

Abstract
This paper describes the phosphine-catalyzed annulation of methyl allenoate with various aromatic aldehydes to form 6-aryl-4-methoxy-5,6-dihydro-2-pyrones. In this reaction, the addition of an alcohol was necessary to induce dihydropyrone formation, with the optimal agent being methanol. Moreover, the addition of n-butyllithium suppressed the formation of the non-cyclized product, leading to the exclusive isolation of the dihydropyrone. This method provides an efficient, one-step route toward disubstituted dihydropyrones from simple, stable starting materials.
5,6-Dihydro-2-pyrone moieties are present in many biological metabolites, some of which exhibit desirable medicinal properties,1 such as antitumor,1q antifungal,1l anti-HIV,1p anti-hepatitis C,1q,1s and antibiotic1c activity. As a result of their prevalence in medicinally useful compounds, there are many reported syntheses of dihydropyrones, several of which have been used successfully to prepare natural products. One such approach involves standard acid-catalyzed lactonization, following construction of the carbon framework.2 For example, the nucleophilic opening of epoxides by propiolic acid dianions and subsequent alkyne-to-alkene transformation provides substrates suitable for lactonization.3 An alternative approach toward dihydropyrones is the preparation of related six-membered oxygen-containing heterocycles and subsequent functional group manipulation to install the carbonyl group.4 These two methods are, however, quite inefficient because they require the use of several sequential reactions; a single-step, multicomponent coupling protocol would be desirable.5 A more attractive methodology lies in the hetero-Diels–Alder reaction between Brassard’s diene and an aldehyde.6 Although this approach leads to the dihydropyrone core in a single step, reasonable yields have been achieved only when using the expensive Eu(hfc)3 catalyst. In addition, the diene itself requires extremely careful handling, and is unstable during storage. A resin-bound derivative of Brassard’s diene has been reported as being stable for extended periods, but its reactions come with reduced yields and other limitations inherent to solid-bound reagents.7 The various other routes toward 5,6-dihydro-2-pyrone moieties suffer similarly from low efficiencies.8
In addition to exhibiting biologically important functions, 4-alkoxy-5,6-dihydro-2-pyrones are also synthetically useful: they can be efficiently transformed into hydroxypyrazoles,9a reduction of the carbonyl group provides a hydroxylated anomeric carbon for further functionalization,9c,9d and they are converted to disubstituted 2,3-dihydro-4-pyrones upon addition of a nucleophile followed by acid hydrolysis.9e The synthetic utility of 5,6-dihydro-2-pyrones has also been demonstrated in several syntheses of polyether ionophore antibiotics.9b
Considering the many versatile and efficient methodologies that have emerged featuring phosphine-catalyzed annulations of electron-deficient allenes,10 we suspected that it might be possible to synthesize disubstituted dihydropyrones from allenoates. Previously, we reported the preparation of dioxanes and pyrones through nucleophilic phosphine-catalyzed additions of allenoates to aldehydes as effective electrophiles (Figure 1).11
Figure 1.

Divergent pathways for phosphine-catalyzed annulations of aldehydes with 2,3-butadienoates.
Upon the addition of a phosphine to 2,3-butadienoate 1, a phosphonium dienolate having s-cis stereochemistry, 2′, is formed favorably as a result of the stabilizing Coulombic interaction between the partially anionic carbonyl group and the phosphonium center (Scheme 1).12 Dioxanes result when an aldehyde adds to this s-cis-dienolate, followed by the addition of a second molecule of aldehyde and subsequent 6-endo-trig cyclization.11a Use of a more sterically bulky phosphine destabilizes the s-cis-phosphonium dienolate and shifts the equilibrium in favor of the s-trans isomer 2.11b Upon addition of an aldehyde, the intermediate β-phosphonium enoate having an E-alkene geometry, 3, is formed, allowing intramolecular lactonization. The alkoxide released from the allenoate abstracts a proton to form an alcohol and initiate a series of proton transfers that form the pyrone product with regeneration of the phosphine catalyst.11b In a different scenario, if the alkoxide generated upon lactonization adds conjugatively to the β-phosphonium enoate 4, 4-alkoxy-5,6-dihydro-2-pyrones can be formed after β-elimination of the phosphine catalyst. The presence of sterically bulky phosphines, which are necessary for the generation of s-trans-phosphonium dienolates, is believed, however, to impede such Michael addition of the alkoxide. Therefore, we needed an alternative means of influencing the equilibrium and inducing the formation of the s-trans-phosphonium dienolate 2; we hypothesized that adding an external alcohol—to act as an electron donor toward the phosphonium ion—would overcome the Coulombic attraction in the s-cis-isomer. This process would allow a smaller phosphine to act as the catalyst and increase the concentration of the Michael donor.
Scheme 1.

Possible reaction pathways
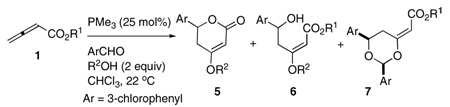
Our initial experiments with alcohol additives were successful, displaying a modest induction of 4-alkoxy-5,6-dihydro-2-pyrone (Table 1).13 The added alcohol presumably provided the necessary disruption of the phosphonium–alkoxide electrostatic interaction in addition to possibly hydrogen bonding to the dienolate alkoxide. Varying the nature of the alcohol allowed us to fine-tune the nucleophilicity of the requisite Michael donor. The addition of isopropanol and ethanol resulted primarily in dioxane products, presumably because the equilibrium favored the s-cis-phosphonium dienolate diastereoisomer (entries 1 and 2). We also observed the non-cyclized product 6 when using ethanol as the additive. The addition of methanol provided the highest total mass recovery and a significant reduction in the yield of the dioxane product, albeit with a corresponding increase in the amount of undesired non-cyclized product 6 (entry 3). Suspecting that the enhanced nucleophilicity of methanol might have been responsible for the formation of the non-cyclized product, we hoped to encourage cyclization by employing commercially available halogenated ethanol derivatives exhibiting increased acidity and diminished nucleophilicity (entries 4–9). Encouragingly, the relatively poorer nucleophilicities of 2-fluoroethanol and 2-chloroethanol provided the desired dihydropyrone product 5 with significantly reduced formation of the non-cyclized product 6 (entries 4 and 5). Although it was gratifying that these alcohols provided no dioxane product 7, the mismatch between the ester and the added alcohols led to a mixture of either MeO or XCH2CH2O groups incorporated in the dihydropyrone product 5. The addition of 2,2-dichloroethanol or 2,2,2-trichloroethanol led to no apparent consumption of the starting material, presumably because of each alcohol’s excessive acidity (entries 6 and 7).14 The use of matched 2-chloroethyl and 2-fluoroethyl alcohols and allenoates provided single dihydropyrones 5 and non-cyclized products 6, but still with significantly lower mass recoveries compared with that obtained using methanol (entries 8 and 9). Therefore, for subsequent experiments we chose to use methanol as the additive to induce dihydropyrone formation, but we needed to find a way to suppress the formation of the non-cyclized product.
Table 1.
Survey of Alcohol Additives for the Reactions of Various Allenoates With 3-Chlorobenzaldehydea
 | ||||
|---|---|---|---|---|
| entry | R1 | R2 | pKa of R2OH in H2Ob | yieldc % (5:6d:7) |
| 1 | iPr | iPr | 16.5 | 54 (16:0:84) |
| 2 | Et | Et | 16.0 | 70 (23:21:55) |
| 3 | Me | Me | 15.54 | 78 (54:39:7) |
| 4 | Me | CH2CH2F | 14.2, 13.92 | 49 (85:15:0)e |
| 5 | Me | CH2CH2Cl | 14.31 | 55 (90:10:0)e |
| 6 | Me | CH2CHCl2 | 12.89 | trace |
| 7 | Me | CH2CCl3 | 12.24, 11.8 | 0 |
| 8 | CH2CH2Cl | CH2CH2Cl | 14.31 | 41 (81:19:0) |
| 9 | CH2CH2F | CH2CH2F | 14.2 | 54 (90:10:0) |
See the Supporting Information for a detailed experimental procedure.
Data from reference 15.
Isolated yields.
The alkene stereochemistry was exclusively E (NOESY NMR spectroscopy).
Both methoxide and the 2-haloethoxide were incorporated as β-substituents (OR2) in 5 and 6.
We suspected that the non-cyclized product 6 resulted from protonation of the benzylic alkoxide 3 and regeneration of the catalyst through Michael addition of the alkoxide without lactonization (Scheme 2).16 Considering that the intermediate alkoxide 3 was necessary for lactonization, we reasoned that protonation of the benzylic alkoxide by methanol might be inhibiting the desired lactonization. We hypothesized that addition of a base, allowing generation of methoxide in situ, would encourage progress along the desired pathway and decrease the production of the non-cyclized product 6.
Scheme 2.

Formation of the dihydropyrones 5 and the noncyclized coupling products 6
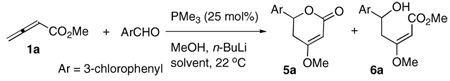
With this concept in mind, we added n-butyllithium to generate lithium methoxide in situ (Table 2). Although we observed no non-cyclized product under these basic conditions, the dramatic reduction in mass recovery necessitated a switch to dichloromethane as solvent (entry 1). An added bonus of switching the solvent was complete suppression of the dioxane product 7 (entry 2). Use of a 1:1 mixture of methanol and lithium methoxide led to the promising generation of the dihydropyrone in 71% yield with no non-cyclized product formed (entry 3). A decrease in the concentration of methanol provided a corresponding decrease in the mass recovery, suggesting that the presence of methanol was critical (entries 4 and 5). Not surprisingly, however, an increase in the concentration of methanol (vs. methoxide) induced the formation of more of the non-cyclized product (entry 6). Decreasing the concentration of n-butyllithium was also detrimental (entry 7). Higher concentrations of methoxide at a constant free methanol concentration (entries 8 and 9) and higher concentrations of 1:1 mixtures of methanol and methoxide (entries 10 and 11) both led to decreasing product formation with increasing additive concentration.
Table 2.
Effect of the Number of Equivalents of MeOH and n-BuLia
 | |||||
|---|---|---|---|---|---|
| entry | solvent | MeOH (eq) | n-BuLi (eq) | yield (%)b | |
| 5a | 6ac | ||||
| 1 | CHCl3 | 2.0 | 1.0 | 37 | - |
| 2 | CH2Cl2 | 2.0 | 0.0 | 51 | 18 |
| 3 | CH2Cl2 | 2.0 | 1.0 | 71 | - |
| 4 | CH2Cl2 | 1.5 | 1.0 | 61 | - |
| 5 | CH2Cl2 | 1.0 | 1.0 | 49 | - |
| 6 | CH2Cl2 | 3.0 | 1.0 | 62 | 11 |
| 7 | CH2Cl2 | 1.25 | 0.25 | 58 | 4 |
| 8 | CH2Cl2 | 3.0 | 2.0 | 64 | - |
| 9 | CH2Cl2 | 4.0 | 3.0 | 52 | |
| 10 | CH2Cl2 | 4.0 | 2.0 | 66 | - |
| 11 | CH2Cl2 | 6.0 | 3.0 | 59 | |
See the Supporting Information for a detailed experimental procedure.
Isolated yields.
Stereochemistry of the alkene was exclusively E (NOESY NMR spectroscopy).
Having determined optimized concentrations of methanol and n-butyllithium, we proceeded to probe the scope of the substrate for the formation of dihydropyrones (Table 3). Benzaldehydes possessing a variety of electron withdrawing substituents provided the desired dihydropyrones in good yields (entries 1–7). Aromatic substitution in the meta position was optimal, as exemplified by the reactions of 3-cyano-, 3-nitro-, and 3-trifluoromethylbenzaldehyde (entries 2, 4, and 6). Substitution at the ortho position, the most sterically hindered site, was also tolerated, as exemplified by the similar yields for the reactions of 4- and 2-trifluoromethylbenzaldehyde (entries 5 and 7). Consistent with the results of previous allene/aldehyde annulations,11 the use of more-electron-rich aromatic aldehydes provided lower yields (entries 8–10). 3-Pyridine carboxaldehyde had similar reactivity, providing the corresponding substituted pyridine in 44% yield.
Table 3.
Syntheses of Disubstituted Dihydropyrones
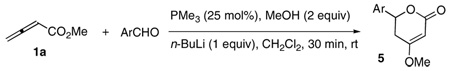 | |||
|---|---|---|---|
| entry | Ar | product | yield (%)a |
| 1 | 4-NCC6H4 | 5b | 68 |
| 2 | 3-NCC6H4 | 5c | 83 |
| 3 | 4-O2NC6H4 | 5d | 40 |
| 4 | 3-O2NC6H4 | 5e | 74 |
| 5 | 4-CF3C6H4 | 5f | 63 |
| 6 | 3-CF3C6H4 | 5g | 74 |
| 7 | 2-CF3C6H4 | 5h | 58 |
| 8 | C6H5 | 5i | 37 |
| 9 | 3-MeC6H4 | 5j | 36 |
| 10 | 3-MeOC6H4 | 5k | 36 |
| 11 | 3-pyridyl | 5l | 44 |
Isolated yields.
In summary, 4-methoxy-6-aryl-5,6-dihydro-2-pyrones can be assembled rapidly in one step from simple, bench-stable starting materials in moderate to good yields. The generation of dihydropyrones through these phosphine-catalyzed annulations of allenoates and aldehydes was facilitated by the addition of alcohols and alkoxides. The presence of an added alcohol allowed preferable formation of the s-trans-phosphonium dienolate and subsequent lactonization after its addition to the aldehyde; the added alkoxide suppressed the formation of a non-cyclized three-component coupling product from the allenoate, aldehyde, and alcohol. We are investigating several applications of this protocol, including intramolecular variants using substrates incorporating aldehyde or alcohol participants, enantioselective processes using chiral phosphines or alcohols, and increasing the product complexity further through the use of α- and/or γ-substituted allenoates.
Supplementary Material
Representative experimental procedures and spectral data for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgment
This study was supported by the NIH (R01GM071779). We thank Dr. Xue-Feng Zhu for performing some initial experiments related to this work.
References
- 1.(a) Page CB, Pinder AR. J. Chem. Soc. 1964:4811. [Google Scholar]; (b) McInnes AG, Yoshida S, Towers GHN. Tetrahedron. 1965;21:2939. [Google Scholar]; (c) Argoudelis AD, Zieserl JF. Tetrahedron Lett. 1966;7:1969. doi: 10.1016/s0040-4039(00)76280-0. [DOI] [PubMed] [Google Scholar]; (d) Cavill GWK, Clark DV, Whitfield FB. Aust. J. Chem. 1968;21:2819. [Google Scholar]; (e) Moss MO, Robinson FV, Wood AB, Paisley HM, Feeney J. Nature. 1968;220:767. doi: 10.1038/220767a0. [DOI] [PubMed] [Google Scholar]; (f) Evans RH, Jr, Ellestad GA, Kunstmann MP. Tetrahedron Lett. 1969;10:1791. doi: 10.1016/s0040-4020(01)82703-4. [DOI] [PubMed] [Google Scholar]; (g) Kirson I, Glotter E, Abraham A, Lavie D. Tetrahedron. 1970;26:2209. [Google Scholar]; (h) Kimura Y, Katagiri K, Tamura S. Tetrahedron Lett. 1971;12:3137. [Google Scholar]; (i) Govindachari TR, Parthasarathy PC. Tetrahedron Lett. 1971;12:3401. [Google Scholar]; (j) Ellestad GA, McGahren WJ, Kunstmann MP. J. Org. Chem. 1972;37:2045. doi: 10.1021/jo00977a044. [DOI] [PubMed] [Google Scholar]; (k) Jewers K, Davis JB, Dougan J, Manchanda AH, Blunden G, Kyi A, Wetchapiana AH. Phytochemistry. 1972;11:2025. [Google Scholar]; (l) Yasui K, Tamura Y, Nakatani T, Kawada K, Ohtani M. J. Org. Chem. 1995;60:7567. [Google Scholar]; (m) Murakami N, Wang W, Aoki M, Tsutsui Y, Sugimoto M, Kobayashi M. Tetrahedron Lett. 1998;39:2349. [Google Scholar]; (n) Boyer FE, Vara Prasad JVN, Domagala JM, Ellsworth EL, Gajda C, Hagen SE, Markoski LJ, Tait BD, Lunney EA, Palovsky A, Ferguson D, Graham N, Holler T, Hupe D, Nouhan C, Tummino PJ, Urumov A, Zeikus E, Zeikus G, Gracheck SJ, Sanders JM, VanderRoest S, Brodfuehrer J, Iyer K, Sinz M, Gulnik SV, Erickson JW. J. Med. Chem. 2000;43:843. doi: 10.1021/jm990281p. [DOI] [PubMed] [Google Scholar]; (o) Fátima Â, Kohn LK, Antônio MA, Carvalho JE, Pilli RA. Bioorg. Med. Chem. 2005;13:2927. doi: 10.1016/j.bmc.2005.02.007. [DOI] [PubMed] [Google Scholar]; (p) Li H, Tatlock J, Linton A, Gonzalez J, Borchardt A, Dragovich P, Jewell T, Prins T, Zhou R, Blazel J, Parge H, Love R, Hickey M, Doan C, Shi S, Duggal R, Lewis C, Fuhrman S. Bioorg. Med. Chem. Lett. 2006;16:4834. doi: 10.1016/j.bmcl.2006.06.065. [DOI] [PubMed] [Google Scholar]
- 2.(a) Spino C, Mayes N, Desfossés H. Tetrahedron Lett. 1996;37:6503. [Google Scholar]; (b) Lokot′ IP, Pashkovskii FS, Lakhvich FA. Chem. Heterocycl. Compd. 2001;37:707. [Google Scholar]
- 3.(a) Carlson RM, Oyler AR. Tetrahedron Lett. 1974;25:2615. [Google Scholar]; (b) Takano S, Kamikubo T, Sugihara T, Ogasawara K. Tetrahedron: Asymmetry. 1992;3:853. [Google Scholar]
- 4.(a) Dujardin G, Rossignol S, Brown E. Synthesis. 1998:763. [Google Scholar]; (b) Harris JM, O’Doherty GA. Tetrahedron Lett. 2000;41:183. [Google Scholar]; (c) Yamashita Y, Saito S, Ishitani H, Kobayashi S. J. Am. Chem. Soc. 2003;125:3793. doi: 10.1021/ja028186k. [DOI] [PubMed] [Google Scholar]
- 5.(a) Midland MM, Graham RS. J. Am. Chem. Soc. 1984;106:4294. [Google Scholar]; (b) Togni A. Organometallics. 1990;9:3106. [Google Scholar]; (c) Midland MM, Koops RW. J. Org. Chem. 1990;55:5058. [Google Scholar]; (d) Du H, Zhao D, Ding K. Chem. Eur. J. 2004;10:5964. doi: 10.1002/chem.200400515. [DOI] [PubMed] [Google Scholar]; (e) Fan Q, Lin L, Liu J, Huang Y, Feng X, Zhang G. Org. Lett. 2004;6:2185. doi: 10.1021/ol049364c. [DOI] [PubMed] [Google Scholar]
- 6.(a) Midland MM, Graham RS. J. Am. Chem. Soc. 1984;106:4294. [Google Scholar]; (b) Togni A. Organometallics. 1990;9:3106. [Google Scholar]; (c) Midland MM, Koops RW. J. Org. Chem. 1990;55:5058. [Google Scholar]; (d) Du H, Zhao D, Ding K. Chem. Eur. J. 2004;10:5964. doi: 10.1002/chem.200400515. [DOI] [PubMed] [Google Scholar]; (e) Fan Q, Lin L, Liu J, Huang Y, Feng X, Zhang G. Org. Lett. 2004;6:2185. doi: 10.1021/ol049364c. [DOI] [PubMed] [Google Scholar]
- 7.Pierres C, George P, van Hijfte L, Ducep J-B, Hibert M, Mann A. Tetrahedron Lett. 2003;44:3645. [Google Scholar]
- 8.(a) Reid EB, Ruby WR. J. Am. Chem. Soc. 1951;73:1054. [Google Scholar]; (b) Dugger RW, Heathcock CH. J. Org. Chem. 1980;45:1181. [Google Scholar]; (c) Fan R, Hudlicky T. Tetrahedron Lett. 1989;30:5533. [Google Scholar]; (d) Schlessinger RH, Gillman KW. Tetrahedron Lett. 1999;40:1257. [Google Scholar]; (e) Audrain H, Jørgensen KA. J. Am. Chem. Soc. 2000;122:11543. [Google Scholar]; (f) Reddy MVR, Brown HC, Ramachandran PV. J. Organomet. Chem. 2001;624:239. [Google Scholar]
- 9.(a) Ayoub MT, Shandala MY, Gussba Bashi GM. J. Chem. Soc. Perkin Trans. 1981;1:697. [Google Scholar]; (b) Faul MM, Huff BE. Chem. Rev. 2000;100:2407. doi: 10.1021/cr940210s. [DOI] [PubMed] [Google Scholar]; (c) Ghosh AK, Wang Y, Kim JT. J. Org. Chem. 2001;66:8973. doi: 10.1021/jo010854h. [DOI] [PubMed] [Google Scholar]; (d) Mulzer J, Öhler E. Chem. Rev. 2003;103:3753. doi: 10.1021/cr940368c. [DOI] [PubMed] [Google Scholar]; (e) Winkler JD, Oh K. Org. Lett. 2005;7:2421. doi: 10.1021/ol050702z. [DOI] [PubMed] [Google Scholar]
- 10.(a) Zhang C, Lu X. J. Org. Chem. 1995;60:2906. [Google Scholar]; (b) Zhu G, Chen Z, Jiang Q, Xiao D, Cao P, Zhang X. J. Am. Chem. Soc. 1997;119:3836. [Google Scholar]; (c) Xu Z, Lu X. Tetrahedron Lett. 1997;38:3461. [Google Scholar]; (d) Xu Z, Lu X. J. Org. Chem. 1998;63:5031. [Google Scholar]; (e) Xu Z, Lu X. Tetrahedron Lett. 1999;40:549. [Google Scholar]; (f) Kumar K, Kapur A, Ishar MPS. Org. Lett. 2000;2:787. doi: 10.1021/ol000007l. [DOI] [PubMed] [Google Scholar]; (g) Kumar K, Kapoor R, Kapur A, Ishar MPS. Org. Lett. 2000;2:2023. doi: 10.1021/ol0000713. [DOI] [PubMed] [Google Scholar]; (h) Ung AT, Schafer K, Lindsay KB, Pyne SG, Amornraksa K, Wouters R, Van der Linden I, Biesmans I, Lesage ASJ, Skelton BW, White AH. J. Org. Chem. 2002;67:227. doi: 10.1021/jo010864i. [DOI] [PubMed] [Google Scholar]; (i) Liu B, Davis R, Joshi B, Reynolds DW. J. Org. Chem. 2002;67:4595. doi: 10.1021/jo016154u. [DOI] [PubMed] [Google Scholar]; (j) Du Y, Lu X, Yu Y. J. Org. Chem. 2002;67:8901. doi: 10.1021/jo026111t. [DOI] [PubMed] [Google Scholar]; (k) Lu C, Lu X. Org. Lett. 2002;4:4677. doi: 10.1021/ol0270733. [DOI] [PubMed] [Google Scholar]; (l) Wang. J-C, Krische MJ. Angew. Chem., Int. Ed. 2003;42:5855. doi: 10.1002/anie.200352218. [DOI] [PubMed] [Google Scholar]; (m) Zhu X-F, Lan J, Kwon O. J. Am. Chem. Soc. 2003;125:4716. doi: 10.1021/ja0344009. [DOI] [PubMed] [Google Scholar]; (n) Du Y, Lu X. J. Org. Chem. 2003;68:6463. doi: 10.1021/jo034281f. [DOI] [PubMed] [Google Scholar]; (o) Kuroda H, Tomita I, Endo T. Org. Lett. 2003;5:129. doi: 10.1021/ol020198n. [DOI] [PubMed] [Google Scholar]; (p) Jung C-K, Wang J-C, Krische MJ. J. Am. Chem. Soc. 2004;126:4118. doi: 10.1021/ja049377l. [DOI] [PubMed] [Google Scholar]; (q) Wurz RP, Fu GC. J. Am. Chem. Soc. 2005;127:12234. doi: 10.1021/ja053277d. [DOI] [PubMed] [Google Scholar]; (r) Zhao G-L, Shi M. Org. Biomol. Chem. 2005;3:3686. doi: 10.1039/b510572b. [DOI] [PubMed] [Google Scholar]; (s) Zhu X-F, Henry CE, Kwon O. Tetrahedron. 2005;61:6276. [Google Scholar]; (t) Tran YS, Kwon O. Org. Lett. 2005;7:4289. doi: 10.1021/ol051799s. [DOI] [PubMed] [Google Scholar]; (u) Wilson JE, Fu GC. Angew. Chem. Int. Ed. 2006;45:1426. doi: 10.1002/anie.200503312. [DOI] [PubMed] [Google Scholar]; (v) Nair V, Biju AT, Mohanan K, Suresh E. Org. Lett. 2006;8:2213. doi: 10.1021/ol0604623. [DOI] [PubMed] [Google Scholar]; (w) Lu X, Lu Z, Zhang X. Tetrahedron. 2006;62:457. [Google Scholar]; (x) Jean L, Marinetti A. Tetrahedron Lett. 2006;47:2141. [Google Scholar]; (y) Virieux D, Guillouzic A-F, Cristau H-J. Tetrahedron. 2006;62:3710. [Google Scholar]; (z) Scherer A, Gladysz JA. Tetrahedron Lett. 2006;47:6335. [Google Scholar]; (aa) Xia Y, Liang Y, Chen Y, Wang M, Jiao L, Huang F, Liu S, Li Y, Yu Z-X. J. Am. Chem. Soc. 2007;129:3470. doi: 10.1021/ja068215h. [DOI] [PubMed] [Google Scholar]; (bb) Mercier E, Fonovic B, Henry CE, Kwon O, Dudding T. Tetrahedron Lett. 2007;48:3617. [Google Scholar]; (cc) Castellano S, Fiji HDG, Kinderman SS, Watanabe M, de Leon P, Tamanoi F, Kwon O. J. Am. Chem. Soc. 2007;129:5843. doi: 10.1021/ja070274n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (dd) Wallace DJ, Sidda RL, Reamer RA. J. Org. Chem. 2007;72:1051. doi: 10.1021/jo062170l. [DOI] [PubMed] [Google Scholar]; (ee) Henry CE, Kwon O. Org. Lett. 2007;9:3069. doi: 10.1021/ol071181d. [DOI] [PubMed] [Google Scholar]; (ff) Cowen BJ, Miller SJ. J. Am. Chem. Soc. 2007;129:10988. doi: 10.1021/ja0734243. [DOI] [PubMed] [Google Scholar]; (gg) Tran YS, Kwon O. J. Am. Chem. Soc. 2007;129:12632. doi: 10.1021/ja0752181. [DOI] [PMC free article] [PubMed] [Google Scholar]; (hh) Sriramurthy V, Barcan GA, Kwon O. J. Am. Chem. Soc. 2007;129:12928. doi: 10.1021/ja073754n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Zhu X-F, Henry CE, Wang J, Dudding T, Kwon O. Org. Lett. 2005;7:1387. doi: 10.1021/ol050203y. [DOI] [PubMed] [Google Scholar]; (b) Zhu X-F, Schaffner A-P, Li RC, Kwon O. Org. Lett. 2005;7:2977. doi: 10.1021/ol050946j. [DOI] [PubMed] [Google Scholar]; (c) Dudding T, Kwon O, Mercier E. Org. Lett. 2006;8:3643. doi: 10.1021/ol061095y. [DOI] [PubMed] [Google Scholar]
- 12.Zhu X-F, Henry CE, Kwon O. J. Am. Chem. Soc. 2007;129:6722. doi: 10.1021/ja071990s. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The immediate β-elimination of the phosphine, following the conjugate addition of the alkoxide, was a faster reaction relative to the synthesis of the 2-pyrone. The former reactions were complete within 30 min at room temperature (cf. 48 h at 60 °C for the pyrone). No reaction occurred in the absence of a phosphine catalyst.
- 14.The zwitterions formed after the addition of trimethylphosphine to enones can be protonated and isolated as a phosphonium salts following the addition of hydrochloric acid. See: Stewart IC, Bergman RG, Toste FD. J. Am. Chem. Soc. 2003;125:8696. doi: 10.1021/ja035232n.
- 15.(a) Table of pKa values compiled by D. H. Ripin and D. A. Evans, for Chem 206, accessed online at http://daecr1.harvard.edu/pdf/evans_pKa_table.pdf. (b) Table of pKa values compiled by R. Williams, W. P. Jencks, and F. H. Westheimer, accessed online at www.webqc.org/pkaconstants.php.Dalby KN, Kirby AJ, Hollfelder F. J. Chem. Soc., Perkin Trans. 1993;2:1269.(d) Calculated using Advanced Chemistry Development (ACD/Labs) Software (v. 8.14) for Solaris (1994–2007 ACD/Labs); error reported as ±0.10.
- 16.The non-cyclized product 6 did not convert into the dihydropyrone 5 under the reaction conditions.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative experimental procedures and spectral data for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.