPT inhibits the reverse reaction by intercalating between the base pairs at the DNA cleavage site, thus stabilizing the ternary DNA cleavage complex consisting of the drug, DNA, and the enzyme. Collision of the DNA replication fork with the single-stranded DNA break then results in a double-stranded break and cancer cell apoptosis. Two clinically useful CPT derivatives, topotecan (Hycamtin®) and irinotecan (Camptosar®), are presently in clinical use as anticancer agents.3 However, lactone ring hydrolysis of the CPT system produces the hydroxy carboxylate which has a high affinity to human serum albumin protein, and this limits the efficacy of CPT derivitaves.4 As a consequence, there is interest in developing metabolically stable non-CPT Top1 inhibitors as novel anticancer drugs.5
As a strategy to enhance stability, replacement of the E-ring present in CPT with an aromatic ring has been investigated. The resulting compounds are collectively referred to “aromathecins” as opposed to camptothecins. We have previously shown that aromathecin 2a is a weak Top1 inhibitor with low cytotoxicity.6 Based on the crystal structure of the CPT-stabilized DNA-Top1 cleavage complex, which revealed that the crucial 20-OH of CPT is hydrogen bonded with Asp533,7 introduction of an additional hydrogen bond donor to the E-ring of the aromathecins capable of interacting with Asp533 would be expected to increase the Top1 inhibitory potency. Therefore, aromathecin 2b was considered. Interestingly, a review of the literature revealed that compound 2b had been previously isolated from Camptotheca acuminata as a rare natural product (0.000006% yield) named 22-hydroxyacuminatine.8 Although 2 b exhibited cytotoxicity toward P388 and KB cell lines, it has remained to be determined whether this cytotoxicity is due to Top1 inhibition. This question prompted us to investigate the total synthesis of 22-hydroxyacuminatine9 as well as its biological properties. We also wanted to establish a flexible route that would open up a way to synthesize analogues for investigation of structure-activity relationships.
Results and Discussion
Bromide 2c was envisioned as a precursor to 22-hydroxyacuminatine (2b) in that a variety of different transformations are possible with the bromide, which is considered as an advantage in terms of potential analogue syntheses. By analogy to the synthesis of CPT10 and aromathecin 2a,6 the bromide 2c was dissected in the D-ring to give fragments 311 and 4 (Scheme 1).
Scheme 1.
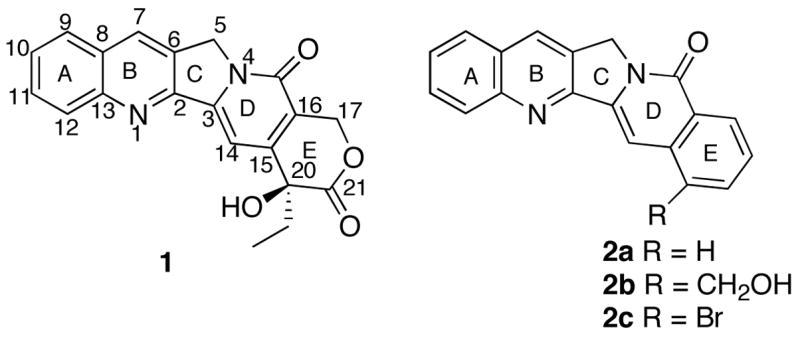
Retrosynthetic Analysis of Aromathecin 2c
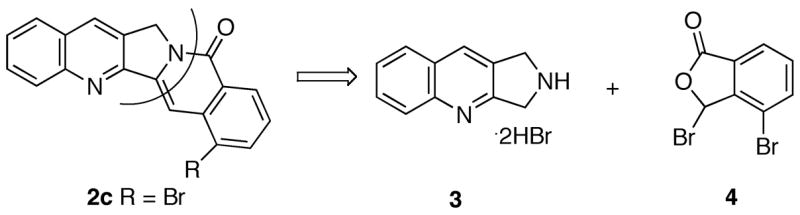
The synthesis of fragment 4 is presented in Scheme 2. Methylation of commercially available benzoic acid derivative 5 provided methyl ester 6a. Oxidation of the methyl group in 6a to the corresponding aldehyde proved to be problematic. No reaction was observed with CrO3/Ac2O/HOAc12 or ceric ammonium nitrate (CAN)13 or selenium dioxide (SeO2),14 presumably due to the steric hindrance. Consistent with this hypothesis, oxidation of 5 with CrO3/Ac2O/HOAc resulted in overoxidation of the methyl group to a carboxylic acid followed by cyclization to the anhydride. On the other hand, Étard reaction of methyl ester 6a with chromyl chloride15 provided lactone 7 instead of the expected aldehyde derivative.16 When less than two equivalents of chromyl chloride was used, another product 6b was also generated from the Étard reaction. Radical bromination of lactone 7 gave the requisite bromide 4 uneventfully.
Scheme 2.
Synthesis of Bromide 4
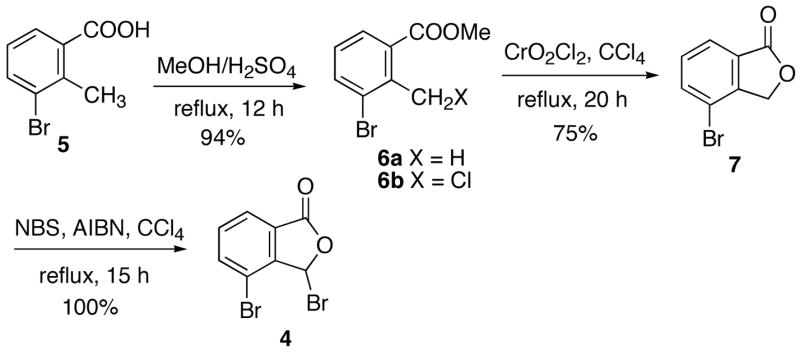
Coupling of fragments 3 11 and 4 afforded bromide 2c in 20% yield (Scheme 3). Treatment of 2c with n-BuLi to undergo bromine-lithium exchange to give the corresponding anion 8, followed by trapping with paraformaldehyde, would give 22-hydroxyacuminatine (2b). However, the product formed from the reaction was 11, while 2b was not formed at all. Employment of excess n-BuLi (2 equivalents) for the reaction did not result in the formation of any 2b. This result indicated that the bromine-lithium exchange occurred at a faster rate than deprotonation at C-5 (see CPT numbering system in structure 1). However, the resulting anion 8 evidently undergoes rapid proton transfer involving the 5 position to give benzylic anion 9. The 1H NMR spectrum of the product 11 is interesting in that the methine proton next to the hydroxymethyl side chain is presented as a doublet instead of the expected triplet. This unusual 1H NMR feature is presumably due to intramolecular hydrogen bond formation between the hydroxyl group and the lactam oxygen, which results in the H1-C2-C3-H4 dihedral angle being close to 90°. This dihedral angle was found to be 91.6° after the structure was energetically minimized by AM1 in Gaussian03 (Figure 1).17
Scheme 3.
Synthesis of 2c and Its Reaction with n-BuLi
Figure 1.
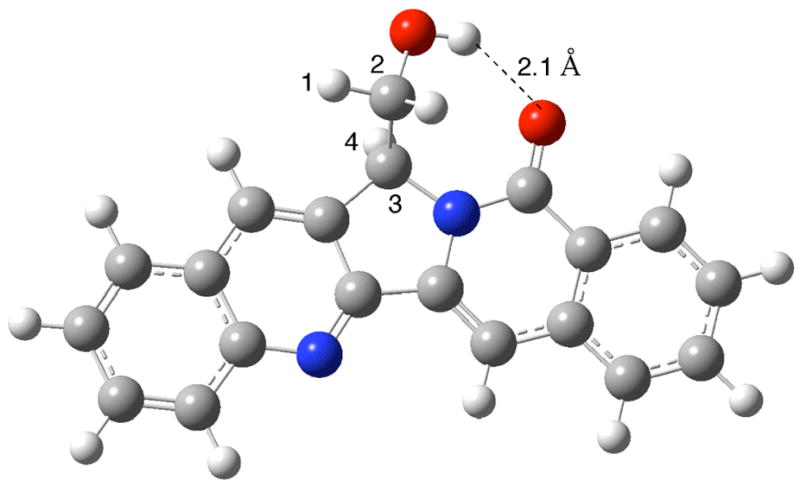
AM1 Optimized structure of 11.
The failure to transform 2c to 2b suggests that it might be advantageous to install the one-carbon unit before the coupling reaction. Therefore, the possible intermediate 14 with a cyano group at the desired position was considered, which would require the preparation of nitrile 13 as one of the building blocks. To this end, Rosenmund-von Braun reaction18 of bromide 7 with CuCN in refluxing DMF gave cyanide 12 in quantitative yield leaving the lactone intact (Scheme 4). No reaction was observed at a temperature of 120 °C or lower. Radical bromination of the new lactone 12 delivered bromide 13, which reacted with the tricyclic amine hydrobromide 3 to yield cyanide 14. DIBAL-H reduction followed by hydrolysis and further DIBAL-H reduction of the resulting aldehyde afforded 22-hydroxyacuminatine (2b) in 67% yield.
Scheme 4.
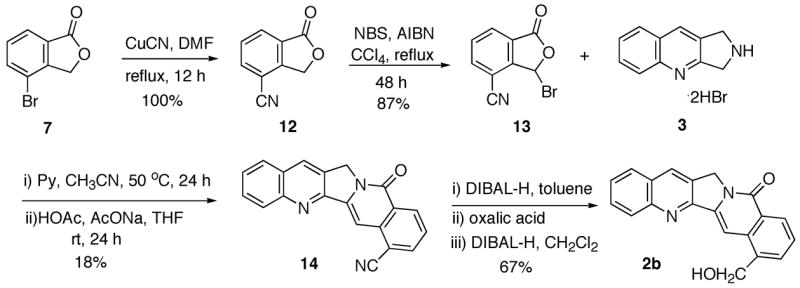
Synthesis of 22-Hydroxyacuminatine (2b)
Due to the structural similarity of 22-hydroxyacuminatine (2b) with CPT (1) and luotonin A (15), both of which are Top1 inhibitors exhibiting Top1-dependent growth inhibition in yeast,19 a Top1-mediated DNA cleavage assay20 was performed to examine the Top1 inhibitory potency. In this assay, only marginal DNA cleavage was observed with 2b, indicating that it is a weak Top1 inhibitor. To further verify whether the reported cytotoxicity of 2b8 is due to the weak Top1 inhibition, the cytotoxicity of 2b in a P388 leukemia cell line and its Top1-deficient subclone P388/CPT4521 was investigated.22 The IC50 observed for 22-hydroxyacuminatine (2b) in P388 cells is 35.5 ± 0.5 μM, while in P388/CPT45 cells (a Top1-deficient cell line) it is 43.0 ± 1.0 μM. The IC50 in P388 cells is roughly an order of magnitude higher than the originally reported value.8 More importantly, these values demonstrate that the cytotoxicity of 2b is not due to Top1 inhibition. It seems unlikely that differences between the camptothecins and 22-hydroxyacuminatine (2b) in their hydrogen bonding to the protein are responsible for the observed difference in Top1 inhibitory activities, since docking and energy minimization of 2b in the ternary complex indicates that, like the lactone form of the camptothecins,7,23 the hydroxyl group of 2b can form a direct hydrogen bond with the Asp533 side chain carboxylate of Top1 (Figure 2). On the other hand, the biological evaluation results are consistent with the hypothesis that π-π stacking is more important than hydrogen bonding interactions in determining inhibitor binding in the ternary cleavage complex,24,25 and the results are also in agreement with an earlier report that compound 2a does not exhibit its cytotoxicity by poisoning Top1 in yeast.26 The inefficient π-π stacking between 22-hydroxyacuminatine (2b) and the neighboring base pairs is most likely due to the nonpolar nature of the E-ring, where no heteroatoms are included or are directly attached, resulting in an electrostatically neutral potential surface (Figure 3) that does not complement the electrostatics of the adjacent DNA base pairs as well as the E ring of camptothecin does. According to the energy-minimized hypothetical model, the E ring of 22-hydroxyacuminatine is not well positioned to overlap with the adjacent base pairs (see Figure 2). This further demonstrates that the electronic properties of the E-ring of the aromathecins and camptothecins are critical determinants of enzyme inhibitory activity.27
Figure 2.
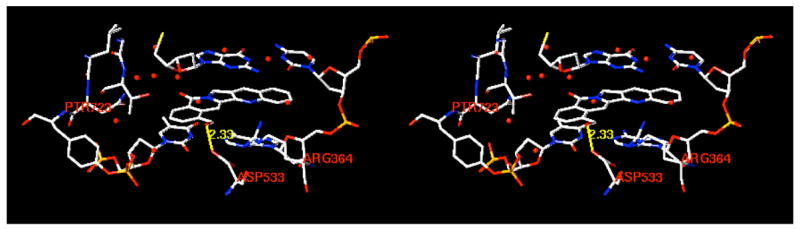
Hypothetical model of the binding of 22-hydroxyacuminatine (2b) in the Top1-DNA-inhibitor ternary complex. The cleaved DNA strand is on the left, while the uncleaved strand is on the right. The DNA minor groove side is facing forward, while the major groove is on the back. The model is programmed for walleyed (relaxed) viewing.
Figure 3.
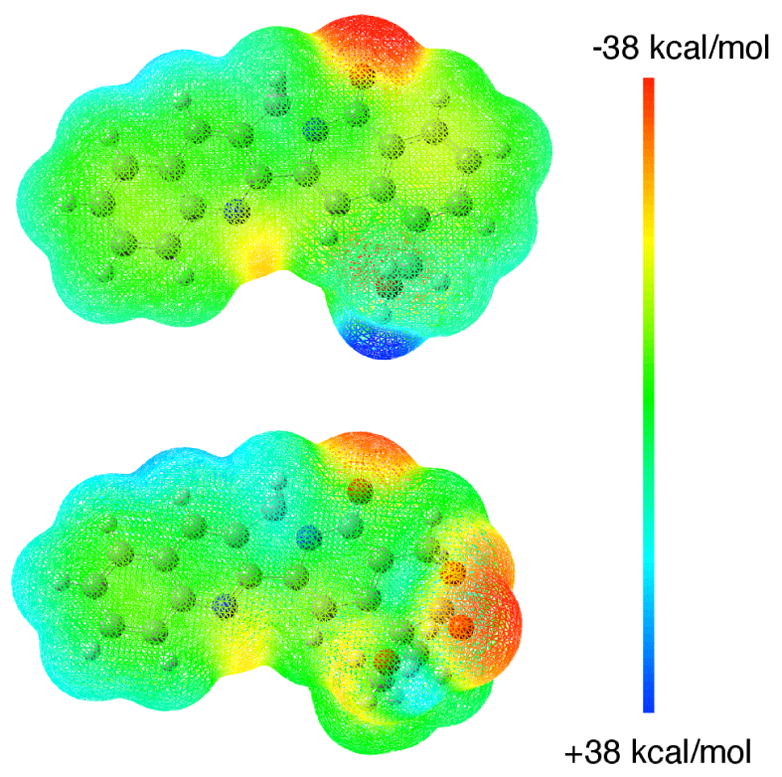
Electrostatic potential surfaces of 22-hydroxyacuminatine (top) and camptothecin (bottom) mapped on their total electron densities calculated at the HF/6–31G** level of theory. Both electrostatic potential surface maps are scaled to a range of −38 to +38 kcal/mol.
In conclusion, a highly convergent total synthesis of 22-hydroxyacuminatine (2b) has been achieved. The biological properties of 22-hydroxyacuminatine were characterized, demonstrating that Top1 inhibition is not a significant factor in its cytotoxicity despite its structural resemblance to CPT.
Experimental Section
22-Hydroxyacuminatine (2b)
DIBAL-H (1.0 M in CH2Cl2, 57 μL, 57 μmol) was added to a stirred solution of nitrile 14 (11.7 mg, 38 μmol) in toluene (3 mL) at −78 °C. The resulting reaction mixture was stirred at −78 °C for 3 h, when MeOH (0.1 mL) was added to quench the reaction. The reaction mixture was poured into a 1.0 M solution of oxalic acid (5 mL) and stirred at room temperature for 2 h. Solid Na2CO3 was added to neutralize the oxalic acid at 0 °C and the reaction mixture was extracted with CHCl3 (3 × 30 mL). The combined organic layers were washed with H2O (2 × 5 mL) and brine (2 × 5 mL). The organic solution was dried over Na2SO4, filtered, concentrated and the residue was subjected to a flash column chromatography, eluting with CHCl3, yielding a light yellow solid 9.0 mg (76%): 1H NMR (300 MHz, CDCl3) δ 10.52 (s, 1 H), 8.97 (s, 1 H), 8.81 (d, J = 8.4 Hz, 1 H), 8.38 (s, 1 H), 8.29 (t, J = 8.4 Hz, 1 H), 8.22 (d, J = 8.4 Hz, 1 H), 7.91 (d, J = 7.8 Hz, 1 H), 7.82 (t, J = 7.8 Hz, 1 H), 7.71 (t, J = 7.8 Hz, 1 H), 7.64 (t, J = 7.8 Hz, 1 H), 5.39 (s, 2 H). DIBAL-H (1.0 M in CH2Cl2, 58 μL, 58 μmol) was added to a stirred solution of aldehyde obtained as above (9.0 mg, 29 μmol) in CH2Cl2 (2 mL) at −78 °C. The reaction mixture was stirred at −78 °C for 2 h, when MeOH (0.1 mL) was added to quench the reaction at −78 °C. A saturated solution of potassium sodium tartrate (5 mL) was added and the solution was stirred at room temperature for 1 h. CHCl3 (3 × 20 mL) was used to extract the product. The combined organic layers were washed with H2O (2 × 5 mL) and brine (2 × 5 mL). The organic solution was dried over Na2SO4, filtered, concentrated and the residue was subjected to a flash column chromatography, eluting with CHCl3 and CHCl3-MeOH (10:1), yielding a light yellow solid 6.0 mg (67%): mp >255 °C (dec) [lit.8, mp 258–260 °C (dec)]. 1H NMR (500 MHz, DMSO-d6) δ 8.66 (s, 1 H), 8.29 (d, J = 7.5 Hz, 1 H), 8.19 (d, J = 8.5 Hz, 1 H), 8.11 (d, J = 7.5 Hz, 1 H), 7.83 (td, J = 8.5, 1.5 Hz, 1 H), 7.81 (d, J = 7.0 Hz, 1 H), 7.73 (s, 1 H), 7.69 (td, J = 8.0, 1.5 Hz, 1 H), 7.57 (t, J = 7.5 Hz, 1 H), 5.51 (t, J = 5.0 Hz, 1 H), 5.36 (s, 2 H), 4.95 (d, J = 5.0 Hz, 2 H); ESIMS m/z (rel intensity) 315 (MH+, 100); HRESIMS m/z calcd for C20H14N2O2 + H 315.1134, found 315.1136. Anal. (C20H14N2O2·1.0H2O) C, H, N.
7-Bromo-12H-5,11a-diazadibenzo[b,h]fluoren-11-one (2c)
A solution of bromide 4 (193 mg, 0.66 mmol) in THF (7 mL) was added to a stirred suspension of tricyclic amine hydrobromide 3 (220 mg, 0.66 mmol) and Et3N (3.0 mL) in THF (7 mL). added to dilute the reaction mixture, which was then washed with water (2 × 10 mL) and brine (2 × 10 mL). The organic solution was dried over anhydrous Na2SO4, filtered, and concentrated, yielding a brown residue, which was used for the next operation without further purification. The product obtained as above was dissolved in acetic acid (7 mL) and sodium acetate (0.7 g) was added to the reaction mixture, which was then stirred at room temperature for 36 h. Then acetic acid was removed under reduced pressure and the residue was dissolved in CHCl3 (100 mL), and washed with saturated NaHCO3 (2 × 10 mL), water (2 × 10 mL) and brine (2 × 10 mL). The organic solution was dried over Na2SO4, filtered, concentrated and the residue was subjected to silica gel (40 g) flash column chromatography, eluting with CHCl3, yielding a light yellow solid (49 mg, 20%): mp > 270 °C (dec). 1H NMR (300 MHz, CDCl3) δ 8.50 (d, J = 8.4 Hz, 1 H), 8.37 (s, 1 H), 8.31 (d, J = 8.4 Hz, 1 H), 8.10 (s, 1 H), 7.97 (d, J = 7.5 Hz, 1 H), 7.91 (d, J = 7.8 Hz, 1 H), 7.82 (t, J = 7.2 Hz, 1 H), 7.64 (t, J = 6.9 Hz, 1 H), 7.40 (t, J = 7.8 Hz, 1 H), 5.37 (s, 2 H); ESIMS m/z (rel intensity) 365 (81BrMH+, 100), 363 (79BrMH+, 100). Anal. (C19H11N2OBr) C, H, N.
3,4-Dibromo-3H-isobenzofuran-1-one (4)
A mixture of lactone 7 (906 mg, 4.27 mmol), NBS (818 mg, 4.7 mmol) and AIBN (71 mg, 0.43 mmol) in CCl4 (40 mL) was heated at reflux for 15 h. CCl4 was removed under reduced pressure. The residue was extracted with hot n-hexane (3 × 100 mL). Removal of hexane yielded a white powder (1.25 g, 100%): mp 89–90 °C. 1H NMR (300 MHz, CDCl3) δ 7.89 (d, J = 7.8 Hz, 1 H), 7.87 (d, J = 7.8 Hz, 1 H), 7.50 (t, J = 7.8 Hz, 1 H), 7.22 (s, 1 H); 13C NMR (75 MHz, CDCl3) δ 166.2, 147.3, 138.7, 132.6, 126.3, 125.0, 117.8, 74.5; CIMS m/z (rel intensity) 295 (81Br81BrMH+, 29), 293 (81Br79BrMH+, 63), 291 (79Br79BrMH+, 31), 100 (100). Anal. (C8H4Br2O2) C, H, N
Methyl 3-Bromo-2-methylbenzoate (6a)
Concentrated H2SO4 (0.5 mL) was added to a stirred solution of 3-bromo-2-methylbenzoic acid (5) (2.15 g, 10 mmol) in MeOH (20 mL) at room temperature. The resulting mixture was then heated under reflux for 12 h. Methanol was partially removed under reduced pressure and Et2O (150 mL) was added to dilute the residue, which was then washed with a saturated solution of NaHCO3 (2 × 10 mL), H2O (2 × 10 mL) and brine (2 × 10 mL). The organic solution was dried over anhydrous Na2SO4, filtered and concentrated, yielding a colorless liquid oil (2.15 g, 94%): 1H NMR (300 MHz, CDCl3) δ 7.64 (d, J = 7.8 Hz, 1 H), 7.60 (d, J = 7.8 Hz, 1 H), 7.00 (t, J = 7.8 Hz, 1 H), 3.98 (s, 3 H), 2.55 (s, 3 H); 13C NMR (75 MHz, CDCl3) δ 167.8, 138.7, 135.9, 132.6, 129.1, 127.1, 126.9, 52.2, 20.6; IR (film) 2997, 2951, 1725, 1434, 1285, 1255, 1096, 1010, 753 cm−1; EIMS m/z (rel intensity) 230 (81BrM+, 19), 228 (79BrM+, 19), 89 (78), 63 (100). Anal. (C9H9BrO2) C, H, Br.
3-Bromo-phthalide (7)
A solution of methyl ester 6a (510 mg, 2.23 mmol) in CCl4 (3 mL) was placed in a round-bottomed flask immersed in an ice-cold bath. A solution of chromyl chloride (0.36 mL, 4.45 mmol) in CCl4 (3 mL) was added over a period of 1 h. The resulting mixture was stirred at room temperature for 2 h and then slowly heated to reflux for 20 h. The reaction mixture was cooled to room temperature and poured into to an ice-cold saturated solution of Na2SO3 (5 mL) at 0 °C. EtOAc (3 × 30 mL) was used to extract the product. The combined organic layers were washed with H2O (2 × 10 mL) and brine (2 × 10 mL). The organic solution was dried over anhydrous Na2SO4, filtered and concentrated, yielding a colorless powder (356 mg, 75%): mp 88–89 °C. 1H NMR (300 MHz, CDCl3) δ 7.67 (d, J = 7.8 Hz, 1 H), 7.63 (d, J = 7.8 Hz, 1 H), 7.31 (t, J = 7.8 Hz, 1 H), 5.03 (s, 2 H); 13C NMR (75 MHz, CDCl3) δ 169.4, 146.2, 136.3, 130.5, 127.4, 124.0, 116.1, 69.4; EIMS m/z (rel intensity) 214 (81BrM+, 31), 212 (79BrM+, 30), 185 (100), 183 (98), 157 (27), 155 (29), 75(63). Anal. (C8H5BrO2) C, H, Br
12-Hydroxymethyl-12H-5,11a-diaza-dibenzo[b,h]fluoren-11-one (11)
n-BuLi (16 μL, 2.5 M in hexane, 0.04 mmol) was added to a stirred solution of bromide 2c (10 mg, 0.027 mmol) in THF (2 mL) at −78 °C. The resulting dark red solution was stirred at −78 °C for 15 min when a suspension of paraformaldehyde (4 mg, 0.135 mmol) in THF (0.5 mL) was added. The reaction mixture was then allowed to warm to room temperature and stirred at room temperature for 1 h. H2O (2 mL) was added to quench the reaction. CHCl3 (50 mL) was added to dilute the reaction mixture, which was then washed with H2O (2 × 5 mL) and brine (2 × 5 mL). The organic solution was dried over anhydrous Na2SO4, filtered and concentrated. The residue was subjected to silica gel flash column chromatography, eluting with CHCl3-MeOH (20:1), yielding a light yellow solid (2.5 mg, 29%): mp > 250 °C (dec). 1H NMR (300 MHz, CDCl3) δ 8.53 (d, J = 8.1 Hz, 1 H), 8.32 (s, 1 H), 8.25 (d, J = 8.7 Hz, 1 H), 7.92 (d, J = 7.8 Hz, 1 H), 7.73–7.83 (m, 4 H), 7.58–7.66 (m, 2 H), 5.95 (d, J = 7.5 Hz, 1 H), 4.38 (d, J = 12.3 Hz, 1 H), 4.05 (dd, J = 12.6, 7.5 Hz, 1 H); 13C NMR (75 MHz, CDCl3) δ 163.0, 149.1, 139.6, 137.4, 133.1, 131.2, 130.7, 129.9, 129.5, 128.8, 128.5, 128.2, 127.9 (2 C), 127.6 (2 C), 127.1, 102.3, 66.8, 66.0; ESIMS m/z (rel intensity) 315 (MH+, 100); HREIMS m/z calcd for C20H14N2O2 314.1055, found 314.1046.
3-Cyano-phthalide (12)
A mixture of bromide 7 (1.0 g, 4.72 mmol) and CuCN (634 mg, 7.1 mmol) in anhydrous DMF (15 mL) was heated under reflux for 12 h. The reaction mixture was cooled to room temperature and quenched by slow addition of brine (20 mL). EtOAc (3 × 60 mL) was used to extract the product. The combined organic layers were washed with H2O (2 × 20 mL) and brine (2 × 20 mL). The organic solution was dried over anhydrous Na2SO4, filtered and concentrated. The residue was subjected to silica gel flash column chromatography, eluting with n-hexane-EtOAc (4:1), yielding a white powder (750 mg, 100%): mp 173–174 °C. 1H NMR (300 MHz, CDCl3) δ 8.13 (d, J = 7.5 Hz, 1 H), 7.97 (d, J = 7.8 Hz, 1 H), 7.69 (t, J = 7.5 Hz, 1 H), 5.45 (s, 2 H); 13C NMR (75 MHz, CDCl3) δ 168.8, 149.3, 137.3, 130.1 (2 C), 127.4, 114.9, 107.4, 68.6; IR (film) 3077, 2235, 1770, 1267, 1020, 754 cm−1; CIMS m/z (rel intensity) 160 (MH+, 100). Anal. (C9H5NO2) C, H, N.
3-Bromo-4-cyano-3H-isobenzofuran-1-one (13)
A mixture of lactone 12 (705 mg, 4.43 mmol), NBS (868 mg, 4.88 mmol) and AIBN (73 mg, 0.443 mmol) in CCl4 (30 mL) was heated under reflux for 60 h. CCl4 was removed under reduced pressure and the residue was extracted with Et2O. Evaporation of Et2O resulted in a residue that was subjected to silica gel flash column chromatography, eluting with n-hexane-EtOAc (4:1), yielding a white powder 922 mg (87%): mp 118–119 °C. 1H NMR (300 MHz, CDCl3) δ 8.16 (d, J = 7.8 Hz, 1 H), 8.05 (d, J = 7.8 Hz, 1 H), 7.78 (t, J = 7.8 Hz, 1 H), 7.50 (s, 1 H); 13C NMR (75 MHz, CDCl3) δ 177.7, 150.5, 138.8, 131.8, 130.2, 125.6, 113.8, 108.3, 71.9; CIMS m/z (rel intensity) 240 (81BrMH+, 18), 238 (79BrMH+, 20), 158 (100).
7-Cyano-12H-5,11a-diazadibenzo[b,h]fluoren-11-one (14)
A solution of bromide 13 (150 mg, 0.63 mmol) in CH3CN (2 mL) was added to a stirred suspension of tricyclic amine hydrobromide 3 (105 mg, 0.31 mmol) and pyridine (1.5 mL) in CH3CN (2 mL). The resulting mixture was stirred at room temperature for 24 h. CHCl3 (100 mL) was added to dilute the reaction mixture, which was then washed with water (2 × 10 mL) and brine (2 × 10 mL). The organic solution was dried over anhydrous Na2SO4, filtered, and concentrated, yielding a brown residue, which was used for the next operation without further purification. The product obtained as above was dissolved in acetic acid (5 mL) and sodium acetate (0.5 g) was added to the reaction mixture, which was then heated at 50 °C for 24 h. Then acetic acid was removed under reduced pressure and the residue was dissolved in CHCl3 (100 mL) and washed with saturated NaHCO3 (2 × 10 mL), water (2 × 10 mL) and brine (2 × 10 mL). The organic solution was dried over Na2SO4, filtered, concentrated and the residue subjected to silica gel (40 g) flash column chromatography, eluting with CHCl3, yielding a light yellow solid (18 mg, 18%): mp > 262 °C (dec). 1H NMR (300 MHz, CDCl3) δ 8.73 (d, J = 8.4 Hz, 1 H), 8.36 (s, 1 H), 8.25 (d, J = 8.4 Hz, 1 H), 8.05 (dd, J = 8.4, 1.2 Hz, 1 H), 7.98 (s, 1 H), 7.90 (d, J = 7.5 Hz, 1 H), 7.81 (td, J = 7.2, 1.2 Hz, 1 H), 7.63 (td, J = 7.2, 1.2 Hz, 1 H), 7.59 (t, J = 7.8 Hz, 1 H), 5.37 (s, 2 H); ESIMS m/z (rel intensity) 310 (MH+, 100). Anal. (C20H11N3O·0.75H2O) C, H, N.
Molecular Modeling Procedure
The structure of the ternary complex, containing topoisomerase I, DNA, and topotecan, was downloaded from the Protein Data Bank (PDB code 1K4T). One molecule of PEG and the topotecan carboxylate form were deleted. All of the atoms were then fixed according to the Sybyl atom types. Hydrogens were added and minimized using MMFF94s force field and MMFF94 charges. The structure of the 22-hydroxyacuminatine, constructed in Sybyl and energy minimized with the Tripos force field and Gasteiger-Hückel charges, was overlapped with the structure of topotecan using the A, B, C rings, and the structure of topotecan was then deleted. The new whole complex was subsequently subjected to energy minimization using MMFF94s force field with MMFF94 charges. During the energy minimization, the structure of the 22-hydroxyacuminatine was allowed to move, while the structures of the protein, nucleic acids, and the surrounding water molecules were frozen. The energy minimization was performed using the Powell method with a 0.05 kcal/mol Å energy gradient convergence criterion and a distance-dependent dielectric function.
Supplementary Material
Elemental analysis data and 1H NMR spectra of compounds 2b, 2c, 11, and 39. This material is available free of charge at http://pubs.acs.org
Acknowledgments
This work was made possible by the National Institutes of Health (NIH) through support of this work with Research Grant UO1 CA89566. The in vitro testing was conducted through the Developmental Therapeutics Program, DCTD, NCI under Contract NO1-CO-56000. This research was conducted in a facility constructed with support from Research Facilities Improvement Program Grant Number C06-14499 from the National Center for Research Resources of the National Institutes of Health.
References
- 1.Thomas CJ, Rahier NJ, Hecht SM. Camptothecin: Current Perspectives. Bioorg Med Chem. 2004;12:1585–1604. doi: 10.1016/j.bmc.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 2.Wang JC. Cellular Roles of DNA Topoisomerases: A Molecular Perspective. Nat Rev Mol Cell Biol. 2002;3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 3.Kollmannsberger C, Mross K, Jakob A, Kanz L, Bokemeyer C. Topotecan - A Novel Topoisomerase I Inhibitor: Pharmacology and Clinical Experience. Oncology. 1999;56:1–12. doi: 10.1159/000011923. [DOI] [PubMed] [Google Scholar]
- 4.Burke TG, Mi ZH. The Structural Basis of Camptothecin Interactions with Human Serum-Albumin - Impact on Drug Stability. J Med Chem. 1994;37:40–46. doi: 10.1021/jm00027a005. [DOI] [PubMed] [Google Scholar]
- 5.Meng LH, Liao AY, Pommier Y. Non-Camptothecin DNA Topoisomerase I Inhibitors in Cancer Therapy. Curr Top Med Chem. 2003;3:305–320. doi: 10.2174/1568026033452546. [DOI] [PubMed] [Google Scholar]
- 6.Fox BM, Xiao X, Antony S, Kohlhagen G, Pommier Y, Staker BL, Stewart L, Cushman M. Design, Synthesis, and Biological Evaluation of Cytotoxic 11-Alkenylindenoisoquinoline Topoisomerase I Inhibitors and Indenoisoquinoline-Camptothecin Hybrids. J Med Chem. 2003;46:3275–3282. doi: 10.1021/jm0300476. [DOI] [PubMed] [Google Scholar]
- 7.Staker BL, Feese MD, Cushman M, Pommier Y, Zembower D, Stewart L, Burgin AB. Structures of Three Classes of Anticancer Agents bound to the Human Topoisomerase I-DNA Covalent Complex. J Med Chem. 2005;48:2336–2345. doi: 10.1021/jm049146p. [DOI] [PubMed] [Google Scholar]
- 8.Lin LZ, Cordell GA. Quinoline Alkaloids from Camptotheca acuminata. Phytochemistry. 1989;28:1295–1297. [Google Scholar]
- 9.Ma Z, Lee DYW. Total Synthesis of the Cytotoxic Alkaloid 22-Hydroxyacuminatine. Tetrahedron Lett. 2004;45:6721–6723. [Google Scholar]
- 10.Corey EJ, Crouse DN, Anderson JE. Total Synthesis of Natural 20(S)-Camptothecin. J Org Chem. 1975;40:2140–2141. doi: 10.1021/jo00902a034. [DOI] [PubMed] [Google Scholar]
- 11.Zalkow LH, Nabors JB, French K, Bisarya SC. Studies in Synthesis of Camptothecin - Efficient Synthesis of 2,3-Dihydro-1H-Pyrrolo 3,4-B Quinoline. J Chem Soc (C) 1971:3551–3554. doi: 10.1039/j39710003551. [DOI] [PubMed] [Google Scholar]
- 12.Lieberman SC, Connor R. p-Nitrobenzaldehyde. Org Syn Coll Vol II. 1943:441–443. [Google Scholar]
- 13.Trahanovsky WS, Young LB. Controlled Oxidation of Organic Compounds with Cerium(IV). II. The Oxidation of Toluenes. J Org Chem. 1966;31:2033–2035. [Google Scholar]
- 14.Plobeck N, Delorme D, Wei ZY, Yang H, Zhou F, Schwarz P, Gawell L, Gagnon H, Pelcman B, Schmidt R, Yue SY, Walpole C, Brown W, Zhou E, Labarre M, Payza K, St-Onge S, Kamassah A, Morin PE, Projean D, Ducharme J, Roberts E. New Diarylmethylpiperazines as Potent and Selective Nonpeptidic Delta Opioid Receptor Agonists with Increased In Vitro Metabolic Stability. J Med Chem. 2000;43:3878–3894. doi: 10.1021/jm000228x. [DOI] [PubMed] [Google Scholar]
- 15.Hartford WH, Darrin M. The Chemistry of Chromyl Compounds. Chem Rev. 1958;58:1–61. [Google Scholar]
- 16.Wheeler OH. Etard Reaction. I. Its Scope and Limitation with Substituted Toluenes. Can J Chem. 1958;36:667–671. [Google Scholar]
- 17.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JJA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03. Revision B.05. Pittsburg: [Google Scholar]
- 18.Ellis GP, Romney-Alexander TM. Cyanation of Aromatic Halides. Chem Rev. 1987;87:779–794. [Google Scholar]
- 19.Cagir A, Jones SH, Gao R, Eisenhauer BM, Hecht SM. Luotonin A. A naturally occurring human DNA topoisomerase I poison. J Am Chem Soc. 2003;125:13628–13629. doi: 10.1021/ja0368857. [DOI] [PubMed] [Google Scholar]
- 20.Kohlhagen G, Paull K, Cushman M, Nagafuji P, Pommier Y. Protein-Linked DNA Strand Breaks Induced by NSC 314622, a Novel Noncamptothecin Topoisomerase I Poison. Mol Pharmacol. 1998;54:50–58. doi: 10.1124/mol.54.1.50. [DOI] [PubMed] [Google Scholar]
- 21.Antony S, Kohlhagen G, Agama K, Jayaraman M, Cao S, Durrani FA, Rustum YM, Cushman M, Pommier Y. Cellular Topoisomerase I Inhibition and Antiproliferative Activity by MJ-III-65 (NSC 706744), an Indenoisoquinoline Topoisomerase I Poison. Mol Pharmacol. 2005;67:523–530. doi: 10.1124/mol.104.003889. [DOI] [PubMed] [Google Scholar]
- 22.Xiao X, Miao Z-H, Antony S, Pommier Y, Cushman M. Dihydroindenoisoquinolines Funtion as Prodrugs of Indenoisoquinolines. Bioorg Med Chem Lett. 2005;15:2795–2798. doi: 10.1016/j.bmcl.2005.03.101. [DOI] [PubMed] [Google Scholar]
- 23.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, Jr, Stewart L. The Mechanism of Topoisomerase I Poisoning by a Camptothecin Analog. Proc Natl Acad Sci USA. 2002;99:15387–15392. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiao X, Cushman M. An Ab Initio Quantum Mechanics Calculation that Correlates with Ligand Orientation and DNA Cleavage Site Selectivity in Camptothecin-DNA-Topoisomerase I Ternary Cleavage Complexes. J Am Chem Soc. 2005;127:9960–9961. doi: 10.1021/ja042485n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao X, Antony S, Pommier Y, Cushman M. On the Binding of Indeno[1,2- c]isoquinolines in the DNA-Topoisomerase I Cleavage Complex. J Med Chem. 2005;48:3231–3238. doi: 10.1021/jm050017y. [DOI] [PubMed] [Google Scholar]
- 26.Cheng KJ, Rahier NJ, Eisenhauer BM, Gao R, Thomas SJ, Hecht SM. 14-Azacamptothecin: A Potent Water-Soluble Topoisomerase I Poison. J Am Chem Soc. 2005;127:838–839. doi: 10.1021/ja0442769. [DOI] [PubMed] [Google Scholar]
- 27.Xiao X, Cushman M. The Effect of E-ring Modifications in Camptothecin on Topoisomerase I Inhibition: A Quantum Mechanics Treatment. J Org Chem. 2005;70:9584–9587. doi: 10.1021/jo0513360. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Elemental analysis data and 1H NMR spectra of compounds 2b, 2c, 11, and 39. This material is available free of charge at http://pubs.acs.org