

Abstract
We present a simple and robust approach that uses the overall rotational diffusion tensor as a structural constraint for domain positioning in multidomain proteins and protein-protein complexes. This method offers the possibility to use NMR relaxation data for detailed structure characterization of such systems provided the structures of individual domains are available. The proposed approach extends the concept of using long-range information contained in the overall rotational diffusion tensor. In contrast to the existing approaches, we use both the principal axes and principal values of protein’s rotational diffusion tensor to determine not only the orientation but also the relative positioning of the individual domains in a protein. This is achieved by finding the domain arrangement in a molecule that provides the best possible agreement with all components of the overall rotational diffusion tensor derived from experimental data. The accuracy of the proposed approach is demonstrated for two protein systems with known domain arrangement and parameters of the overall tumbling: the HIV-1 protease homodimer and Maltose Binding Protein. The accuracy of the method and its sensitivity to domain positioning is also tested using computer-generated data for three protein complexes, for which the experimental diffusion tensors are not available. In addition, the proposed method is applied here to determine, for the first time, the structure of both open and closed conformations of Lys48-linked di-ubiquitin chain, where domain motions render impossible accurate structure determination by other methods. The proposed method opens new avenues for improving structure characterization of proteins in solution.
Introduction
Structural organization of multidomain proteins and protein-protein complexes is a subject of constant interest in structural biology. Structural characterization of these systems, however, presents a significant challenge, because the existing methods for structure determination, X-ray crystallography and nuclear magnetic resonance (NMR), rest on the assumption of a unique conformation and, therefore, could be inadequate when applied to inherently flexible systems. Indeed, domain motions, naturally occurring in a multidomain protein in solution, are completely restricted in crystals. Moreover, packing forces could result in a positioning of protein domains in a crystal structure that might not represent the physiologically relevant conformation (see examples in refs. 1–4). NMR has an obvious advantage in that molecules can be studied in their native milieu. The challenges for NMR characterization of multidomain systems are due to the following factors: (1) the conventional NMR approaches based on the nuclear Overhauser effect (NOE) are of limited applicability because of the scarcity of close interatomic contacts between the domains, and (2) the interdomain NOEs that are observed are often averaged by domain motions, which could render these data uninterpretable.
The introduction of long-range, orientational constraints derived from molecular alignment5,6 or anisotropy of molecular tumbling7,8 opened new avenues for structure characterization of macromolecular systems. The orientation-sensitive NMR measurements (spin relaxation, residual dipolar couplings) are uniquely suited for providing structural information about domain organization within a protein, due to the availability of multiple reporter groups (e.g. N-H bonds) with well-defined orientation within each domain. Thus, the concept of orienting domains in a protein based on orientation of the principal axes of diffusion or alignment tensors of the whole molecule “reported” by the individual domains, proposed in refs. 1, 9 and schematically illustrated in Fig. 1, proved indispensable for structure characterization of multidomain proteins. Provided the structures of individual domains in the monomeric state are available and no significant backbone perturbations occur in the context of the multidomain system, this approach allows proper orientation of the domains in the molecule by a simple rigid-body rotation. Demonstrated examples range from determining interdomain orientation in proteins1,3,4,9–15 to monitoring conformational changes in these systems induced by ligand binding1,16 or pH conditions.12,14,15 Despite these advances in determining interdomain orientation in a protein, accurate domain positioning remains a significant challenge, due to the deficiency of the information on the interdomain contacts. This problem can be circumvented by docking approaches17–20 combining information on interdomain orientation with the interface mapping; the obvious limitation of these methods when applied to multidomain systems is that they imply the existence of a single conformation. Moreover, docking methods are based on surface complementarity and, naturally, have limited applicability when the contact surfaces change in the course of domain movement. It has also been proposed to complement NMR with small-angle scattering data (X-rays or neutrons) in order to address the domain positioning problem.21,22
Figure 1.

Schematic illustration of the proposed concept of domain positioning based on the rotational diffusion tensor. Provided the structures of the individual domains are available, the interdomain orientation is determined first, and domains are oriented by a rigid-body rotation that aligns the corresponding principal axes (shown as sticks) of the overall diffusion tensor of the complex “reported” by the individual domains, as detailed elsewhere.1,2,10,14 This procedure, however, does not define the relative domain positioning in the molecule. For example, although the interdomain orientation is the same for the domain arrangements (a) through (d), only in (a) is the overall rotational diffusion tensor (red ellipse) consistent with both the magnitude and orientation of the experimental diffusion tensor. In these drawings a dashed ellipse schematically represents the experimentally determined diffusion tensor, whereas a solid red ellipse represents the actual diffusion tensor for a given protein shape/structure. Because the NMR relaxation data sense not only the rate of tumbling but also the orientation of the rotation axes relative to each domain, the relaxation-based approach should be self-sufficient for proper positioning of the domains within a molecule.
The overall tumbling properties of a molecule in solution reflect its size and shape, and therefore contain important information about the domain arrangement within the molecule. Recently developed NMR methods allow accurate measurement of rotational diffusion tensors of proteins.23,24 In addition, computational approaches are now available for reliable prediction of these tensors directly from protein structures.25,26 As mentioned above, the existing structural approaches based on diffusion or alignment tensors so far have been focused on determining interdomain orientation. The interdomain distances and relative domain positioning with respect to each other have remained undetermined. This information, critical for complete structure characterization of multidomain systems, is encoded both in the principal values and in the orientation of the overall rotational diffusion tensor. However, it has not been utilized thus far.
Here we extend the concept of using long-range information contained in the rotational diffusion tensor beyond the existing approaches, in order not only to determine the interdomain orientation but also to properly position individual domains relative to each other within a protein or protein complex. We present a simple and robust method of arranging domains in a multidomain system based on its overall rotational diffusion tensor. Applications of this method to various proteins demonstrate that, given the structures of individual protein domains, the proposed approach provides a complete characterization of domain organization (orientation and positioning) of a protein, thus fully utilizing the informational content of the overall diffusion tensor.
Methods
In the method proposed here, individual domains are positioned with respect to each other such that the resulting rotational diffusion tensor provides the best possible match to the experimentally obtained diffusion tensor, as schematically illustrated in Fig. 1. For theoretical calculation of the overall diffusion tensor for a given protein structure (domain arrangement) we use an ellipsoidal representation of the protein shape, based on the principal component analysis (PCA) of solvent-accessible protein surface, implemented in our computer program ELM.26 The diffusion tensor of such an ellipsoid is then computed using exact equations.27,28 Comparison with experimental data (NMR, fluorescence) shows that this method provides good accuracy in predicting protein rotational diffusion tensors and is sufficiently fast to be implemented in an iterative search program.26 This algorithm is implemented in an in-house Matlab program that, for a given structure, compares all components of the overall diffusion tensor, Dcalc, calculated using PCA, with those of the experimentally measured diffusion tensor, Dexp, and searches for the domain arrangement that minimizes the value of the target function:
| (1) |
It should be noted that such a criterion simultaneously matches both the principal values and principle axes of Dcalc and Dexp, because the rates and the preferred axes of molecular tumbling both depend on the domain positioning within a molecule. No additional terms (like, e.g. penalty for protein-protein overlap) are currently included in the target function, Eq. 1, thus the method outlined here should be considered a proof-of-principle rather than a complete structure determination procedure. The current implementation uses simplex method of exhaustive search in a three-dimensional space of Cartesian coordinates describing the relative position of one domain with respect to the other. The ability to determine relative orientation of the domains independently of this procedure1,2,10,14 simplifies the search, as only translational degrees of freedom associated with the relative domain positioning are involved.
To evaluate the accuracy of the proposed method and its sensitivity to domain positioning we applied it here to five protein systems with known domain arrangement. For two of these proteins, HIV-1 protease and maltose binding protein (MBP), the overall rotational diffusion had been characterized experimentally. For HIV-1 protease we used the NMR solution structure published by Yamazaki et al.29 (PDB code 1BVG) and the components of the rotational diffusion tensor (Table 1) determined by Tjandra et al.30 For MBP we used the NMR structure derived by Mueller et al.31 (PDB code 1EZP). The orientation of MBP’s rotational diffusion tensor with respect to the molecular reference frame of the protein was not available from the published 15N relaxation study.3 Thus, to obtain a complete overall diffusion tensor of MBP we reanalyzed raw NMR relaxation data from that paper3 (see Supporting Information).
Table 1.
Comparison of the diffusion tensors derived from experimental data (“exp.”) and calculated for the resulting structures (“calc.”). Shown are the principal components of the tensors and the angles between the corresponding principal axes, as indicated.
| Protein | MBP | HIV-1 protease | Ub2, closed | Ub2, open | ||||
|---|---|---|---|---|---|---|---|---|
| exp. | calc. | exp. | calc. | exp. | calc. | exp. | calc. | |
| Dx [107 s−1] | 0.81 | 0.79 | 1.33 | 1.34 | 1.53 | 1.60 | 1.53 | 1.61 |
|
| ||||||||
| Dy [107 s−1] | 0.83 | 0.84 | 1.41 | 1.41 | 1.73 | 1.61 | 1.73 | 1.63 |
|
| ||||||||
| Dz [107 s−1] | 1.08 | 1.08 | 1.85 | 1.87 | 2.20 | 2.28 | 2.20 | 2.27 |
|
| ||||||||
| Angles
| ||||||||
| Xexp to Xcalc | 1.74° | 9.65° | 5.31° | 16.73° | ||||
| Yexp to Ycalc | 1.24° | 9.68° | 11.95° | 27.66° | ||||
| Zexp to Zcalc | 1.31° | 0.86° | 10.83° | 22.01° | ||||
The testing of the proposed approach also included three protein complexes with unknown overall diffusion tensor parameters: the barnase-barstar complex (PDB code 1BRS32); G120R mutant of human growth hormone (hGH) in complex with the extracellular domain of the growth hormone receptor hGHbp (PDB code 1A2233); and a complex of protein Z with an in vitro selected affibody (PDB code 1LP134). The rotational diffusion tensors for these complexes (Supporting Information) were predicted from their atom coordinates using the PCA-based method.26 These computer-generated diffusion tensors were then used to reconstruct the complexes and to explore the values of the target function, Eq. 1, as a function of the relative displacement of the domains from their original position. Thus these three complexes serve as control examples of protein systems where the diffusion tensors are “known exactly”.
Finally, to demonstrate the utility of our method in the case of domain motions, we analyzed Lys48-linked di-ubiquitin system, where mutual domain positioning of ubiquitin domains was not characterized thus far. For the individual ubiquitin (Ub) domains in di-ubiquitin (Ub2) we used NMR solution structure of monomeric Ub determined by Cornilescu at al.35 (PDB code 1D3Z, where we clipped the flexible C-terminus beyond Arg72). As shown earlier12,14 the backbone structure of each Ub domain in Ub2 is practically identical to that of an isolated Ub. The complete rotational diffusion tensor of Ub2 and the mutual domain orientations at pH 6.8, were obtained previously.15
Domain alignment based on the orientations of the principal vectors (axes) of the overall rotational diffusion tensor has already been described in detail earlier.1,2,4,10,14,15 Therefore here we address only the problem of domain positioning. Domains in all proteins analyzed here (except for Ub2 which is a special case discussed below) are considered immobile and their mutual arrangement in the molecule is known. Thus, in this study we keep the mutual domain orientations in the above complexes fixed (as determined in the original structures 29,31–34 or for Ub2 in ref. 15) and adjust only their relative positioning, by a simple translation of one domain with respect to the other in order to minimize the target function.
Ubiquitin units in Ub2 exhibit significant reorientations (on a ~10 ns time scale) between two distinct Ub2 conformations, closed and open, with the former being predominantly populated (90%) at neutral pH.15 As shown in refs. 15 and 36, domain motions in Ub2 can be described by a simple model of interconversion (exchange) between the two states, while the overall rotational diffusion tensor of the protein remains practically the same for both open and closed states. Thus, in the present analysis we simultaneously positioned Ub domains for both open and closed conformations of Ub2, and the corresponding contributions to the target function from the two conformations of the chain were weighted by their occupation probabilities.
It should also be noted that hydration of proteins in aqueous solution has a pronounced effect on their diffusion properties. We have shown previously that a monolayer of water molecules covering the surface of a protein can account reasonably well for the available experimental data on rotational diffusion of various proteins.26 Thus, to model the hydration layer effect we assumed that the surfaces of all proteins in this study are covered with a monolayer of water molecules, i.e. the thickness of the hydration layer in the PCA-based calculations was set to 2.8 Å.26
Results and Discussion
The key idea of the proposed approach is to find the domain arrangement that provides the best possible agreement with the overall rotational diffusion tensor derived from experimental data. This is achieved via an exhaustive search featuring computation of the overall diffusion tensor at each step (see Methods). Until recently, such an approach was impractical because the existing algorithms for prediction of the rotational diffusion tensor from protein structure25 were too slow to be implemented in an efficient search algorithm. The development of fast methods for diffusion tensor prediction,26,37 allowing a more than two orders of magnitude speedup in calculations, made it possible to use protein diffusion tensor as an experimental constraint in protein structure determination.
Application to HIV-1 Protease and Maltose Binding Protein
To demonstrate the accuracy of the proposed method we reconstructed the mutual domain arrangement in two different protein systems, the HIV-1 protease and maltose binding protein, for which the three-dimensional structures and experimental diffusion tensors are available (see Methods). HIV-1 protease is a natural homodimer complex. MBP is a single-chain protein that folds into the N-terminal and C-terminal domains comprising residues 6–109 and 264–309 (N-terminal part) and 114–258 and 316–370 (C-terminal part). For each of these proteins, we let the search algorithm find the relative positions of their domains that give the best agreement with the experimentally obtained diffusion tensors. As shown in Fig. 2, the proposed method reproduces the domain arrangement in these proteins with remarkable accuracy (see also Supporting Information). The only difference between the original HIV-1 protease structure and the structure obtained by our method is in a small displacement: both HIV-1 domains in the fitted structure are shifted by about 0.5 Å with respect to the original structure (Fig. 2a, b). The root-mean-square deviation (RMSD) of 0.36 Å between the original and best-fit structures is within the precision range of the original ensemble of NMR structures that has a RMSD of 0.6 Å. The principal values and the orientation of the principal axes frame of the overall diffusion tensor are both reproduced with very high accuracy (Table 1).
Figure 2.
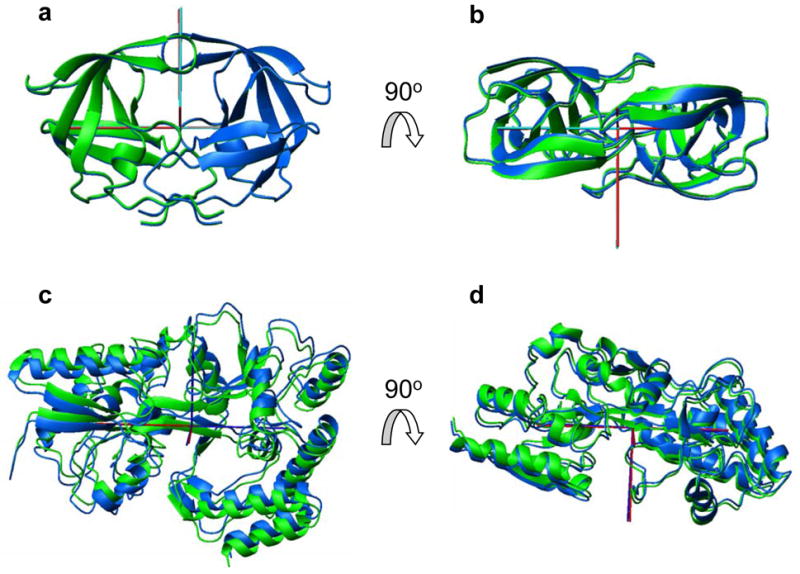
Comparison of the structures of (a, b) HIV-1 protease and (c, d) maltose binding protein determined using the proposed method with their NOE-based NMR structures. Shown is the backbone of the original (green) and fitted (blue) structures. To emphasize the relative shift of the domains, these structures are positioned here such that their centers coincide and the relative orientation of the diffusion tensors axes is as obtained from the analysis. Their superimposition minimizing the RMSD between the original and fitted structures is shown in Supporting Information. Red rods represent the principal axes of the experimentally obtained diffusion tensor, cyan rods correspond to the principal axes of the diffusion tensors calculated for the resulting structures. All molecular drawings in this paper were made using MolMol.46
For MBP, the shift between the original and fitted domain positions (Fig. 1c, d) is larger (about 2 Å) although the agreement between the principal values of the experimental and back-calculated rotational diffusion tensors is comparable to that for HIV-1 protease, and even significantly better for the orientations of the diffusion tensor axes (see Table 1). Nevertheless, the RMSD of 1.3 Å between the original and fitted MBP structures is still within the experimental precision of the ensemble of NMR structures, which has the RMSD of 2.3 Å. The greater discrepancy in domain positioning for MBP can be due to several factors. First, the experimental MBP structure has greater structural noise compared to HIV-1 protease. Second, MBP is a 370 residue protein that is about twice as big as the HIV-1 homodimer (2 × 99 residues). Thus, larger absolute shifts (δR) in domain positions for MBP could result in smaller relative changes, δD/D, in the overall rotational diffusion tensor (recall that D ∞ R−3, where R is the “size” of the protein, hence δD/D ∞ δR/R). Last but not least, these results could reflect differences in the accuracy and precision of the experimentally determined rotational diffusion tensors for the two proteins. In fact, the diffusion tensors derived from NMR relaxation data are prone to errors originating from the experimental noise as well as from the assumptions made in the data analysis.14,24,38 For instance, the derivation of MBP’s diffusion tensor assumed a single rigid structure of the protein, which could be an oversimplification if domain motions are present.
In order to test the performance of our method in the ideal case when the diffusion tensor components are known “exactly”, we repeated the same procedure, this time using the diffusion tensor predicted based on the original structure (instead of the experimental tensor) to guide the assembly of the corresponding proteins. In addition to HIV-1 protease and MBP, we applied this procedure to three protein complexes (see Methods and Supporting Information), for which the experimental diffusion tensors were not available. In all these “ideal” cases, the resulting fitted structures were in excellent agreement with the original structures, with the RMSD values below 0.01 Å (Supporting Information). These results clearly demonstrate the accuracy of the domain positioning approach based on the “true” rotational diffusion tensor.
Sensitivity to domain positioning and convergence
The values of the target function (χ2, Eq. 1) for HIV-1 protease and MBP, plotted as a function of domain’s displacement (Fig. 3a and Fig. 3b, respectively) clearly demonstrate a well-pronounced single minimum corresponding to the optimal domain’s position, which thus justifies the obtained domain arrangement and the stability of the fitting procedure. This conclusion is further supported by similar χ2 profiles obtained for the other three protein complexes (see Methods and Supporting Information). In all the cases studied here, displacing one of the domains within a cube with the side of 20 Å centered at the original domain’s position, resulted in the convergence to the original structure. Thus, even adopting a very conservative point of view, we can conclude that the proposed method, based on the target function in Eq. 1, ensures convergence to a single domain arrangement even when the initial domain displacement from the optimal domain arrangement is as big as ±10 Å (an interval comparable to the size of a typical protein domain).
Figure 3.
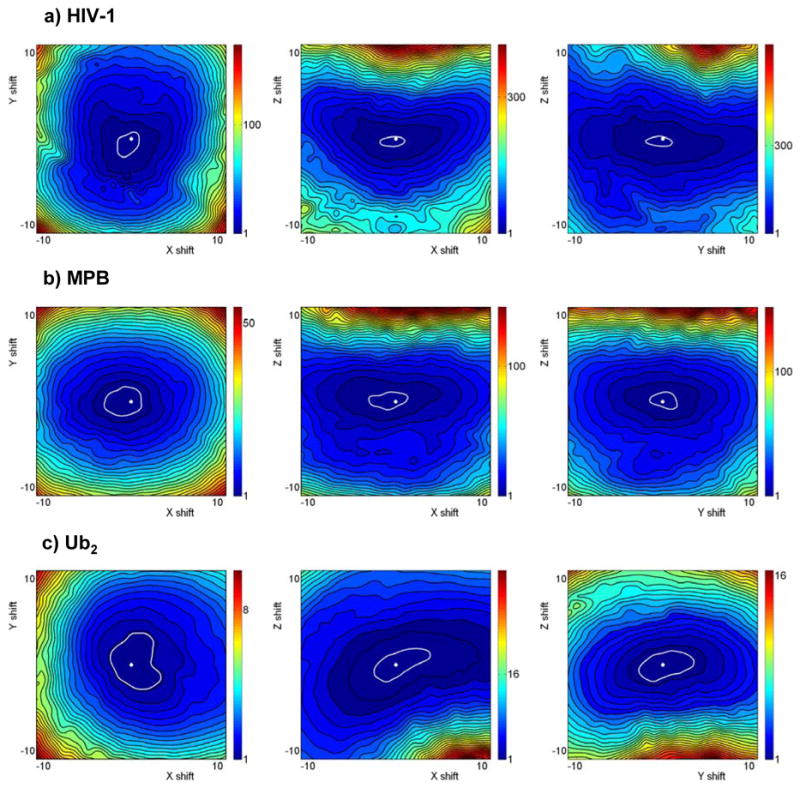
The dependence of the normalized target function, , on domain displacements (in Å) along the principal axes of the experimentally determined diffusion tensors: (a) for the HIV-1 protease, (b) for MBP, and (c) for Ub2 at pH 6.8. White dots in the center of each panel mark the location of the χ2-minimum corresponding to the fitted domain positions. White perimeters mark 68% confidence intervals obtained by the chi;2 boundary method.47 The estimated confidence intervals (in Å) along the X, Y, Z-axes, respectively, are for HIV-1: [−1.8, 1], [−2, 1], [−1, 0.3]; for MBP: [−3.1, 1.3], [−1.7, 1.7], [−1, 1.1]; and for Ub2: [−2.5, 4], [−3.9, 3.7], [−2, 1.9].
Comparing the dependence of the target function, χ2, on domain displacements along the principal axes of the diffusion tensor (Fig. 3), it is evident that the sharpest minima correspond to shifts along the Z-axis of the tensor, while noticeably broader minima are associated with domain shifts along the other two axes, X and Y. To quantify this observation, in all examples shown in Fig. 3 the estimated confidence intervals in the Z-direction are approximately a factor of 2 narrower than those in the X and Y directions; the latter being comparable with each other (see caption to Fig. 3). Note that the principal components of the diffusion tensor are ordered such that Dx≤Dy≤Dz. A shift of one of the domains along the Z-axis, i.e. in the direction of the largest component of the tensor, causes elongation or shortening in the shape of the molecule, hence primarily affects the anisotropy, A=2Dz/(Dx+Dy), of the tensor, whereas shifts in the perpendicular directions (X or Y) will primarily alter its rhombicity, R=3A(Dy−Dx)/[2Dz(A−1)], which reflects deviations from the axial symmetry of the molecule. The sharper χ2-minimum in the Z-direction (Fig. 3) suggests that the anisotropy of the overall diffusion tensor is more sensitive to domain positioning than its rhombicity. This observation is in agreement with our computational analysis26 of a large set of monomeric proteins, which suggests that the anisotropies of diffusion tensors are usually better defined than their rhombicities.
Di-ubiquitin chain structure
The results presented above encouraged us to apply the proposed method to Lys48-linked di-ubiquitin, where the actual structures of the conformers were unknown. At neutral conditions, Ub2 exists in dynamic equilibrium between two conformations, referred to here as “closed” and “open”, which differ in their occupation probabilities (90% and 10%, respectively) and the relative orientations of Ub domains.15 Based on the spectroscopic data,12 the closed conformation of Ub2 is characterized by a well-defined Ub/Ub interface formed by the hydrophobic surfaces (residues Leu8, Ile44, Val70) of both ubiquitins, while no such interface was detected in the open conformation. In a previous study we determined the relative orientations of Ub domains in both Ub2 conformers based on their orientation with respect to the principal axes frame of the overall diffusion tensor.15 We have demonstrated that in the case of Ub2 the overall tumbling and domain reorientations can be considered statistically independent, to a first approximation, and thus deconvoluted from each other. This is possible because the overall shape of the molecule and hence the overall diffusion tensor remain practically the same for both open and closed states of Ub2. That analysis however provided no information about relative positions of the domains with respect to each other. The method proposed here allows us to properly position Ub domains by matching the experimentally determined overall rotational diffusion tensor. Fig 3c shows the profile of χ2 as a function of the relative domain positioning in Ub2. The resulting structures are shown in Fig. 4a, b, and the comparison between the diffusion tensors derived from the experimental data and calculated for the fitted structures is presented in Table 1. It should be mentioned that the difference between the experimental and best-fit tensors for Ub2 is somewhat bigger than for the HIV-1 protease and MBP control examples. This reflects a reduced experimental precision in the diffusion tensor of Ub2, due to several reasons. First, in order to avoid self-association of Ub2 the experiments were performed at low protein concentrations (250 μM),12 which could have affected the precision of the experimental relaxation data. Second, Ub2 exhibits large-amplitude domain reorientations on a time scale comparable to the overall tumbling. Thus, despite the separability of the corresponding correlation functions,15,36 the contributions from the overall and domain motions to the measured relaxation rates are convoluted, which inevitably reduces the precision in the derived diffusion tensor. Moreover, due to its low occupation probability (~10%) the open conformation of Ub2 is less well sampled than the closed conformation. The reduced precision and accuracy of interdomain orientation in the open conformation would then explain the bigger angles between the principal axes of the experimental and best-fit diffusion tensors for this conformation (Table 1). It should be mentioned here that the Ub2 structures in Fig. 4a, b represent an averaged conformation of the chain in each particular state, because any smaller-amplitude interdomain motion present in each of these states would inevitably be averaged out as a result of an oversimplification introduced by the two-state approximation made in the relaxation data analysis.36
Figure 4.
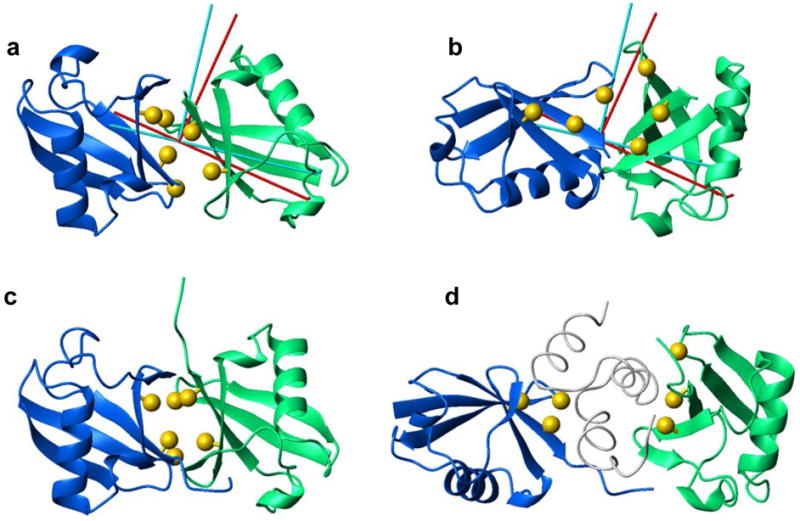
Structures of the (a) closed and (b) open conformations of Lys48-linked Ub2 at pH6.8 obtained using the proposed approach and their comparison with the existing structural data: (c) the crystal structure 42 of Ub2 at pH 4.5 and (d) the solution structure of the Ub2/UBA complex 39 at pH 6.8 (the UBA domain is shown as a thin grey ribbon). The distal domain is colored blue, the proximal domain is green. The location of the hydrophobic patch residues Leu8, Ile44, and Val70 of both Ub domains is shown in ball-and-stick representation (Cβ atoms, colored gold). In panels (a) and (b), the rods represent the principal axes of the experimentally determined diffusion tensor (red) and the diffusion tensor calculated for the resulting structures (cyan). Flexible C-terminal residues 73–76 are not shown in (a), (b). Note that no constraints related to direct interdomain interaction were included in the calculations shown here.
Verification of di-ubiquitin structures
Unlike the control examples considered above (including HIV-1 protease and MBP), the actual structure of Ub2 in solution is unknown. However, several lines of evidence suggest that our results are in agreement with the existing experimental data. First, the structure of the closed conformation (Fig. 3a) agrees with the chemical shift perturbation data 12 (not included in the calculations here) indicating that residues Leu8, Ile44, and Val70 form the interdomain interface in Ub2. Mutations in these residues have been shown to significantly weaken the closed conformation of Ub2.39 Second, the values of the radius of gyration predicted for the Ub2 structures in Fig. 4a, b using CRYSOL40 (17.3–17.9 Å and 17.0–17.6 Å for the closed and open conformers, respectively) are in excellent agreement with the value of 17.4 ± 0.8 Å experimentally determined by small-angle X-ray scattering.41 Third, the derived structures are also in agreement with the paramagnetic relaxation enhancements observed in the proximal Ub domain as a result of site-directed paramagnetic spin labeling of the distal domain.14,15 The reconstructed position of the unpaired electron of the spin label (see ref. 15 for details), based on the closed conformation, is at a distance of 8.2 Å from the Cα atom of residue 48 of the distal Ub, in good agreement with its expected location (Supporting Information). Last but not least, in both Ub2 structures, the flexible C-terminus of the distal Ub (not shown in Fig. 4a, b) is positioned sufficiently close to Lys48 of the proximal Ub in order to form the isopeptide bond between the two Ub domains in Ub2 (note that no constraints were included in the calculation to reflect the presence of the Ub-Ub linker).
It is also instructive to compare the Ub2 structures obtained here with those observed under different conditions: the crystal structure of Ub2 (PDB code 1AAR42) and the Ub2 complex with the C-terminal UBA domain of hHR23a (PDB code 1ZO639). As one can see from comparison of Figs. 4a, b with Figs. 4c, d, these latter structures resemble the closed and open conformations of Ub2 derived by our method. Indeed, a direct superimposition (not shown) gives a fair agreement between the closed conformation of Ub2 in solution obtained here and the Ub2 crystal structure, with the backbone RMSD of 2.4 Å for the elements of secondary structure. Most important for the proposed method is that our calculation placed the two Ub units at a distance very similar to that in the crystal structure, with the centers of mass of the ubiquitins being 23.0 Å apart in Fig. 4a and 23.4 Å apart in Fig. 4c. The difference in domain orientation between the two structures can be represented as a 30° rotation of one of the domains with respect to the other (or 10° rotation for the distal and 21° for the proximal), which is within the precision of the interdomain orientation in the closed conformation.15 It should be pointed out that Ub2 is inherently flexible, and its conformation in solution is pH-dependent.12,15 Therefore, the closed conformation of Ub2 at pH6.8 in solution is not expected to match exactly the structure of the chain in crystals grown at a different pH (pH4.5). The fact that no closed conformation has been observed in solution at pH4.512 suggests that the crystal structure of Ub2 could be a result of crystal packing forces. Taking this into consideration together with the fact that no information about interdomain contacts was included in our calculation, the observed agreement between the two structures is remarkable.
A comparison with the structure of the Ub2/UBA complex (Fig. 4d) reveals structural/mechanistic details of the conformational changes in Ub2 accompanying UBA binding. In particular, the UBA insertion shifted Ub units away from each other compared to their equilibrium positions in the unbound Ub2: the distance between the centers of the ubiquitins increased from 22.3 Å in the open conformation (Fig. 4b) or 23.0 Å in the closed (Fig. 4a) to 35.4 Å in the complex (Fig. 4d). In addition, UBA binding to the open Ub2 conformation is accompanied by a 43° rotation of the distal and 30° of the proximal domain, the total interdomain reorientation angle is 70°. The corresponding UBA-induced reorientations in the closed conformation are 24° for the distal Ub and 46° for the proximal, or 69° total. To further characterize the conformational transition involved in UBA binding, we analyzed these structures using DynDom.43 The analysis indicates that domain rearrangement in the closed conformation of Ub2 (Fig. 4a) upon UBA binding can also be visualized as a combination of a 5.4 Å translation and a 68° rotation of one of the ubiquitins (e.g. proximal) about an axis which is parallel to the Ub/Ub interface (99.7% a closure axis) and goes through the Ub-Ub linker (K48 in the proximal domain), acting as a hinge in the process of interface opening.
The utility of rotational diffusion tensor as a structural constraint
The results presented above demonstrate that the overall rotational diffusion tensor can be used for structure determination of multidomain proteins and protein complexes. The uniqueness of the approach proposed here is based on the diffusion tensor’s sensitivity to the overall shape of the protein and on the availability of a large number of structurally well defined reporter groups within each domain. Because spin relaxation senses both the axes of molecular tumbling and the corresponding rates, the relaxation-based approach could be self-sufficient for proper orientation and positioning of the domains within a molecule. For example, even in the case of spherically-shaped individual domains, when the structures (a), (b), and (d) in Fig. 1 are all characterized by the same overall tumbling time (and the principal values of the diffusion tensor), their rotational diffusion tensors are distinct, when expressed in the same coordinate frame. This is due to the fact that the NMR relaxation data report not only on the rate of tumbling but also on the orientation of the rotation axes relative to each domain.
The approach described here assumes that (1) the structure of the individual domains is known (e.g., is essentially the same as of the isolated domains) and (2) that the domains tumble together as a single moiety and not as completely independent “beads on a flexible string”, i.e. the description of the system with the common overall rotational diffusion tensor is physically meaningful. The validity of these assumptions for a particular multidomain system requires verification, as discussed in details elsewhere 2,14 It is worth pointing out in this regard, that, despite the large-amplitude opening/closing interdomain dynamics in Ub2, to a good approximation the two domains do tumble and orient together, as inferred from 15N relaxation data and residual dipolar couplings 12.
The time scale of interdomain motion is an important factor to be considered when applying the proposed method to flexible systems. If this motion is much faster than the overall tumbling, the latter will be characterized by an averaged (over all available conformations) diffusion tensor, and the information on the relative interdomain orientations will be averaged, too. Therefore Dcalc in Eq. 1 should represent a common diffusion tensor averaged over the diffusion tensors calculated for different interconverting conformations. In the opposite case, when the exchange is comparable to or slower than the overall tumbling (as in the case of Ub2 considered here), the diffusion tensor is not averaged, and it makes sense to characterize each conformation by its diffusion tensor. Thus one has to fit the calculated diffusion tensor for each conformation to its experimental diffusion tensor (available, for example, from relaxation data analysis), and this procedure is expected to yield meaningful structural information about the interconverting states.
The usefulness of the diffusion tensor as a structural constraint could be placed in a wider context, beyond its application to multidomain systems demonstrated here. Because of the sensitivity to the overall size and shape of the protein, the diffusion tensor-based “long-distance” constraints can also be used to improve general structure characterization for monomeric proteins. As demonstrated in ref. 8, the orientation dependence of 15N relaxation rates can be used as orientational constraints for structure refinement of anisotropically tumbling monomeric proteins. The ability to evaluate the diffusion tensor at virtually every step of protein structure calculation26 now opens the possibility to use both the orientation and the principal values of the tensor in order to drive the resulting structure to closely match the actual size and shape of the protein. Combined with the site-specific relaxation data, this could allow more accurate orientation of the individual groups with respect to the overall shape of the protein. In addition, the inclusion of the diffusion tensor as a constraint could help improve the compactness of NMR-derived protein structures,44 which is currently achieved by including a radius of gyration-based term in structure refinement. Unlike the radius of gyration, which is a scalar shape-nonspecific measure of the global size of the molecule, the diffusion tensor contains structural information specific to both the size and shape of a protein.
Conclusions
Here we presented a novel approach to structure characterization of multidomain systems based on structural information encoded in the rotational diffusion tensor. The results demonstrate that the full informational content of the overall rotational diffusion tensor can be used for structure determination of multidomain proteins and protein complexes. This includes not only the orientation of the domains but also their relative positioning within the molecule. The feasibility of this approach is demonstrated here for five protein systems, where our method reproduced their structures with very high accuracy. Detailed testing of the proposed method shows that it ensures convergence of the fitting procedure to a single domain arrangement in those situations when the initial uncertainty in domain positioning is less than or comparable to domain dimensions.
It should be emphasized that the proposed method provides a unique possibility for structure characterization of protein systems with significant domain mobility, which might not be amenable to other, more conventional structural methods, like X-ray crystallography, NOE- and RDC-based NMR methods, and docking approaches. In fact, the application of this method to Lys48-linked di-ubiquitin allowed us to determine, for the first time, the structure of the open and closed conformations of this chain in solution, and obtain a detailed picture of conformational changes in Ub2 induced by ligand binding. Thus, the proposed approach could become the method of choice for structure characterization of inherently flexible systems, like multidomain proteins and weakly bound protein complexes.
The current implementation of the method merely matches the experimental and predicted diffusion tensors and, therefore, the results presented here should be considered as a proof-of-principle. A more accurate, high-resolution structure calculation would require combining diffusion tensor information with other structural constraints (e.g. from NOEs, RDCs, contact surface mapping etc), as well as the proper force field potentials accounting for the van der Waals, electrostatic, and other interactions. This can be achieved by incorporating our approach into the existing structure determination and docking protocols, which should be relatively straightforward and will be our future goal.
The proposed concept could, in principle, be extended to domain positioning based on residual dipolar couplings resulting from molecular alignment. Indeed, in the case of steric forces, the molecular alignment reflects the shape of the molecule, and therefore should be sensitive to the relative positioning (not only orientation) of the domains. Computational tools for predicting molecular alignment based on the structure have been developed.45 The situation is complicated here by the tracelessness of the alignment (Saupe) tensor, which introduces an arbitrary scaling factor into the experimentally determined principal values of the tensor. It should be possible, however, to use ratios (free of this scaling factor) of the principal components of the alignment tensors in a similar way to that described in this paper.
Supplementary Material
Parameters of the overall rotational diffusion tensor obtained for MBP from the experimental data; a figure depicting superimposition of the original and fitted structures of HIV-1 protease and MBP; a figure demonstrating the dependence of the target function on domain arrangement for 1BRS, 1A22, and 1LP1 structures; a table listing the diffusion tensor parameters predicted for these structures accompanied by a figure showing the orientation of the corresponding diffusion tensor axes; and a figure presenting validation of the derived structure of the closed conformation of Ub2 using site-specific spin labeling. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
Supported by NIH grant GM065334 to DF. We thank Prof. Nikolai Skrynnikov for kindly providing the experimental NMR relaxation data for MBP and Dr. Jennifer B. Hall for critical reading of the manuscript. Atom coordinates for the Ub2 conformations have been deposited with the Protein Data Bank (ID codes: 2PEA, 2PE9). The software used in this study will be available from the authors upon request.
References
- 1.Fushman D, Xu R, Cowburn D. Biochemistry. 1999;38:10225–10230. doi: 10.1021/bi990897g. [DOI] [PubMed] [Google Scholar]
- 2.Fushman D, Cowburn D. In: Protein NMR for the Millenium (Biological Magnetic Resonance Vol 20) Krishna NRLB, editor. Kluwer; 2002. pp. 53–78. [Google Scholar]
- 3.Skrynnikov N, Goto N, Yang D, Choy W, Tolman J, Mueller G, Kay L. J Mol Biol. 2000;295:1265–1273. doi: 10.1006/jmbi.1999.3430. [DOI] [PubMed] [Google Scholar]
- 4.Hwang PM, Skrynnikov NR, Kay LE. J Biomol NMR. 2001;20:83–88. doi: 10.1023/a:1011226512421. [DOI] [PubMed] [Google Scholar]
- 5.Tolman JR, Flanagan JM, Kennedy MA, Prestegard JH. Proc Natl Acad Sci U S A. 1995;92:9279–9283. doi: 10.1073/pnas.92.20.9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tjandra N, Bax A. Science. 1997;278:1111–1114. doi: 10.1126/science.278.5340.1111. [DOI] [PubMed] [Google Scholar]
- 7.Bruschweiler R, Liao X, Wright PE. Science. 1995;268:886–889. doi: 10.1126/science.7754375. [DOI] [PubMed] [Google Scholar]
- 8.Tjandra N, Garrett DS, Gronenborn AM, Bax A, Clore GM. Nat Struct Biol. 1997;4:443–449. doi: 10.1038/nsb0697-443. [DOI] [PubMed] [Google Scholar]
- 9.Fischer MWF, Losonczi JA, Weaver LJ, Prestegard JH. Biochemistry. 1999;38:9013–9022. doi: 10.1021/bi9905213. [DOI] [PubMed] [Google Scholar]
- 10.Ghose R, Fushman D, Cowburn D. J Magn Reson. 2001;149:214–217. doi: 10.1006/jmre.2001.2295. [DOI] [PubMed] [Google Scholar]
- 11.Clore GM, Bewley CA. J Magn Reson. 2002;154:329–335. doi: 10.1006/jmre.2001.2489. [DOI] [PubMed] [Google Scholar]
- 12.Varadan R, Walker O, Pickart C, Fushman D. J Mol Biol. 2002;324:637–647. doi: 10.1016/s0022-2836(02)01198-1. [DOI] [PubMed] [Google Scholar]
- 13.Varadan R, Assfalg M, Haririnia A, Raasi S, Pickart C, Fushman D. J Biol Chem. 2004;279:7055–7063. doi: 10.1074/jbc.M309184200. [DOI] [PubMed] [Google Scholar]
- 14.Fushman D, Varadan R, Assfalg M, Walker O. Progress NMR Spectroscopy. 2004;44:189–214. [Google Scholar]
- 15.Ryabov Y, Fushman D. Proteins. 2006;63:787–796. doi: 10.1002/prot.20917. [DOI] [PubMed] [Google Scholar]
- 16.Evenas J, Tugarinov V, Skrynnikov NR, Goto NK, Muhandiram R, Kay LE. J Mol Biol. 2001;309:961–974. doi: 10.1006/jmbi.2001.4695. [DOI] [PubMed] [Google Scholar]
- 17.Dominguez C, Boelens R, Bonvin AM. J Am Chem Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 18.Clore GM, Schwieters CD. J Am Chem Soc. 2003;125:2902–2912. doi: 10.1021/ja028893d. [DOI] [PubMed] [Google Scholar]
- 19.van Dijk ADJ, Fushman D, Bonvin AM. Proteins. 2005;60:367–381. doi: 10.1002/prot.20476. [DOI] [PubMed] [Google Scholar]
- 20.van Dijk AD, Kaptein R, Boelens R, Bonvin AM. J Biomol NMR. 2006;34:237–244. doi: 10.1007/s10858-006-0024-8. [DOI] [PubMed] [Google Scholar]
- 21.Mattinen ML, Paakkonen K, Ikonen T, Craven J, Drakenberg T, Serimaa R, Waltho J, Annila A. Biophys J. 2002;83:1177–1183. doi: 10.1016/S0006-3495(02)75241-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grishaev A, Wu J, Trewhella J, Bax A. J Am Chem Soc. 2005;127:16621–16628. doi: 10.1021/ja054342m. [DOI] [PubMed] [Google Scholar]
- 23.Dosset P, Hus JC, Blackledge M, Marion D. J Biomol NMR. 2000;16:23–28. doi: 10.1023/a:1008305808620. [DOI] [PubMed] [Google Scholar]
- 24.Walker O, Varadan R, Fushman D. J Magn Reson. 2004;168:336–345. doi: 10.1016/j.jmr.2004.03.019. [DOI] [PubMed] [Google Scholar]
- 25.Garcia de la Torre J, Huertas ML, Carrasco B. J Magn Reson. 2000;B147:138–146. doi: 10.1006/jmre.2000.2170. [DOI] [PubMed] [Google Scholar]
- 26.Ryabov YE, Geraghty C, Varshney A, Fushman D. J Am Chem Soc. 2006;128:15432–15444. doi: 10.1021/ja062715t. [DOI] [PubMed] [Google Scholar]
- 27.Perrin F. J Phys Radium. 1934;5:497–511. [Google Scholar]
- 28.Perrin F. J Phys Radium. 1936;7:1–11. [Google Scholar]
- 29.Yamazaki T, Hinck AP, Wang YX, Nicholson LK, Torchia DA, Wingfield P, Stahl SJ, Kaufman JD, Chang CH, Domaille PJ, Lam PY. Protein Sci. 1996;5:495–506. doi: 10.1002/pro.5560050311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tjandra N, Wingfield P, Stahl S, Bax A. J Biomol NMR. 1996;8:273–284. doi: 10.1007/BF00410326. [DOI] [PubMed] [Google Scholar]
- 31.Mueller GA, Choy WY, Yang D, Forman-Kay JD, Venters RA, Kay LE. J Mol Biol. 2000;300:197–212. doi: 10.1006/jmbi.2000.3842. [DOI] [PubMed] [Google Scholar]
- 32.Buckle AM, Schreiber G, Fersht AR. Biochemistry. 1994;33:8878–8889. doi: 10.1021/bi00196a004. [DOI] [PubMed] [Google Scholar]
- 33.Clackson T, Ultsch MH, Wells JA, de Vos AM. J Mol Biol. 1998;277:1111–1128. doi: 10.1006/jmbi.1998.1669. [DOI] [PubMed] [Google Scholar]
- 34.Hogbom M, Eklund M, Nygren PA, Nordlund P. Proc Natl Acad Sci U S A. 2003;100:3191–3196. doi: 10.1073/pnas.0436100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cornilescu G, Marquardt JL, Ottiger M, Bax A. J Amer Chem Soc. 1998;120:6836–6837. [Google Scholar]
- 36.Ryabov YE, Fushman D. J Am Chem Soc. 2007;129:3315–3327. doi: 10.1021/ja067667r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ortega A, Garcia de la Torre J. J Am Chem Soc. 2005;127:12764–12765. doi: 10.1021/ja053080l. [DOI] [PubMed] [Google Scholar]
- 38.Fushman D, Ghose R, Cowburn D. J Am Chem Soc. 2000;122:10640–10649. [Google Scholar]
- 39.Varadan R, Assfalg M, Raasi S, Pickart C, Fushman D. Mol Cell. 2005;18:687–698. doi: 10.1016/j.molcel.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 40.Svergun DI, Barberato C, Koch MHJ. J Appl Cryst. 1995;28:768–773. [Google Scholar]
- 41.Tenno T, Fujiwara K, Tochio H, Iwai K, Morita EH, Hayashi H, Murata S, Hiroaki H, Sato M, Tanaka K, Shirakawa M. Genes Cells. 2004;9:865–875. doi: 10.1111/j.1365-2443.2004.00780.x. [DOI] [PubMed] [Google Scholar]
- 42.Cook WJ, Jeffrey LC, Carson M, Zhijian C, Pickart CM. J Biol Chem. 1992;267:16467–16471. doi: 10.2210/pdb1aar/pdb. [DOI] [PubMed] [Google Scholar]
- 43.Hayward S, Berendsen HJ. Proteins. 1998;30:144–154. [PubMed] [Google Scholar]
- 44.Kuszewski J, Gronenborn AM, Clore GM. J Am Chem Soc. 1999;121:2337–2338. [Google Scholar]
- 45.Zweckstetter M, Bax A. J Biomol NMR. 2002;23:127–137. doi: 10.1023/a:1016316415261. [DOI] [PubMed] [Google Scholar]
- 46.Koradi R, Billeter M, Wuthrich K. J Mol Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 47.Press WH, Teukolsky SA, Vetterling WT, Flannery BP. Numerical Recipes in C. Cambridge University Press; NY: 1992. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Parameters of the overall rotational diffusion tensor obtained for MBP from the experimental data; a figure depicting superimposition of the original and fitted structures of HIV-1 protease and MBP; a figure demonstrating the dependence of the target function on domain arrangement for 1BRS, 1A22, and 1LP1 structures; a table listing the diffusion tensor parameters predicted for these structures accompanied by a figure showing the orientation of the corresponding diffusion tensor axes; and a figure presenting validation of the derived structure of the closed conformation of Ub2 using site-specific spin labeling. This material is available free of charge via the Internet at http://pubs.acs.org.