

Abstract
Specific interactions of human melanocortin-4 receptor (hMC4R) with its non-peptide and peptide agonists were studied using alanine-scanning mutagenesis. The binding affinities and potencies of two synthetic small-molecule agonists (THIQ, MB243) were strongly affected by substitutions in transmembrane α-helices (TM) 2, 3, 6, and 7 (residues Glu100 Asp122, Asp126, Phe261, His264, Leu265, and Leu288). In addition, I129A mutation primarily affected binding and potency of THIQ, while F262A, W258A, Y268A mutations impaired interactions with MB243. By contrast, binding affinity and potency of the linear peptide agonist NDP-MSH were substantially reduced only in D126A and H264A mutants. 3D models of receptor-ligand complexes with their agonists were generated by distance geometry using the experimental, homology-based, and other structural constraints, including interhelical H-bonds and two disulfide bridges (Cys40-Cys279, Cys271-Cys277) of hMC4R. In the models, all pharmacophore elements of small-molecule agonists are spatially overlapped with the corresponding key residues (His6, D-Phe7, Arg8 and Trp9) of the linear peptide: their charged amine groups interact with acidic residues from TM2 and TM3, similar to His6 and Arg6 of NDP-MSH; their substituted piperidines mimic Trp9 of the peptide and interact with TM5 and TM6; while the D-Phe aromatic rings of all three agonists contact with Leu133, Trp258, and Phe261 residues.
Melanotropins, which include melanocyte-stimulating hormones (α-, β-, and γ-MSH) and adrenocorticotropic hormone (ACTH), are the products of proteolytic cleavage of the 31-36 kDa precursor, pro-opiomelanocortin (1). α-MSH (Ac-Ser1-Tyr2-Ser3-Met4-Glu5-His6-Phe7-Arg8-Trp9-Gly10-Lys11-Pro12-Val13-NH2) shares with all melanotropins the central core tetrapeptide ‘His6-Phe7-Arg8-Trp9’, which is essential for its biological activity (2). These neuropeptides exert their function through five subtypes of melanocortin receptors (MCRs), which have been cloned and characterized (1, 3). MCRs belong to the G protein-coupled receptor (GPCR) superfamily (4) and are positively coupled to cAMP-generation by adenylate cyclase via the stimulatory Gs-proteins. They are involved in regulation of multiple physiological functions, such as pigmentation (MC1R), adrenal cortical steroidogenesis (MC2R), exocrine secretion (MC5R), energy homeostasis, penile erection (MC3R and MC4R) and many others (1, 5, 6).
The MC4R subtype is regarded as a potential drug target, because it is involved in feeding and sexual behavior (7-9). Mammals with a defective MC4R gene, which is expressed in the brain, are characterized by obese phenotype and increased food intake (10-12). AGRP acts as an inverse agonist, reducing the elevated basal activity of MC4R (13-17). Pharmacological studies indicate that activation of the MC4R in rodents modulates erectile function (9). Consequently research efforts have been focused on the development of potent and MC4R-selective agonists as potential anti-obesity drugs or as treatments for sexual dysfunction (18, 19). On the other hand, MC4R antagonists that block the satiety-inducing effect of α-MSH could be helpful for treatment of anorexia or cancer cachexia (20).
Recently a number of small-molecule MC4R agonists and antagonists have been synthesized using “privileged structures” formerly employed in other GPCR ligands (21, 22). In particular, THIQ and MB243 (Figure 1) were identified as potent MC4R agonists with >100 fold selectivity over MC1R, MC3R and MC5R (18, 19).
Figure 1.

Structures of hMC4R small-molecule agonists.
The binding affinities, potencies, and selectivities of the ligands can be interpreted, understood, and applied for rational drug design only in the context of 3D structures of receptor-ligand complexes. However, among all GPCRs, a crystal structure has been determined only for rhodopsin, and only in its inactive state (23, 24). In the absence of direct crystallographic data, other GPCRs can be modeled simply by homology from the rhodopsin template. Indeed, homology modeling is a well-established technique (25) that currently works best for GPCRs (26). However, several challenges remain. First, the active, agonist-bound conformation of GPCRs differs from the inactive state, especially in the position of transmembrane helix (TM) 6 (27). Second, the sequence alignment of rhodopsin and other GPCRs is not obvious in the region of the nonregular loops and at helical distortions, such as the α-aneurisms (the insertion of an extra-residue in a helical turn)) present in TM2 and TM5, in the rhodopsin structure, which may be absent in other GPCRs. Finally, ligand docking is a complicated problem (28).
These difficulties can be resolved by obtaining additional experimental data probing interactions of the specific receptors and their ligands and by using distance geometry calculations to assist homology modeling by satisfying target receptor-specific experimental and other structural constraints (29-32). The experimental constraints can be determined using a variety of techniques (31, 32). For example, the experimental studies of the effects of numerous mutations on binding of natural antagonists to hMC4R allowed modeling of the complex of the inactive conformation of hMC4R with AGRP and agouti protein (33). Moreover, we have recently calculated the model of active conformation of the μ-opioid receptor, using distance constraints from the crystal structure of the inactive conformation of rhodopsin (24) together with a large set of experimental constraints that are compatible with active states in several GPCRs (31). This model of the active conformation of μ-receptor can be used as a structural template for the modeling of other rhodopsin-like GPCRs that apparently share common activation mechanism and similar active conformations (34, 35).
In the present study, mutagenesis data are used similarly to model the active state of the hMC4R receptor in complex with agonists by distance geometry calculations with experimental, hydrogen-bonding, and homology-based constraints. More specifically, we have identified hMC4R residues required for activity of two small-molecule, peptidomimetic agonists, THIQ and MB243, by examining the effects of thirteen mutations in the TM domain on agonist binding and ligand-induced receptor-mediated cAMP accumulation. A homology model of the active hMC4R state was generated, based on our previous experimental and modeling studies of the inactive hMC4R (33) and of the μ-opioid receptor in the active state (31) and was used for docking of the two small-molecule agonists into the receptor model, guided by our mutagenesis data. Subsequently, a complex of hMCR4 with its linear peptide agonists, α-MSH and NDP-MSH (36) was calculated assuming the structural overlap of pharmacophoric elements of peptide and nonpeptide ligands.
Experimental Procedures
Cell transfection and culture
The wild type hMC4R and all mutants were expressed from the eukayotic expression vector pcDNA3.1 (Invitrogen, Carlsbad, CA). The hMC4R mutants had previously been constructed during the course of other studies (33, 37, 38) using Quick Change Site-Directed Mutagenesis Kit (stragene, La Jolla, CA). The presence of desired mutations and the integrity of the entire receptor sequence were confirmed by sequencing performed in the University of Michigan Biochemistry core. Large scale plasmid preparations were made using Qiagen Plasmid Maxi Kit (Qiagen, Valencia, CA). HEK 293 cells were transiently transfected in a 10 cm dish with 5 µg of receptor plasmid DNA using 20 µl of Lipofectamine Reagent (Invitrogen, Carlsbad, CA). After 24 hours the cells were trypsinized and aliquoted into 24-well plates and grown in Dulbecco’s modified Eagles medium (DMEM) containing 4.5 g/100 ml glucose, 10% fetal calf serum, 1 mM sodium pyruvate.
Ligand binding
125I-NDP-MSH was prepared by the chloramine-T method as previously described (38). NDP-MSH was purchased from Peninsula Laboratories, Inc. The specific activity of 125I-NDP-MSH was 618-727 Ci/mmol. Binding experiments were performed using 0.35 nM of radioligand. Binding assays were performed as previously described (38). 3 × 105 cells were planted on 24 well plates and cultured for ∼17-19 h before the experiments. IC50 values of NDP-MSH were determined from the inhibition of radioligand binding by increasing concentrations of non-labeled ligand. The highest non-labeled ligand concentration used was 10-6 M. Nonspecific binding was determined in the presence of cold ligand at 10-5 M. Nonspecific binding was less than 5% of specific binding. IC50 values are reported as the mean ± standard error. To calculate Ki values the equation Ki = Kd = IC50— [radioligand] was used (39). Experiments were repeated at least three times using duplicate wells on different days.
3′, 5′- adenosine monophosphate (cAMP) measurement
cAMP measurements were performed using a competitive binding assay kit (TRK 432, Amersham; Arlington Heights, IL) as previously described (38). 2 × 105 cells were planted on 24 well plates and cultured for ∼17-19 h before the experiments. THIQ (18) and MB243 (19) were synthesized by Merck & Co., Inc. The mean value of the data was fit to a sigmoid curve with a variable slope factor using the non-linear least squares regression in Graphpad Prism (Graphpad Software). EC50 values are reported as mean ± the standard error. Experiments were repeated at least three times using duplicate wells on different days.
Modeling of melanocortin receptor-agonist complexes
The inactive state of hMC4R (SwissProt accession code P32245) has recently been modeled from the rhodopsin crystal structure (24) by iterative distance geometry refinement (33), an approach described and tested previously (29-31, 40). This modeled structure of hMC4R remained very close to the rhodopsin template (r.m.s.d. 1.31 Å for 198 Cα-atoms of TM domain). The homology modeling of the agonist-bound state of hMC4R (residues 29-321), was accomplished here similarly using the program DIANA (41), which calculates a set of structures satisfying the imposed distance and angle constraints. Two different structural templates were used: the model of the inactive conformation of hMC4R (33) and the model of the active conformation of μ-opioid receptor, which we have previously described (31). The template of the active conformation of μ-opioid receptor, which deviates from the rhodopsin structure mostly in the position of TM6 (r.m.s.d. 2.2 Å for 212 Cα-atoms of TM domain), was chosen because it adequately reproduces helical shifts, and especially the large movement of TM6 upon receptor activation. All constraints for the seven-α-helical TM domain were taken from the active μ-receptor model, while the constraints for loops were taken from the model of inactive hMC4R. Based on mutagenesis and modeling results we have previously proposed that in MCRs a β-hairpin is formed in EL1, as in bacterial rhodopsins, an additional short β-strand in the N-terminal segment is attached to EL3 by a disulfide bond between Cys40 and Cys279, while another important disulfide bond, Cys271-Cys277, connects EL3 to TM6 (33). An alterative conformation of EL3 was also used with TM6 extended by three residues at the extracellular end. Sequence alignment of MCRs and rhodopsin (Figure 2) has been described previously (33). The alignment assumes the disappearance of anα-aneurism in TM2 that is present in rhodopsin, but conservation of theα-aneurism found in TM5 of the rhodopsin crystal structure, consistent with previous mutagenesis studies (33).
Figure 2.

Sequence alignments of bovine rhodopsin with human MC4R. Underlined characters represent residues fromα-helices, mutated residues are colored by red. The most conserved residue in each TMH (1.50, 2.50, 3.50, 4.50, 5.50, 6.50, 7.50, in the nomenclature of reference 78) of rhodopsin-like GPCR is indicated with an asterisk.
During distance geometry calculations of the ligand-free receptor with DIANA (41), the spatial positions of all TM helices were restrained using the following upper distance constraints (specified in Supplemental Materials): (a) the Cβ…Cβ distances from the templates with allowed deviations of 0.5 Å, (b) a set of 54 H-bonds specific for hMC4R (O…O, N…O distances of 2.9 Å,), and (c) constraints for two disulfide bonds (S γ...Sγ, Cβ...Cβ, Cβ…Sγ distances of 2.04, 4.20, 3.05 Å, respectively), linking Cys40-Cys279 and Cys271-Cys277; (c) constraints (N…O distances of 3.5 Å) for two artificial Zn2+-binding centers previously designed in hMC4R (42) between TM2 and TM3, I104H-Asp122 and I125H-Glu100, which enabled receptor activation by Zn2+-ions. The implementation of these constraints retained the structure of the templates, but allowed small spatial adjustments of allα-helices during the calculations. The dihedral angles of receptor residues were generally taken as in the template, with allowed deviations of 30°. Side chain rotamers were taken from the model of the inactive hMC4R (33), with the exception of several residues that rotate upon receptor activation (31), such as the conserved Asp126, Leu140, Arg147, Tyr212, Trp258 and several additional residues (Leu133, Arg165, Phe184, Met208, Met215). The standard target function weights and minimization protocol were applied (41). The pairwise r.m.s.d. between the ten best calculated models of the hMC4R was <0.7 Å (for 282 Cα-atoms).
3D-structures of the agonist ligands THIQ and MB243 were generated with QUANTA (Accelrys), and low-energy conformers of the ligands (within 3 kcal/mol of the lowest energy conformation) were used for docking. Both ligands were first docked manually into the binding pocket of hMC4R to satisfy the following criteria: (1) a similar spatial arrangement of common pharmacophore groups in THIQ and MB243; (2) interaction of most functionally important groups of the ligands (especially, the central aromatic ring and N-terminal N+ group) with the corresponding receptor residues found to be most important for binding of the small molecule agonists (Glu100, Asp122, Phe261, and His264), as described in Results; (3) minimization of steric overlaps, and maximization of receptor-ligand H-bonds and ionic and aromatic-aromatic interactions (conformers of several side-chains in the binding pocket were also adjusted). The structures of the complexes were then refined using the standard docking tool (DOCK) from QUANTA.
Large linear peptide agonists, NDP-MSH andα-MSH, were included in the distance geometry calculations together with the receptor. Therefore, separate modeling of the isolated peptides and their docking was not required. The following types of constraints were used for the complex: (1) those within the receptor, taken exactly as in the ligand-free form (above); (2) 7 H-bonds between the receptor and ligand identified in the initial model of the complex and during its iterative refinement (see Results); (3) Cβ-Cβ upper limit constraints between ligand residues 4-10 and 5-10 of 4.5 and 7.5 Å, respectively, that correspond to 4-10 disulfide and 5-10 lactam bridges in the highly potentα-MSH analogues [Cys4-Cys10]-α-MSH (43) and MT-II (Ac-Nle4-cyclo[Asp5, Lys10]α-MSH(4-11)NH2) (44), respectively; and (4) torsion angle constraints in the central portion of the peptides (His6- L,D-Phe7-Arg8-Trp9), chosen to mimic the bound conformers of the small-molecule agonists determined as described above. Side chain conformers of His6 (ϰ1 ∼ -60°) and (L, D)Phe7 (ϰ1 ∼ 180°) were chosen to mimic orientations of D-Tic and D-Phe aromatic rings of THIQ. The iterative distance geometry refinement removed steric overlaps and maximized the number of H-bonds between the receptor and ligands. Final energy minimization of all hMC4R-agonist complexes was performed using the CHARMm (45) potential with ε=10, and using the adopted-basis Newton-Raphson method (50 iterations).
Results
Interactions of agonists with hMC4R residues
In order to understand how agonists interact with hMC4R, we studied the effects of point mutations on the binding affinity and potency of the peptide agonist NDP-MSH and two small-molecule agonists, THIQ and MB243. Alanine-scanning mutagenesis was done in thirteen positions in the area of the putative ligand binding pocket (16, 33, 37, 38, 46), whose mutation has been shown to affect binding of linear peptide agonists to MC4R and MC1R (37, 38, 46). The mutated residues include Trp258, which is highly conserved in rhodopsin-like GPCRs and is presumed to be involved in activation mechanism (31), five residues that are generally conserved in MCRs (Glu100, Asp122, Asp126, Phe261, H264), four residues conserved in most MCRs, except MC2R (Phe184, Phe262, Leu265, Leu288) and three more variable residues (Ile129, Leu133, Tyr268). The residues were individually replaced by alanine to uncover the role of the assessed side chains in their interaction with small-molecule agonists. The results are summarized in Table 1, where IC50 values are determined from competition binding assay of NDP-MSH against its radioactive analogues, and EC50 values are defined as receptor-mediated accumulation of cAMP induced by corresponding agonists.
Table 1.
The effect of hMC4R mutations on the agonist binding and agonist-induced cAMP accumulation.
| Mutant | NDP-MSH | THIQ | MB243 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| HMC4R | IC50, nM | EC50, nM | Ratioa | IC50, nM | EC50, nM | Ratioa | IC50, nM | EC50, nM | Ratioa |
| Wild type | 2.30±0.30 | 1.70±0.61 | 1 | 2.55±0.41 | 1.75±0.33 | 1 | 30.3±5.4 | 17.21±3.4 | 1 |
| 2 | |||||||||
| E100A | 6.71±0.65b | 12.41±3.42 | 7.3 | 93.5±6.3 | 92±4 | 53 | >1000 | >1000 | >100 |
| D122A | 9.32±2.10b | 5.01±0.32 | 2.9 | 110±22 | 105±10 | 60 | >1000 | 418±30 | 24 |
| D126A | >1000b | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >100 |
| I129A | 1.60±0.30 | 2.14±0.15 | 1.2 | 29.9±3.1 | 15.7±4.5 | 9 | 68.5±8.3 | 58.1±5.3 | 3.4 |
| L133A | 5.10±2.00 | 0.84±0.44 | 0.49 | 8.5±2.2 | 3.74±1.74 | 2.1 | 71.3±10.2 | 20.1±1.5 | 1.2 |
| F184A | 2.94±0.61 | 1.03±0.23 | 0.6 | 1.18±0.89 | 1.32±0.53 | 0.75 | 14.5±6.8 | 4.72±0.73 | 0.27 |
| W258A | 4.01±0.13 | 2.94±0.53 | 1.7 | 9.55±2.6 | 6.24±1.26 | 3.6 | 785±23 | 357±25 | 21 |
| F261A | 3.80±0.60 | 3.35±0.21 | 2.0 | 126±11 | 107±20 | 61 | >1000 | >1000 | >100 |
| F262A | 2.30±1.70 | 1.71±0.42 | 1 | 18.9±3.4 | 7.26±1.14 | 4.1 | 520±64 | 176±15 | 10.2 |
| H264A | >1000b | 134±7 | 79 | N.D. | 19.6±2.8 | 11.2 | N.D. | 477±30 | 27.7 |
| L265A | 5.83±1.04 | 6.84±1.72 | 4.0 | 26.8±6.6 | 51.3±6.3 | 29 | 262±31 | 231±15 | 13.4 |
| Y268A | 1.87±0.90b | 3.03±0.22 | 1.8 | 6.94±2.21 | 4.93±2.51 | 2.8 | 433±85 | 153±16 | 8.9 |
The wild type hMC4R binds NDP-MSH and THIQ with ∼10 fold higher affinity (IC50 = 1.7 nM and 1.2 nM, respectively) than MB243 (IC50 = 16 nM). The IC50 and EC50 values are similar for each of the three agonists at the wild type receptor. All thirteen mutations alter potencies but do not significantly affect the efficacies of the agonists.
The changes in IC50 and EC50 values for NDP-MSH were also parallel in the majority of tested mutants, with the notable exception of H264A and D126A mutations, which led to significant reduction of NDP-MSH binding affinity such that no specific binding was detected by the filtration method.
The binding affinities and potencies of the small molecule agonists are affected by substitutions of the majority of mutated residues, which are located in TM3, TM6 and TM7 (Table 1). This suggests that these side chains are involved in the direct contact with the small-molecule agonists, although indirect effects cannot be excluded. The relative contributions of the replaced side chains in the ligand-receptor interactions are reflected in the degree of change in the binding affinity in the mutants. Our data indicate that the most important interaction likely involve charged residues (Glu100, Asp122, Asp126, and His264), residues from TM6 (Phe261 Leu265,) and from TM7 (Leu288), as their mutations decrease IC50 and EC50 of both agonists more than 10 fold. These residues are relatively conserved for MCRs 1, 3, 4, and 5 and therefore are not likely to be involved in the specific interactions with MC4R that provide >100 fold selectivity of these agonists over MC1R, MC3R, and MC5R (18,19). To reveal the ligand-receptor interactions responsible for the selectivity of these agonists would require additional mutations in other sites of the binding pocket, particularly, in the positions of variable residues such as Leu44, Val46, Ile103, Ile125, Tyr268, which interact with these agonists in our modeled receptor-ligand complexes (see below).
The binding affinities and potencies of small agonists are more sensitive to point mutations than those of peptide agonists: receptor activation by the linear peptide agonist, NDP-MSH, is strongly affected only by D126A and H264A mutations (potency decreased more than 80 fold), and, to a lesser extent, by the E100A mutation. This may be due to more extensive and redundant interactions between the receptor and peptide and perhaps due to peptide flexibility, which allows adjustment inside the modified receptor. In addition, the lower affinity agonist MB243 is much more affected by substitutions of Trp258 and Phe262, than the higher affinity agonist THIQ. These data can guide receptor-ligand docking.
Modeling active conformation of hMC4R
It is accepted that active and inactive states of GPCR are different and that agonists can preferentially bind to the active conformation . However, an experimental structure has been obtained only for the inactive state of bovine rhodopsin with bound inverse agonist, 11-cisretinal (23, 24). Recently we have developed an active state model for the μ-opioid receptor, based on the crystal structure of rhodopsin and a set of experimental constraints (disulfides, metal binding clusters, etc.) that facilitate activation (31). This model was applied here as a template for modeling of hMC4R (see Experimental Procedures). After calculations, the active state models of hMC4R and μ-opioid receptor superimpose well in the TM domain (r.m.s.d. 1.6 Å for 205 Cα-atoms).
A comparison of models for the active and inactive states of hMC4R reveals the structural changes that accompany activation. Overall r.m.s.d. of these models is 2.4 Å for all common 282 Cα atoms, but decreases to 1.8 Å (for 166 Cα atoms) after excluding TM6 and interhelical loops, which undergo movement during activation. TM6 shifts outward and rotates counterclockwise (viewed from the extracellular side) during activation, moving its intracellular end away from TM3 and toward TM5. As a result of this and other changes, the receptor structure tightens near its extracellular surface, but opens up at the cytoplasmic side, providing a cavity for binding of the Gαs subunit. In the active state, several side chains (Asp126, Leu133, Leu140, Arg147, Arg165, Phe184, Met208,Tyr212, Met215, Trp258) change their orientation, which affects the geometry of the binding pocket and alters interhelical H-bonds, especially between residues from TMs 3, 6, 7, exactly as observed for the μ-receptor (31). The most significant changes can be described as follows. First, the concerted movements of Trp258 (ϰ2 rotation from ∼90° to ∼20°), Asp126, and Leu133 (ϰ1 rotation from ∼180° to ∼-60°) residues affect the shape of the ligand binding pocket at its bottom, making it more suitable for agonist. Second, the counterclockwise rotation of TM6 brings His264 into the binding pocket, shifts Phe261, Leu265, and Tyr268, and moves Phe262 toward the lipid. This explains the larger effect of mutations involving His264, Phe261, Leu265, and Tyr288 residues and the relatively minor effect of F262A substitution on agonist binding affinities and potencies (Table 1). Third, the rotating Arg147 residue (from the conserved DRY motif in TM3) breaks an H-bond with the adjacent Asp146, but forms new H-bonds with Tyr212 (TM5) and Tyr302 (TM7), while the released Asp146 residue forms a new H-bond with Arg165 from IL2. This affects the shape of the intracellular binding cavity for Gαs.
A distinct feature of the melanocortin receptors is the deletion of the β-hairpin from EL2, together with a highly conserved disulfide bond connecting this β-hairpin to TM3, features generally found in the rhodopsin family of GPCRs. Instead, the structure can be stabilized by two additional disulfide bridges, connecting EL3 with the N-terminus (Cys40-Cys279), or EL3 with TM6 (Cys271-Cys277) (33). The deletion of the β-hairpin from EL2 creates a large, elongated ligand binding cavity containing several acidic residues. The walls of this cavity are formed by Leu44, Val46 (N-terminus), Phe51 (TM1), Glu100, Ile103, and Ile104 (TM2), Asp122, Ile125, Asp126, Ile129, Cys130, and Leu133 (TM3), Ser175 and Phe184 (TM4), Val193, Cys196 and Met200(TM5), Trp258, Phe261, His264, Leu265, and Tyr268 (TM6), Phe284, Leu288, and Met292 (TM7), Ser188and Ser190 (EL2), and Met281 (EL3) residues, many of which were probed in our mutagenesis experiments. This cavity can be filled by the N-terminal region of the receptor or by natural peptide agonists (α, β, γ-MSH).
Docking of small-molecule agonists into active state model of hMC4R
Both peptidomimetic agonists have a common tripeptide scaffold, with 4-Cl or 4-F derivatives of D-Phe occupying the central position, an N-terminal part with charged amino groups, and a C-terminal portion formed by 4,4-disubstituted piperidines. Both ligands have limited flexibility, with rotations allowed around several single bonds (Figure 1). One of the agonists, THIQ, has been extensively studied by crystallography, NMR in solution, theoretical calculations, and analysis of structure-activity relationships of its analogues (18, 48). THIQ has two nearly identical crystal structures, with an extended polypeptide backbone and piperidine and cyclohexane groups in the chair conformation (48). The crystal structure resembles the tentative biologically active conformation (18). In these conformations the 4-chlorophenyl group and piperidine ring are close and interact with each other, while the aromatic ring of D-Tic is oriented in the opposite direction, and the 1,2,4-triazol ring is highly exposed to the environment. After local energy minimization, one of the crystal structures has the lowest energy, while the other has ΔE=0.8 kcal/mol. Our theoretical conformational analysis of THIQ identified 12 clusters of conformers with ΔE < 3 kcal/mol, all of which were used for docking.
The docking of THIQ was guided by placement of its key pharmacophore elements of the ligand in contact with receptor residues that were found to be most important for binding affinities and potency, i.e. Glu100, Asp122, Asp126, Phe261, Leu265, His264, and Leu288 (Table 1). Two “anchor” pharmacophore groups were the aromatic halogen-substituted ring of the central D-Phe residue and the positively charged amine nitrogen (Figure 1). Both groups are present in either natural melanocortin peptides or synthetic agonists (18, 19, 49). In the model of the complex, the central aromatic ring of D-Phe(4-Cl) contacts with Phe261 and Leu288, the amine group was placed near Glu100, Asp122, and Asp126 and the 1,2,4-triazole ring of the peptidomimetic interacts with His264. The D-Phe(4-Cl) aromatic ring was arranged between TM3 and TM6, in a position and orientation that is similar to that of the tyramine segment of opioid ligands in the μ-opioid receptor (31). The elongated binding cavity can accept only the extended conformations of the ligand with ϰ1 of D-Phe ∼180°. Conformers of THIQ that are more compact or have ±60° ϰ1 conformers of D-Phe can not be docked without steric overlap with the receptor.
A number of extended low-energy conformers of THIQ could be fitted to the cavity, including the crystallographic structures. These conformers had slightly different orientations of D-Tic and the 1,2,4-triazole ring. The best fitting conformation of THIQ (ψ1 = 59, φ2 = 79°, ψ2 = -141°, ϰ1 = 180°, ω2 = 180°, ϰ2 = -70°, ϰ3 = -71°, and ϰ4 = 64°) was not of lowest energy in isolation (ΔE=1.1 kcal/mol). However, it formed energetically favorable H-bonds with the receptor (N+ to Asp122 and Asp126 and 1,2,4-triazol to His264), unlike the crystal structures. It is quite possible that the ligand retains some residual flexibility around ψ1, φ2, ϰ2, ϰ3 and ϰ4 dihedral angles when bound to the receptor. Such flexibility helps to explain high binding affinity and potency of either L-Tic or D-Tic in THIQ analogue (18).
Receptor-bound THIQ has multiple favorable interactions with hMC4R residues (Figure 3). For example, the side-chain of the central D-Phe(4-Cl) residue occupies the bottom of the binding cavity and interact with Ile129, Leu133, Trp258, Phe261 and Leu288 residues, most of which are important for binding and activation by THIQ (Table 1). The D-Tic of THIQ is situated between Leu44, Val46, preceding TM1, and residues from TM2 (Glu100, Ile103, Ile104), TM3 (Asp122, Ile125, Asp126) and TM7 (Asn285). The charged amino group of D-Tic forms ionic interactions with several acidic residues from TMs 2 and 3 including the most functionally important Asp126, with which it also forms an H-bond. Moreover, Asp126 forms an H-bond with the backbone amide group of D-Phe, which restrains the position of the ligand inside the binding pocket. The triazole part of THIQ occupies a spot between TMs 3-7. Its piperidine ring forms contacts with Phe184(TM4) and Phe284(TM7), the cyclohexyl ring is situated between Val193, Cys196 (TM5) and Leu265 (TM6), and the polar triazole group interacts with Leu265, His264 and Tyr268 (TM6).
Figure 3.
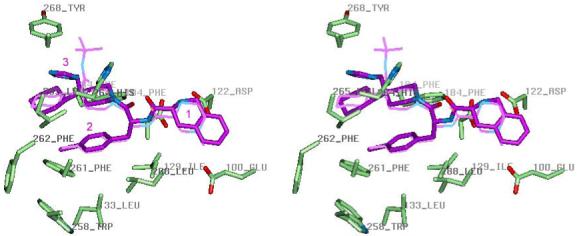
Stereoview of small-molecule agonist THIQ (purple) inside the binding pocket of the hMC4R active conformation model. The mutated residues of hMC4R are shown in the licorice representation colored by element. MB243 is shown in thin purple line for comparison. The central part of both ligands, a 4-substituted D-Phe2, occupies the bottom of the cavity formed by Ile129, Leu133, Trp258, Phe261, and Leu288, the positively charged amino group of the first residue is close to negatively charged Glu100, Asp122, and Asp126, and Asp126 also forms and H-bond with the backbone amide group of D-Phe2; while the third residue mimic contacts with Phe184 Leu265, and Tyr268, with its polar group interacting with His264.
A similar analysis was carried out for the THIQ analogue with L-stereoisomer of the central Phe residue. The extended low-energy conformation of L-Phe-THIQ does not fit the binding pocket due to changes in backbone angles (φ2 = -78°, ψ2 = 142°), resulting in the reorientation of the first peptide bond, a broken receptor-ligand H-bond, and steric clashes between the shifted ligand and TM6 (Tyr268). This may explain the low affinity and potency observed for the L-Phe analogue of THIQ to hMC4R (18). Interestingly, L-Phe-THIQ demonstrates higher affinity toward hMC3R and hMC5R, but does not exhibit agonist properties in these receptors (18). The increased binding affinity of L-Phe-THIQ to other MCRs may be related to the substitution of Tyr268 (TM6) in hMC4R by Ile in MC3R or Met in MC5R. At the same time, the reorientation of the first peptide group in L-Phe-THIQ breaks the H-bond between the functionally important Asp126 and NH of L-Phe and forces the rotation of Asp126 to a position appropriate for the inactive receptor state (ϰ1 ∼ 180°). This Asp126 reorientation may be responsible for the loss of agonistic activity of this ligand. Furthermore, substitution of D-Phe by the larger aromatic side chain of D-Nal(2’) also results in the loss of agonism (18). In our model of the active state of hMC4R, D-Nal(2’) exhibits some hindrances with the indole ring of Trp258 from TM6. However, these hindrances disappear in the inactive conformation of hMC4R, where the indole ring of Trp258 changes its orientation, in coordination with rotation of Leu133 from TM3. This is consistent with the observed antagonistic properties of D-Nal(2’)-THIQ (18).
The lowest-energy conformer of MB243 was structurally more compact than that of THIQ, and does not fit the binding pocket. Only an extended conformation (ΔE=1.85 kcal/mol) with ψ1 = 158°, φ2 = 76°, ψ2 = -135°, ϰ1 = 179°, ω2 = 178°, ϰ2 = -175° ϰ3 = 54°, which resembles the bound conformation of THIQ, could be docked into the model of hMC4R (Figure 4). The higher energy of the receptor-bound conformation of MB243 is consistent with 10-fold lower binding affinity, relative to THIQ. Both small agonists interact with the same residues of receptor, with only relatively minor differences, such as the appearance of an H-bond between methylated amine of piperazine of MB243 and Glu100 in addition to the ionic interaction betwen second N+ of piperazine and Asp122, Asp126, and formation of H-bond between tert-butylamide group of MB243 and His264, while THIQ forms only one H-bond between its N+ and Asp126 and interacts with His264 by its 1,2,4-triazole group. These differences may cause the more pronounced effects of E100A and H264A mutations on the potency of MB243, relative to THIQ (Table 1). MB243 in the receptor retains some rotational flexibility of ψ1, φ2, and ϰ2 dihedral angles. In particular, the piperazine can be oriented in the receptor with its N-methyl group facing either the middle or the extracellular part of the pocket. Therefore, different substituents at the N-methyl group of piperazine ring can be adopted in the binding pocket with rather similar affinity (19). The space around N-methyl-piperazine is reduced in other MCR subtypes, due to replacements of Leu44, Val46, Ile103 and Ile125 residues by Val, Ile, Met and Phe, respectively in hMC3R, or by Met, Ile, Thr, Phe, respectively in hMC5R. This may explain the lower affinity and potency of MB243 toward these receptors (19). On the other hand, the antagonistic properties of the L-Phe analogue of MB243 toward all three MCRs (19) could be explained by the broken H-bond between the important Asp126 and the first peptide bond of ligand, and the forced reorientation of Asp126, similar to the situation described above for the L-Phe analogue of THIQ.
Figure 4.
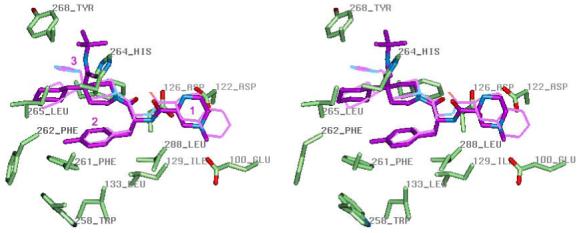
Stereoview of small-molecule agonist MB243 (purple) inside the binding pocket of the hMC4R active conformation model. The mutated residues of hMC4R are shown in the licorice representation colored by element. THIQ is shown in thin purple line for comparison. See legend of Figure 3 for the description of the illustrated ligand-receptor interactions.
Distance geometry modeling of receptor-peptide complexes
Finally, we calculated the complex of hMC4R with peptide agonist NDP-MSH. The initial model of the complex was generated assuming that “message” residues of the peptide (His6-D-Phe7-Arg8-Trp9) would mimic the corresponding pharmacophore elements of THIQ: His6 of the peptide was positioned similar to D-Tic of THIQ; guanidium group of Arg8 formed ionic interactions with the functionally important Asp126similar to the positively charged amine of D-Tic; D-Phe7 was overlapped with D-Phe(4-Cl) of THIQ, and Trp8 mimicked the C-terminal group of THIQ. The subsequent distance geometry refinement of the complex (see Experimental Procedures) helped to maximize the set of peptide-receptor H-bonds and adjust geometry of the peptide and conformers of surrounding receptor side-chains. During the calculations, we also used constraints between residues 4-10, and 5-10, which were introduced to mimic disulfide and lactam bridges from cyclic peptide agonists, [Cys4-Cys10]-αMSH (43) and MT-II (44), respectively.
This computational approach defined the structural details of the receptor-bound conformation of bothα-MSH and NDP-MSH (Table 2) and confirmed their proposed arrangement inside the receptor (Figure 5). The backbone conformation and ϰ1 angles in fragment 3-12 of both peptides and their position inside the receptor were determined unequivocally (r.m.s.d. 1.2 Å for Cα-atoms 3-12 of peptides in 10 superposed receptor models). Both peptide ligands demonstrate a β-hairpin-like structure with a reverse turn spanning His6 and L- or D-Phe7. In this conformation the phenyl ring of L- or D-Phe7 interacts with the indole ring of Trp9, forming a hydrophobic patch, while charged Glu5 and Arg8 are located on different sides of the β-hairpin.
Table 2.
The dihedral angles (°) for calculated receptor bound conformations of fragments 2-12 of α-MSH and NDP-MSH.
| Dihedral angles |
Residues |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Tyr2 | Ser3 | Met4/ Nle4 |
Glu5 | His6 | LPhe7/ DPhe7 |
Arg8 | Trp9 | Gly10 | Lys11 | Pro12 | |
| α-MSH | |||||||||||
| ω | 178 | 175 | 179 | -176 | 171 | -174 | 172 | 173 | 177 | 168 | -179 |
| φ | -96 | -170 | -155 | -83 | -55 | 89 | -67 | -131 | -163 | -121 | -74 |
| ψ | 106 | 105 | 159 | -172 | 131 | 129 | 88 | 65 | -144 | 78 | 127 |
| Χ1 | 173 | 175 | -34 | -62 | -76 | -159 | -63 | -78 | -174 | ||
| Χ2 | 109 | -145 | 81 | -159 | 135 | -67 | 130 | -178 | |||
| Χ3 | -76 | -92 | 143 | -169 | |||||||
| Χ4 | -179 | 66 | |||||||||
| NDP-MSH | |||||||||||
| ω | 178 | 169 | -175 | -170 | 174 | 178 | 177 | 169 | 173 | 176 | -179 |
| φ | -174 | -170 | -155 | -103 | -73 | 157 | 60 | -92 | -162 | -156 | -63 |
| ψ | 125 | 114 | 153 | -170 | 86 | -22 | 52 | 88 | -107 | 74 | 122 |
| Χ1 | -166 | 178 | -73 | -73 | -80 | 159 | -55 | -60 | -175 | ||
| Χ2 | 80 | -177 | 102 | -114 | 99 | -57 | 110 | 173 | |||
| Χ3 | -72 | -63 | 177 | -179 | |||||||
| Χ4 | 121 | 70 | |||||||||
α-MSH, Ac-Ser1-Tyr2-Ser3-Met4-Glu5-His6-Phe7-Arg8-Trp9-Gly10-Lys11-Pro12-Val13-NH2
NDP-MSH, [Nle4, -Phe7-]α-MSH
Figure 5.
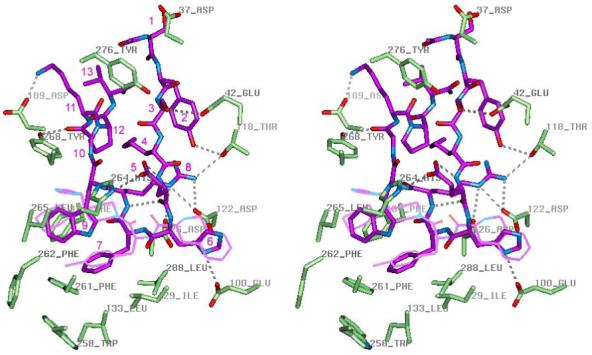
Stereoview of peptide agonist NDP-MSH (purple) inside the binding pocket of the hMC4R active conformation model. The mutated hMC4R residues and residues forming H-bonds with peptide are shown in the licorice representation colored by element. THIQ is shown in thin purple line for comparison. The receptor-bound conformation of NDP-MSH represents a β-hairpin-like structure with a reverse turn spanning His6 and D-Phe7, aromatic rings of D-Phe7 and Trp9 forming stacking interactions, and polar residues Glu5 and Arg8 located on different sides of the β-hairpin. Inside the binding pocket, NDP-MSH forms at least nine H-bonds with polar groups of the receptor (shown by dashed lines): Tyr2 hydroxyl with Thr118 hydroxyl, carboxylate group of Asp5 and backbone carbonyl of Trp9 with His264, imidazole of His6 with Glu100, guanidinium group of Arg8 with Asp122 and Asp126 carboxylate groups and hydroxyl of Thr118, backbone amide group of D-Phe7 with Asp126 carboxylate, backbone carbonyl of Gly10 with Tyr268 hydroxyl, and amine group of Lys11 with Asp189 carboxylate. Important hydrophobic interactions are formed between aromatic rings of D-Phe7 and Trp9 of peptide agonist and non-polar binding pocket residues , Ile129, Leu133, Phe184, Leu265, Trp258, Phe261, Leu288.
In the binding pocket of hMC4R eitherα-MSH or NDP-MSH forms at least 7 H-bonds with receptor residues: His264 (TM6) with the carboxylate group of Asp5 and carbonyl of Trp9; Glu100 (TM2) with the imidazole group of His6; Asp122 and Asp126 with the guanidium group of Arg8, Asp126 (TM3) with the amide group of L,D-Phe7; Asp189 (EL2) with the amine group of Lys11. These H-bonds were included in calculations of receptor-peptide complexes. Several additional H-bonds were also found in many calculated structures: Asp37 (N-terminus) with Ser1; Thr118 (EL1) with Tyr2; Glu42 (N-terminus) with Ser3, Thr118 (EL1) with Arg8, Tyr268 (TM6) with carbonyl of Gly10. His6 of the peptide ligands is buried between polar side chains of Glu100, Asp122 and Asn285 and can form alternative H-bonds with these residues, while Arg8 is water exposed and can form ionic interactions with both Asp122 and Asp126. The phenyl ring of L,D-Phe7 of peptides occupies the bottom of the cavity between Ile129, Cys130, Leu133, Trp258, Phe261 and Leu288, similar to D-Phe(4-Cl) of THIQ. The slightly different orientation for D-Phe7 relative to its L-enatiomer inside the pocket may be responsible for the greater potency and prolonged activity of NDP-MSH, relative toα-MSH (36, 50) and for the larger effect of W258A mutation on the binding affinity and potency ofα-MSH relative to NDP-MSH (37). Trp9 of both peptides interacts with Leu265, His264, and Phe284 of the receptor, similar to the C-terminal 4,4’-substituted piperidine of THIQ. Such an arrangement of NDP-MSH inside the binding pocket explains the substantial effects of E100A, D126A, H264A, and L265A mutations on the potency and affinity of this peptide (Table 1).
Discussion
In this study we have identified residues of hMC4R that may interact with two small molecule agonists and peptide agonists by examining the effects of thirteen mutations on ligand binding and activation of the receptor. This allowed the development of putative 3D models of complexes of hMC4R with either THIQ or MB243, based on current data, SAR of small-molecule agonists (18, 19), and our previous modeling studies of inactive (antagonist-bound) and active (agonist-bound) states of different GPCRs (31, 33). Subsequently, the models of hMC4R with two linear peptide agonists, NDP-MSH andα-MSH, were calculated by distance geometry assuming that the most functionally significant side-chains of the peptide (His6, D-Phe7, Arg8 and Trp9) are spatially overlapped with the corresponding pharmacophore groups of THIQ. The resulting models with peptide agonists were different in atomic details from those in earlier studies (16, 47, 51-53). The proposed peptide-receptor complexes were consistent with the notion that the core residues of flexible peptides reproduced the receptor-bound structure of the more conformationally rigid small molecule agonists. The peptide flexibility was also restrained during calculation by packing interactions and H-bonds with the receptor, and by several intramolecular cross-linking constraints taken from related bioactive cyclic peptides.
The most important pharmacophore element of all melanocortin agonists is an aromatic ring of the central D-Phe residue (18, 19, 49, 54). It is inserted at the bottom of the binding cavity in close proximity to the conserved Trp258 residue that triggers activation of GPCRs (31, 55). This region is occupied by the polyene chain of retinal in rhodopsin (24) or by the tyramine portion of opioid ligands (31). Some interesting details of receptor-ligand interactions can be observed upon superimposition of hMC4R models in complex with the antagonist AGRP (inactive state), with the peptide agonist NDP-MSH, and with small-molecule agonist THIQ (Figure 6). Three key pharmacophore groups of these ligands are similar: (a) phenyl rings of Phe112 of AGRP, D-Phe7 of NDP-MSH, and D-Phe(4-Cl) of THIQ; (b) side chain of Arg111 of AGRP, and charged groups of His6 and Arg8 in NDP-MSH, and N+ group of D-Tic in THIQ; and (c) phenyl ring of Phe113 of AGRP, indole ring of Trp9 of NDP-MSH, and cyclohexane ring of THIQ. The corresponding groups are not completely overlapped, but they occupy the same areas of space and interact primarily with the same receptor residues.
Figure 6.

A. Superposition of receptor-bound conformation of inverse agonist AGRP (87-132) (yellow) inside the model of hMC4R in the inactive state, and peptide agonist, NDP-MSH (purple) inside the model of hMC4R in the active state; B. Superposition of receptor-bound conformations of peptide agonist NDP-MSH (purple) and small-molecule agonist THIQ (green) inside the model of hMC4R in the active state. Core residues of AGRP (Arg111, Phe112, Phe113) and of NDP-MSH (His6, D-Phe7, Arg8, Trp9) are shown in the licorice representation.
The models of the receptor-agonist complexes help to interpret the observed effects of hMC4R mutations on binding affinity and potency of agonists (Table 1). Some residues whose mutations have been shown to affect activation may be in direct contact with ligands. For example, in the proposed models of hMC4R with small molecule agonists Glu100, Asp122, and Asp126 residues form H-bonds and ionic interactions with the positively charged N+ of THIQ (D-Tic) or the piperazine ring of MB243. In the proposed models of hMC4R with peptide agonists, His6 of both peptide ligands is completely buried in the pocket and can form an H-bond either to Glu100 or to Asn285, while their Arg8 residue is more exposed to water and makes ionic interactions and H-bonds with Asp122 and Asp126. The reduced ionic interactions of Arg8 in water and the flexibility of His6 can explain the smaller effect of E100A and D122A replacements on the potency of NDP-MSH, relative to their effect on potency of small agonists (Table 1). Among all acidic residues of MCRs, Asp126 appears to be the most important for potency of all agonists (>100 fold increase of EC50in D126A mutant). The essential role of Asp126 may be due to its H-bond with the backbone NH group of central D- or L-Phe residues in all agonists (this H-bond forces reorientation of Asp126in the active state). His264 may be more important for NDP-MSH binding because it forms two H-bonds with the peptide (with COO- of Glu5 and backbone C=O of Trp9), but only one H-bond with either THIQ (with 1,2,4-triazole) or MB243 (C=O group). L265A mutation demonstrates a large effect only on the binding affinity and the potency of small-molecule agonists (Table 1) probably because the aliphatic side-chain of Leu265 is more tightly packed with the cyclohexane ring of the peptidomimetics than with the corresponding planar ring of Trp9 of the peptide. It can not be excluded, however, that D126A and H264A mutations may also impair plasma membrane targeting and/or assembly, as radiolabel binding and functional activation is dramatically reduced for these mutants.
Our models are consistent with receptor-bound conformations of agonists that have been proposed previously (18, 48, 56-59). The best-fitting conformation of small agonist, THIQ was similar to the crystal structure of this molecule (see Results). It has also been proposed that aromatic rings of D,L-Phe7 and Trp9 residues of MSH peptide form a continuous cluster on one side of the bound structure, while the positively charged side chains of His6 and Arg8 form an opposite ‘hydrophilic surface’ (56, 57). Indeed, such aromatic and charged clusters are present in our models. These two clusters interact with aromatic residues from TMs 3-7 and negatively charged residues from TMH 2-3, respectively. Furthermore, the bound conformation of NDP-MSH peptide has a type II β-turn formed by His6 and L,D-Phe7 residues, as has been suggested previously based on NMR and computational studies of these peptides in aqueous solution (58, 59). However this β-turn is distorted inα-MSH, where backbone angles of the central L,D-Phe7and Arg8 residues are different (Table 2). The β-hairpin-like structure of the peptides was obtained because the Cβ atoms of residues 4-10 and 5-10 were kept in close proximity to each other (4.5 Å and 7.5 Å, respectively) during distance geometry calculations, thus allowing the cyclization through a disulfide bond in [Cys4, Cys10]-α-MSH (43) or a lactam bridge between Asp5 and Lys10 in MT-II (44). Further, in the calculated structures ofα-MSH and NDP-MSH the Cβ…Cβ distances between residues 3-11 (∼ 8.5 Å), 4-11 (∼ 8.0 Å), 5-8 (∼ 7.5 Å) and 5-11 (∼12 Å), are compatible with formation of the corresponding disulfide or lactam bridges in other cyclic peptide agonists, such as Ac-cyclo[Cys3,Nle10,D-Nal7,Cys11]α-MSH(3-11)NH2, Ac-cyclo[Cys4,D-Nal7,Cys11]α-MSH(4-11)NH (60, 61), Ac-Nle4 2-cyclo[D-Orn5,Glu8]α-MSH(4-11)NH (62) and Ac-Nle4-cyclo[Asp52 ,D-Phe7,Aib10,Lys11]α-MSH(4-11)NH2(57), respectively.
During our calculations of receptor-peptide complexes the side-chains of His6 and (L,D)-Phe7 residues of peptides were restrained in gauche- (ϰ1 ∼ -60°) and trans (ϰ1 ∼ 180°) conformations, respectively, to mimic orientations of the aromatic rings of D-Tic1 and p-Cl-D-Phe2 (ϰ1 ∼ 180°) in the receptor-bound conformations of THIQ. Such rotamers are well-adopted inside the binding pocket of hMC4R. This orientation of the imidazole ring of His6 is consistent with the position of the aromatic ring of the ϰ-constrained (2S,3R)3-phenyl-Pro6 residue of the MT-II analogue, which retains bioactivity (59). On the other hand, the proposed trans rotamer of D-Phe7 places the aromatic ring in a different location than the aromatic ring of the (2R, 3S)Phe-Pro7-MT-II analogue, which is completely inactive (59). These data represent additional evidence supporting the proposed side chain orientations of His6 and D-Phe7 inside the receptor binding pocket.
The proposed models help to explain SAR of the small agonists, for example the importance of the D-stereoisomer of Phe2 for binding and bioactivity of THIQ and MB243, the antagonistic properties of its D-Nal(2’)-analogue, and the high potency of a THIQ analogue with a L-Tic residue (18), as was described in Results. Furthermore, it has recently been proposed that the pharmacophore core ‘His6-D-Phe7-Arg8-Trp9’ of MT-II is arranged in the binding pocket similar to the important structural elements of THIQ, based on the comparison of SARs of THIQ and MT-II (18). The results of our modeling support this conclusion.
The models are also consistent with SAR of the peptide ligands (47, 49, 63, 64). In the models the positively charged Arg8 of peptides is located close to the extracellular surface of the receptor and it participates in the H-bond network with acidic residues from TM3 (Asp122, Asp126). The likely presence of water around Arg8 should decrease the role of ionic interactions with this residue. Indeed, the Arg8→Glu8 substitution in MT-II is tolerated, resulting only in ∼40 fold loss of potency compared to the parent peptide (65). The negatively charged Glu5 of the peptide ligands is located on the opposite side of its β-hairpin-like structure, forming H-bond and ionic interactions with basic His264 in TM6. Interestingly, this acidic residue is shifted from position 5 to position 10 in γ-MSH, which is selective for MC3R. As a result, Asp10 of γMSH can form an ionic interaction with Lys223 (TM5) of hMC3R that in hMC4R is substituted by Ser191. This is consistent with the observed importance of Asp10 for MC3-selectivity (66). Further, the Phe7 residue is most essential for binding and activity of MCR ligands (49, 54). Its aromatic ring is situated on the bottom of the receptor pocket and surrounded by Asp126, Leu133and Trp258 side-chains that rotate during activation in our model. The bulkier D-Nal(2’)7 in peptide analogues overlaps with Leu133 in the active receptor conformation, but it can be accommodated easily in the inactive receptor state, where Leu133 and Trp258 change orientation. This is consistent with the functional antagonism of D-Nal(2’)7-containing peptides in MC4R, but not in MC1R, where Leu133 is substituted by Met (54, 63). Indeed L133M mutation in hMC4R converts D-Nal(2’)7-containing SHU9119 into an agonist (67). Another residue essential for high affinity of the peptide ligands is Trp9(49). It is located in a hydrophobic-aromatic pocket, enclosed by Phe184, Cys196, Met200, Phe261, His264, Leu265, Phe284. This large pocket is suitable for different aromatic substitutions, but not for small or polar side chains. Indeed, potent agonists are those with Trp9, L,D-Nal(2’)9, L-Nal(1’)9 or 3-benzothienylalanine, while ligands with Ala9, His6, and Lys9 lose their activity (65, 68). In contrast, the region around His6 is formed by polar and aliphatic residues (Leu44, Val46, Glu100, Ile103, Asp122, Ile125, Asp126, Asn285) and, therefore, can adopt both polar and aromatic side chains (64, 65, 68).
Our modeling of hMC4R does not include the N-terminal fragment, which can be deleted without loss of receptor activity (69). However, it has been recently suggested that the N-terminal domain could function as a tethered intramolecular ligand, which provides intrinsic constitutive activity of hMC4R responsible for the tonic satiety signal (70). Indeed, the N-terminus (residues 15LWNRSSYRLHSNA27 of hMC4R) can be positioned similar to the natural agonist,α-MSH (residues 1SYSMEHFRWGKPV13) in our model. In this case, their central tetrapeptide fragments (indicated in bold) structurally match each other, and the functionally important Arg18 of the N-terminus, whose mutations are associated with obesity (70), interacts with the functionally important acidic residues from TM3 of hMC4R (Figure 7).
Figure 7.
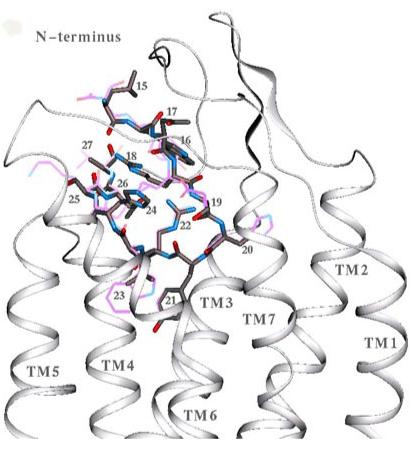
Putative arrangement of the N-terminal fragment (15-27) inside the binding pocket of hMC4R, docked similarly to the peptide agonistα-MSH (1-13). The N-terminal fragment is shown in the licorice representation colored by element,α-MSH is shown by a thin purple line. The central tetrapeptide fragment of the N-terminal sequence 20SYRL structurally overlaps with 6HFRW of the peptide agonist,α-MSH.
In summary, we propose 3D models of complexes of hMC4R with either small molecule agonists (THIQ, MB243), or linear peptides (α-MDH, NDP-MSH), based on our mutagenesis data. The models are consistent with published structure-activity and conformational studies of the ligands. They can be applied in future studies of residues responsible for high selectivity of small-molecule agonists to MC4R using mutations of receptor type-specific ligand contact residues proposed by the model (for example, Leu44, Val46, Ile103, Ile125) to extend the results of this study. The models can also be used for computational analysis of binding of many agonists that were not considered here (71-75)), for finding potential metal binding sites in MCRs (42, 76, 77), and for rational drug design.
The atomic coordinates of the described models are publicly available at http://mosberglab.phar.umich.edu/resources/index.php.
Supplementary Material
Acknowledgment
The authors are grateful to Dr. Jarl E.S. Wikberg for the coordinates of the crystal structure of THIQ, described in a recent paper (48).
Abbreviations
- GPCR
G protein-coupled receptor
- MCR
melanocortin receptor
- TM
transmembrane helix
- EL
extracellular loop
- IL
intracellular loop
- SAR
structure activity relationship
- α-MSH
α-melanocyte stimulating hormone or Ac-Ser1-Tyr2-Ser3-Met4-Glu5-His6-Phe7-Arg8-Trp9-Gly10-Lys11-Pro12-Val13-NH2
- NDP-MSH
[Nle4, D-Phe7]α-MSH
- MT-II
melanotan-II or Ac-Nle4-cyclo[Asp5, Lys10]α-MSH(4-11)NH2
- THIQ
N-[(3R)-1,2,3,4-tetrahydroisoquinolinium-3-ylcarbonyl]-(1R)-1-(4-chlorobenzyl)-2-[4-cyclohexyl-4-(1H-1,2,4-triazol-1ylmethyl)piperidin -1-yl]-2-oxoethylamine
- MB243
(2S)-N-[(1R)-2-[4-cyclohexyl-4-[[(1,1-dimethylethyl)amino]carbonyl]-1-piperidinyl]-1-[(4-fluorophenyl)methyl]-2-oxoethyl]-4-methyl-2-piperazinecarboxamide
- AGRP
Agouti-related protein
Footnotes
This work was supported by NIH grants DK054032 (I.G., M.W.M.), DA03910 (H.I.M.), and the University of Michigan Gastrointestinal Peptide Research Center (NIH Grant P30DK34933)
Supporting Information Available:Table of receptor type-specific distance constraints for the distance geometry refinement of the model of the active conformation of MC4R. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Gantz I, Fong TM. The melanocortin system. Am. J. Physiol. Endocrinol. Metab. 2003;284:E468–74. doi: 10.1152/ajpendo.00434.2002. [DOI] [PubMed] [Google Scholar]
- 2.Hruby VJ, Wilkes BC, Hadley ME, Al-Obeidi F, Sawyer TK, Staples DJ, de Vaux AE, Dym O, Castrucci AM, Hintz MF, Riehm JP, Rao KRJ. alpha-Melanotropin: the minimal active sequence in the frog skin bioassay. J. Med. Chem. 1987;30:2126–2130. doi: 10.1021/jm00394a033. [DOI] [PubMed] [Google Scholar]
- 3.Wikberg JE, Muceniece R, Mandrika I, Prusis P, Lindblom J, Post C, Skottner A. New aspects on the melanocortins and their receptors. Pharmacol. Res. 2000;42:393–420. doi: 10.1006/phrs.2000.0725. [DOI] [PubMed] [Google Scholar]
- 4.Watson S, Arkinstall S. The G-Protein Linked Receptor Facts Book. Academic Press; San Diego: 1994. pp. 1–294. [Google Scholar]
- 5.Haqq AM, Rene P, Kishi T, Khong K, Lee CE, Liu H, Friedman JM, Elmquist JK, Cone RD. Characterization of a novel binding partner of the melanocortin-4 receptor: attractin-like protein. Biochem. J. 2003;376:595–605. doi: 10.1042/BJ20031241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mountjoy KG, Kong PL, Taylor JA, Willard DH, Wilkison WO. Melanocortin receptor-mediated mobilization of intracellular free calcium in HEK293 cells. Physiol. Genomics. 2001;5:11–19. doi: 10.1152/physiolgenomics.2001.5.1.11. [DOI] [PubMed] [Google Scholar]
- 7.MacNeil DJ, Howard AD, Guan X, Fong TM, Nargund RP, Bednarek MA, Goulet MT, Weinberg DH, Strack AM, Marsh DJ, Chen HY, Shen CP, Chen AS, Rosenblum CI, MacNeil T, Tota M, MacIntyre ED, Van der Ploeg LH. The role of melanocortins in body weight regulation: opportunities for the treatment of obesity. Eur. J. Pharmacol. 2002;440:141–157. doi: 10.1016/s0014-2999(02)01425-5. [DOI] [PubMed] [Google Scholar]
- 8.Vergoni AV, Bertolini A. Role of melanocortins in the central control of feeding. Eur. J. Pharmacol. 2000;405:25–32. doi: 10.1016/s0014-2999(00)00538-0. [DOI] [PubMed] [Google Scholar]
- 9.Van der Ploeg LH, Martin WJ, Howard AD, Nargund RP, Austin CP, Guan X, Drisko J, Cashen D, Sebhat I, Patchett AA, Figueroa DJ, DiLella AG, Connolly BM, Weinberg DH, Tan CP, Palyha OC, Pong SS, MacNeil T, Rosenblum C, Vongs A, Tang R, Yu H, Sailer AW, Fong TM, Huang C, Tota MR, Chang RS, Stearns R, Tamvakopoulos C, Christ G, Drazen DL, Spar BD, Nelson RJ, MacIntyre DE. A role for the melanocortin 4 receptor in sexual function. Proc. Natl. Acad. Sci. U. S. A. 2002;99:11381–11386. doi: 10.1073/pnas.172378699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lubrano-Berthelier C, Cavazos M, Dubern B, Shapiro A, Stunff CL, Zhang S, Picart F, Govaerts C, Froguel P, Bougneres P, Clement K, Vaisse C. Molecular genetics of human obesity-associated MC4R mutations. Ann. N. Y. Acad. Sci. 2003;994:49–57. doi: 10.1111/j.1749-6632.2003.tb03161.x. [DOI] [PubMed] [Google Scholar]
- 11.Yeo GS, Lank EJ, Farooqi IS, Keogh J, Challis BG, O’Rahilly S. Mutations in the human melanocortin-4 receptor gene associated with severe familial obesity disrupts receptor function through multiple molecular mechanisms. Hum. Mol. Genet. 2003;12:561–574. doi: 10.1093/hmg/ddg057. [DOI] [PubMed] [Google Scholar]
- 12.Tao YX, Segaloff DL. Functional characterization of melanocortin-4 receptor mutations associated with childhood obesity. Endocrinology. 2003;144:4544–4551. doi: 10.1210/en.2003-0524. [DOI] [PubMed] [Google Scholar]
- 13.Siegrist W, Drozdz R, Cotti R, Willard DH, Wilkison WO, Eberle AN. Interactions of alpha-melanotropin and agouti on B16 melanoma cells: evidence for inverse agonism of agouti. J. Recept. Signal. Transduct. Res. 1997;17:75–98. doi: 10.3109/10799899709036595. [DOI] [PubMed] [Google Scholar]
- 14.Dinulescu DM, Cone RD. Agouti and agouti-related protein: analogues and contrasts. J Biol Chem. 2000;275:6695–6698. doi: 10.1074/jbc.275.10.6695. [DOI] [PubMed] [Google Scholar]
- 15.Nijenhuis WA, Oosterom J, Adan RA. AgRP(83-132) acts as an inverse agonist on the human-melanocortin-4 receptor. Mol. Endocrinol. 2001;15:164–171. doi: 10.1210/mend.15.1.0578. [DOI] [PubMed] [Google Scholar]
- 16.Haskell-Luevano C, Monck EK. Agouti-related protein functions as an inverse agonist at a constitutively active brain melanocortin-4 receptor. Regu.l Pept. 2001;99:1–7. doi: 10.1016/s0167-0115(01)00234-8. [DOI] [PubMed] [Google Scholar]
- 17.Chai BX, Neubig RR, Millhauser GL, Thompson DA, Jackson PJ, Barsh GS, Dickinson CJ, Li JY, Lai YM, Gantz I. Inverse agonist activity of agouti and agouti-related protein. Peptides. 2003;24:603–609. doi: 10.1016/s0196-9781(03)00104-9. [DOI] [PubMed] [Google Scholar]
- 18.Sebhat IK, Martin WJ, Ye Z, Barakat K, Mosley RT, Johnston DB, Bakshi R, Palucki B, Weinberg DH, MacNeil T, Kalyani RN, Tang R, Stearns RA, Miller RR, Tamvakopoulos C, Strack AM, McGowan E, Cashen DE, Drisko JE, Hom GJ, Howard AD, MacIntyre DE, van der Ploeg LH, Patchett AA, Nargund RP. Design and pharmacology of N-[(3R)-1,2,3,4-tetrahydroisoquinolinium- 3-ylcarbonyl]-(1R)-1-(4-chlorobenzyl)- 2-[4-cyclohexyl-4-(1H-1,2,4-triazol-1-ylmethyl) piperidin-1-yl]-2-oxoethylamine (1), a potent, selective, melanocortin subtype-4 receptor agonist. J. Med. Chem. 2002;45:4589–4593. doi: 10.1021/jm025539h. [DOI] [PubMed] [Google Scholar]
- 19.Palucki BL, Park MK, Nargund RP, Ye Z, Sebhat IK, Pollard PG, Kalyani RN, Tang R, Macneil T, Weinberg DH, Vongs A, Rosenblum CI, Doss GA, Miller RR, Stearns RA, Peng Q, Tamvakopoulos C, McGowan E, Martin WJ, Metzger JM, Shepherd CA, Strack AM, Macintyre DE, Van der Ploeg LH, Patchett AA. Discovery of (2S)-N-[(1R)-2-[4-cyclohexyl-4-[[(1,1-dimethylethyl)amino]carbonyl]-1-piperidinyl]-1-[(4-fluorophenyl)methyl]-2-oxoethyl]-4-methyl-2-piperazinecarboxamide (MB243), a potent and selective melanocortin subtype-4 receptor agonist. Bioorg. Med. Chem. Lett. 2005;15:171–175. doi: 10.1016/j.bmcl.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 20.Wisse BE, Schwartz MW, Cummings DE. Melanocortin signaling and anorexia in chronic disease states. Ann. N. Y. Acad. Sci. 2003;994:275–281. doi: 10.1111/j.1749-6632.2003.tb03190.x. [DOI] [PubMed] [Google Scholar]
- 21.Willoughby CA, Hutchins SM, Rosauer KG, Dhar MJ, Chapman KT, Chicchi GG, Sadowski S, Weinberg DH, Patel S, Malkowitz L, Di Salvo J, Pacholok SG, Cheng K. Combinatorial synthesis of 3-(amidoalkyl) and 3-(aminoalkyl)-2-arylindole derivatives: discovery of potent ligands for a variety of G-protein coupled receptors. Bioorg Med. Chem. Lett. 2002;12:93–96. doi: 10.1016/s0960-894x(01)00665-5. [DOI] [PubMed] [Google Scholar]
- 22.Bondensgaard K, Ankersen M, Thogersen H, Hansen BS, Wulff BS, Bywater RP. Recognition of privileged structures by G-protein coupled receptors. J. Med. Chem. 2004;47:888–899. doi: 10.1021/jm0309452. [DOI] [PubMed] [Google Scholar]
- 23.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 24.Li J, Edwards PC, Burghammer M, Villa C, Schertler GF. Structure of bovine rhodopsin in a trigonal crystal form. J. Mol. Biol. 2004;343:1409–1438. doi: 10.1016/j.jmb.2004.08.090. [DOI] [PubMed] [Google Scholar]
- 25.John B, Sali A. Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Res. 2003;31:3982–3992. doi: 10.1093/nar/gkg460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oliveira L, Hulsen T, Lutje Hulsik D, Paiva AC, Vriend G. Heavier-than-air flying machines are impossible. FEBS Lett. 2004;564:269–273. doi: 10.1016/S0014-5793(04)00320-5. [DOI] [PubMed] [Google Scholar]
- 27.Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- 28.Halperin I, Ma B, Wolfson H, Nussinov R. Principles of docking: An overview of search algorithms and a guide to scoring functions. Proteins. 2002;47:409–443. doi: 10.1002/prot.10115. [DOI] [PubMed] [Google Scholar]
- 29.Pogozheva ID, Lomize AL, Mosberg HI. The transmembrane 7 alpha-bundle of rhodopsin: distance geometry calculations with hydrogen bonding constraints. Biophys. J. 1997;72:1963–1985. doi: 10.1016/S0006-3495(97)78842-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fowler C, Pogozheva ID, LeVine H, III, Mosberg HI. Refinement of a homology model of the μ-opioid receptor using distance constraints from intrinsic and engineered zinc-binding sites. Biochemistry. 2004;43:8700–8710. doi: 10.1021/bi036067r. [DOI] [PubMed] [Google Scholar]
- 31.Fowler CB, Pogozheva ID, Lomize AL, Levine H, 3rd, Mosberg HI. Complex of an Active mu-Opioid Receptor with a Cyclic Peptide Agonist Modeled from Experimental Constraints. Biochemistry. 2004;43:15796–15810. doi: 10.1021/bi048413q. [DOI] [PubMed] [Google Scholar]
- 32.Pogozheva ID, Przydzial MJ, Mosberg HI. Homology modeling of opioid receptor-ligand complexes using experimental constraints. AAPS J. 2005;7 doi: 10.1208/aapsj070243., in press.
- 33.Chai B-X, Pogozheva ID, Lai Y-M, Li J-Y, Neubig RR, Mosberg HI, Gantz I. Receptor-antagonist interactions in the complexes of agouti and agouti-related protein with human melanocortin 1 and 4 receptors. Biochemistry. 2005;44:3418–3431. doi: 10.1021/bi0478704. [DOI] [PubMed] [Google Scholar]
- 34.Meng EC, Bourne HR. Receptor activation: what does the rhodopsin structure tell us? Trends Pharmacol Sci. 2001;22:587–593. doi: 10.1016/s0165-6147(00)01825-3. [DOI] [PubMed] [Google Scholar]
- 35.Karnik SS, Gogonea C, Patil S, Saad Y, Takezako T. Activation of G-protein-coupled receptors: a common molecular mechanism. Trends Endocrinol Metab. 2003;14:431–437. doi: 10.1016/j.tem.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 36.Hadley ME, Anderson B, Heward CB, Sawyer TK, Hruby VJ. Calcium-dependent prolonged effects on melanophores of [4-norleucine, 7-D-phenylalanine]-alpha-melanotropin. Science. 1981;213:1025–1027. doi: 10.1126/science.6973820. [DOI] [PubMed] [Google Scholar]
- 37.Yang Y-K, Fong T, Dickinson CJ, Li J-Y, Tota M, Van der Ploeg LTH, Gantz I. Molecular determinants of ligand binding to the human melanocortin-4 receptor. Biochemistry. 2000;39:14900–14911. doi: 10.1021/bi001684q. [DOI] [PubMed] [Google Scholar]
- 38.Yang Y-K, Dickinson C, Haskell-Luevano C, Gantz I. Molecular basis for the interaction of melanocortin peptides with the human melanocortin-1 receptor (α-MSH receptor) J. Biol. Chem. 1997;272:23000–23010. doi: 10.1074/jbc.272.37.23000. [DOI] [PubMed] [Google Scholar]
- 39.DeBlasi A, O’Reilly K, Motulsky HJ. Calculating receptor number from binding experiments using same compound as radioligand and competitor. Trends Pharmacol. Sci. 1989;10:227–229. doi: 10.1016/0165-6147(89)90266-6. [DOI] [PubMed] [Google Scholar]
- 40.Pogozheva ID, Lomize AL, Mosberg HI. Opioid receptor three-dimensional structures from distance geometry calculations with hydrogen bonding constraints. Biophys J. 1998;75:612–634. doi: 10.1016/S0006-3495(98)77552-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Güntert P, Wüthrich K. Improved efficiency of protein structure calculations from NMR data using the program DIANA with redundant dihedral angle constraints. J. Biomol. NMR. 1991;1:447–456. doi: 10.1007/BF02192866. [DOI] [PubMed] [Google Scholar]
- 42.Lagerstrom MC, Klovins J, Fredriksson R, Fridmanis D, Haitina T, Ling MK, Berglund MM, Schioth HB. High affinity agonistic metal ion binding sites within the melanocortin 4 receptor illustrate conformational change of transmembrane region 3. J. Biol. Chem. 2003;278:51521–51526. doi: 10.1074/jbc.M307683200. [DOI] [PubMed] [Google Scholar]
- 43.Sawyer TK, Hruby VJ, Darman PS, Hadley ME. [half-Cys4,half-Cys10]-alpha-Melanocyte-stimulating hormone: a cyclic alpha-melanotropin exhibiting superagonist biological activity. Proc. Natl. Acad. Sci. U. S. A. 1982;79:1751–1755. doi: 10.1073/pnas.79.6.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.al-Obeidi F, Hadley ME, Pettitt BM, Hruby VJ. Design of a new class of superpotent cyclicα-melanotropins based on quenched dynamic simulations. J. Amer. Chem. Soc. 1989;111:3413–3416. [Google Scholar]
- 45.Brooks BR, Bruccoleri ER, Olafson ER, States DJ, Swaminathan S, Karplus M. CHARMM: a program for macromolecular energy, minimization and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 46.Nickolls SA, Cismowski MI, Wang X, Wolff M, Conlon PJ, Maki RA. Molecular determinants of melanocortin 4 receptor ligand binding and MC4/MC3 receptor selectivity. J. Pharmacol. Exp. Ther. 2003;304:1217–27. doi: 10.1124/jpet.102.044974. [DOI] [PubMed] [Google Scholar]
- 47.Haskell-Luevano C, Nikiforovich G, Sharma SD, Yang YK, Dickinson C, Hruby VJ, Gantz I. Biological and conformational examination of stereochemical modifications using the template melanotropin peptide, Ac-Nle-c[Asp-His-Phe-Arg-Trp-Ala-Lys]-NH2, on human melanocortin receptors. J. Med. Chem. 1997;40:1738–1748. doi: 10.1021/jm960845e. [DOI] [PubMed] [Google Scholar]
- 48.Mutulis F, Yahorava S, Mutule I, Yahorau A, Liepinsh E, Kopantshuk S, Veiksina S, Tars K, Belyakov S, Mishnev A, Rinken A, Wikberg JE. New substituted piperazines as ligands for melanocortin receptors. Correlation to the X-ray structure of “THIQ”. J. Med.Chem. 2004;47:4613–4626. doi: 10.1021/jm0311285. [DOI] [PubMed] [Google Scholar]
- 49.Holder JR, Haskell-Luevano C. Melanocortin tetrapeptides modified at the N-terminus, His, Phe, Arg, and Trp positions. Ann. N. Y. Acad. Sci. 2003;994:36–48. doi: 10.1111/j.1749-6632.2003.tb03160.x. [DOI] [PubMed] [Google Scholar]
- 50.Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB, Hadley ME. 4-Norleucine, 7-D-phenylalanine-alpha-melanocyte-stimulating hormone: a highly potent alpha-melanotropin with ultralong biological activity. Proc. Natl. Acad. Sci. U.S.A. 1980;77:5754–5758. doi: 10.1073/pnas.77.10.5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prusis P, Frandberg PA, Muceniece R, Kalvinsh I, Wikberg JE. A three dimensional model for the interaction of MSH with the melanocortin-1 receptor. Biochem. Biophys. Res. Commun. 1995;210:205–10. doi: 10.1006/bbrc.1995.1647. [DOI] [PubMed] [Google Scholar]
- 52.Prusis P, Schioth HB, Muceniece R, Herzyk P, Afshar M, Hubbard RE, Wikberg JE. Modeling of the three-dimensional structure of the human melanocortin 1 receptor, using an automated method and docking of a rigid cyclic melanocyte-stimulating hormone core peptide. J. Mol. Graph. Model. 1997;15:307–317. doi: 10.1016/s1093-3263(98)00004-7., 334.
- 53.Prusis P, Muceniece R, Andersson P, Post C, Lundstedt T, Wikberg JE. PLS modeling of chimeric MS04/MSH-peptide and MC1/MC3-receptor interactions reveals a novel method for the analysis of ligand-receptor interactions. Biochim. Biophys. Acta. 2001;1544:350–357. doi: 10.1016/s0167-4838(00)00249-1. [DOI] [PubMed] [Google Scholar]
- 54.Hruby VJ, Lu D, Sharma SD, Castrucci AL, Kesterson RA, al-Obeidi FA, Hadley ME, Cone RD. Cyclic lactam alpha-melanotropin analogues of Ac-Nle4-cyclo[Asp5, D-Phe7,Lys10] alpha-melanocyte-stimulating hormone-(4-10)-NH2 with bulky aromatic amino acids at position 7 show high antagonist potency and selectivity at specific melanocortin receptors. J Med Chem. 1995;38:3454–3461. doi: 10.1021/jm00018a005. [DOI] [PubMed] [Google Scholar]
- 55.Ruprecht JJ, Mielke T, Vogel R, Villa C, Schertler GF. Electron crystallography reveals the structure of metarhodopsin I. EMBO J. 2004;23:3609–3620. doi: 10.1038/sj.emboj.7600374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prabhu NV, Perkyns JS, Pettitt BM. Modeling of alpha-MSH conformations with implicit solvent. J. Pept. Res. 1999;54:394–407. doi: 10.1034/j.1399-3011.1999.00113.x. [DOI] [PubMed] [Google Scholar]
- 57.Nikiforovich GV, Sharma SD, Hadley ME, Hruby VJ. Studies of conformational isomerism in alpha-melanocyte stimulating hormone by design of cyclic analogues. Biopolymers. 1998;46:155–167. doi: 10.1002/(SICI)1097-0282(199809)46:3<155::AID-BIP3>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 58.Ying J, Kover KE, Gu X, Han G, Trivedi DB, Kavarana MJ, Hruby VJ. Solution structures of cyclic melanocortin agonists and antagonists by NMR. Biopolymers. 2003;71:696–716. doi: 10.1002/bip.10596. [DOI] [PubMed] [Google Scholar]
- 59.Cai M, Cai C, Mayorov AV, Xiong C, Cabello CM, Soloshonok VA, Swift JR, Trivedi D, Hruby VJ. Biological and conformational study of beta-substituted prolines in MT-II template: steric effects leading to human MC5 receptor selectivity. J. Pept. Res. 2004;63:116–131. doi: 10.1111/j.1399-3011.2003.00105.x. [DOI] [PubMed] [Google Scholar]
- 60.Schioth HB, Mutulis F, Muceniece R, Prusis P, Wikberg JE. Discovery of novel melanocortin4 receptor selective MSH analogues. Br. J. Pharmacol. 1998;124:75–82. doi: 10.1038/sj.bjp.0701804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kask A, Mutulis F, Muceniece R, Pahkla R, Mutule I, Wikberg JE, Rago L, Schioth HB. Discovery of a novel superpotent and selective melanocortin-4 receptor antagonist (HS024): evaluation in vitro and in vivo. Endocrinology. 1998;139:5006–5014. doi: 10.1210/endo.139.12.6352. [DOI] [PubMed] [Google Scholar]
- 62.Sugg EE, Castrucci AM, Hadley ME, van Binst G, Hruby VJ. Cyclic lactam analogues of Ac-[Nle4]alpha-MSH4-11-NH2. Biochemistry. 1988;27:8181–8188. doi: 10.1021/bi00421a029. [DOI] [PubMed] [Google Scholar]
- 63.Haskell-Luevano C, Lim S, Yuan W, Cone RD, Hruby VJ. Structure activity studies of the melanocortin antagonist SHU9119 modified at the 6, 7, 8, and 9 positions. Peptides. 2000;21:49–57. doi: 10.1016/s0196-9781(99)00167-9. [DOI] [PubMed] [Google Scholar]
- 64.Nijenhuis WA, Kruijtzer JA, Wanders N, Vrinten DH, Garner KM, Schaaper WM, Meloen RH, Gispen WH, Liskamp RM, Adan RA. Discovery and in vivo evaluation of new melanocortin-4 receptor-selective peptides. Peptides. 2003;24:271–280. doi: 10.1016/s0196-9781(03)00032-9. [DOI] [PubMed] [Google Scholar]
- 65.Bednarek MA, MacNeil T, Kalyani RN, Tang R, Van der Ploeg LH, Weinberg DH. Analogs of lactam derivatives of alpha-melanotropin with basic and acidic residues. Biochem. Biophys. Res. Commun. 2000;272:23–28. doi: 10.1006/bbrc.2000.2589. [DOI] [PubMed] [Google Scholar]
- 66.Oosterom J, Burbach JP, Gispen WH, Adan RA. Asp10 in Lys-gamma2-MSH determines selective activation of the melanocortin MC3 receptor. Eur. J. Pharmacol. 1998;354:R9–11. doi: 10.1016/s0014-2999(98)00491-9. [DOI] [PubMed] [Google Scholar]
- 67.Yang Y, Chen M, Lai Y, Gantz I, Georgeson KE, Harmon CM. Molecular determinants of human melanocortin-4 receptor responsible for antagonist SHU9119 selective activity. J. Biol. Chem. 2002;277:20328–20335. doi: 10.1074/jbc.M201343200. [DOI] [PubMed] [Google Scholar]
- 68.Holder JR, Xiang Z, Bauzo RM, Haskell-Luevano C. Structure-activity relationships of the melanocortin tetrapeptide Ac-His-D-Phe-Arg-Trp-NH2 at the mouse melanocortin receptors. 4. Modifications at the Trp position. J. Med. Chem. 2002;45:5736–5744. doi: 10.1021/jm020296e. [DOI] [PubMed] [Google Scholar]
- 69.Schioth HB, Petersson S, Muceniece R, Szardenings M, Wikberg JE. Deletions of the N-terminal regions of the human melanocortin receptors. FEBS Lett. 1997;410:223–228. doi: 10.1016/s0014-5793(97)00593-0. [DOI] [PubMed] [Google Scholar]
- 70.Srinivasan S, Lubrano-Berthelier C, Govaerts C, Picard F, Santiago P, Conklin BR, Vaisse C. Constitutive activity of the melanocortin-4 receptor is maintained by its N-terminal domain and plays a role in energy homeostasis in humans. J. Clin. Invest. 2004;114:1158–1164. doi: 10.1172/JCI21927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dyck B, Parker J, Phillips T, Carter L, Murphy B, Summers R, Hermann J, Baker T, Cismowski M, Saunders J, ad Goodfellow V. Aryl piperazine melanocortin MC4 receptor agonists. Bioorg. Med. Chem. Lett. 2003;13:3793–3796. doi: 10.1016/s0960-894x(03)00796-0. [DOI] [PubMed] [Google Scholar]
- 72.Herpin TF, Yu G, Carlson KE, Morton GC, Wu X, Kang L, Tuerdi H, Khanna A, Tokarski JS, Lawrence RM, Macor JE. Discovery of tyrosine-based potent and selective melanocortin-1 receptor small-molecule agonists with anti-inflammatory properties. Med. Chem. 2003;46:1123–1126. doi: 10.1021/jm025600i. [DOI] [PubMed] [Google Scholar]
- 73.Pan K, Scott MK, Lee DH, Fitzpatrick LJ, Crooke JJ, Rivero RA, Rosenthal DI, Vaidya AH, Zhao B, Reitz AB. 2,3-Diaryl-5-anilino[1,2,4]thiadiazoles as melanocortin MC4 receptor agonists and their effects on feeding behavior in rats. Bioorg. Med. Chem. 2003;11:185–192. doi: 10.1016/s0968-0896(02)00428-5. [DOI] [PubMed] [Google Scholar]
- 74.Ujjainwalla F, Warner D, Walsh TF, Wyvratt MJ, Zhou C, Yang L, Kalyani RN, MacNeil T, Van der Ploeg LH, Rosenblum CI, Tang R, Vongs A, Weinberg DH, Goulet MT. Design and syntheses of melanocortin subtype-4 receptor agonists: evolution of the pyridazinone archetype. Bioorg. Med. Chem. Lett. 2003;13:4431–4435. doi: 10.1016/j.bmcl.2003.09.026. [DOI] [PubMed] [Google Scholar]
- 75.Richardson TI, Ornstein PL, Briner K, Fisher MJ, Backer RT, Biggers CK, Clay MP, Emmerson PJ, Hertel LW, Hsiung HM, Husain S, Kahl SD, Lee JA, Lindstrom TD, Martinelli MJ, Mayer JP, Mullaney JT, O’Brien TP, Pawlak JM, Revell KD, Shah J, Zgombick JM, Herr RJ, Melekhov A, Sampson PB, King CH. Synthesis and structure-activity relationships of novel arylpiperazines as potent and selective agonists of the melanocortin subtype-4 receptor. J. Med. Chem. 2004;47:744–755. doi: 10.1021/jm0304109. [DOI] [PubMed] [Google Scholar]
- 76.Holst B, Elling CE, Schwartz TW. Metal ion-mediated agonism and agonist enhancement in melanocortin MC1 and MC4 receptors. J. Biol. Chem. 2002;277:47662–47670. doi: 10.1074/jbc.M202103200. [DOI] [PubMed] [Google Scholar]
- 77.Holst B, Schwartz TW. Molecular mechanism of agonism and inverse agonism in the melanocortin receptors: Zn(2+) as a structural and functional probe. Ann. N. Y. Acad. Sci. 2003;994:1–11. doi: 10.1111/j.1749-6632.2003.tb03156.x. [DOI] [PubMed] [Google Scholar]
- 78.Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimentional models and computational probing of structure-function relations in G-protein coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.