

Abstract
Mutations in the gene encoding human ATP:cobalamin adenosyltransferase (hATR) can result in the metabolic disorder known as methylmalonic aciduria (MMA). This enzyme catalyzes the final step in the conversion of cyanocobalamin (vitamin B12) to the essential human cofactor adenosylcobalamin. Here we present the 2.5 Å crystal structure of ATP bound to hATR refined to an Rfree value of 25.2 %. The enzyme forms a tightly associated trimer, where the monomer comprises a five helix bundle and the active sites lie on the subunit interfaces. Only two of the three active sites within the trimer contain the bound ATP substrate, thereby providing examples of apo- and substrate-bound active sites within the same crystal structure. Comparison of the empty and occupied sites indicates that twenty residues at the enzyme’s N-terminus become ordered upon binding of ATP to form a novel ATP-binding site and an extended cleft that likely binds cobalamin. The structure explains the role of 20 invariant residues; six involved in ATP-binding, including Arg190 which hydrogen bonds to ATP atoms on both sides of the scissile bond, 10 required for structural stability and four in positions to interact with cobalamin. The structure also reveals how the point mutations that cause MMA are deficient in these functions.
Keywords: Cobalamin, Vitamin B12, adenosyltransferase, X-ray crystallography, methylmalonic aciduria
Cobalamin functions as an enzymatic cofactor for a variety of metabolic reactions, including methionine synthesis, succinyl-CoA synthesis and fermentation of small molecules (1–3). In mammalian systems, vitamin B12, (cyano-cobalamin, CNCbl), is ingested and converted into two cofactors, methylcobalamin (MeCbl) and adenosylcobalamin (AdoCbl), each of which is required for an essential catalytic activity. MeCbl is the cofactor for methionine synthase, which converts homocysteine to methionine (4, 5). Adenosylcobalamin (AdoCbl) is the cofactor for methylmalonyl CoA mutase (MMCM), which catalyzes the isomerization of methylmalonyl CoA to succinyl CoA and is responsible for normal catabolism of various amino and fatty acids through the TCA cycle (6).
The conversion from CNCbl to the active cofactors requires carrier proteins, membrane transport, cobalt reduction, and exchange of the axial ligand (7–10). Genetic alterations in the branched modification pathway can disrupt either MeCbl or AdoCbl production or both (11, 12). The disruptions at particular points in the pathway can be physiologically distinguished based on enzymatic activities, blood plasma analysis, and medical responsiveness to various cobalamin analogue treatments (13). These inborn errors of vitamin B12 metabolism have been categorized into eight complementation classes, cblABCDEFG and mut (14, 15). Loss of AdoCbl results in methylmalonic aciduria (MMA), a buildup of methylmalonic acid in body fluids, including blood and urine (12, 16). Mutations in three different genetic loci, cblA, cblB and mut, specifically result in AdoCbl deficiency. Methylmalonyl CoA-mutase is encoded at the mut locus (17), cblA was mapped to an uncharacterized enzyme with GTPase activity (18), and the deficient enzyme of the cblB class was identified as ATP:cobalamin adenosyltransferase (ATR) (19). BLAST search of the human genome using a Archaeoglobus fulgidus ATR homolog, review of mutations in the genes of MMA patients, and bacterial complementation studies subsequently revealed the gene encoding the human ATR homolog (hATR), named hMMAB (20, 21). Sequencing of hMMAB in MMA patients revealed thirty-two different types of mutations, including six point mutations. MMA patient cell lines containing hATR mutations show reduced AdoCbl levels in vitro (20, 22, 23).
ATP:Cobalamin adenosyltransferase catalyzes transfer of adenosine from ATP to cobalamin to generate AdoCbl. The PduO-family of adenosyltransfreases has a strong preference for ATP, and other nucleotides are inefficient donors to cobalamin (24–26). Prior to catalytically relevant binding, cob(III)alamin is reduced to cob(II)alamin by single electron reduction, a reaction catalyzed by a reductase such as flavodoxin or methionine synthase reductase (26, 27). If the hATR is preincubated with ATP, the binding of cob(II)alamin triggers a transition to a novel four coordinate cob(II)alamin (28, 29). This is followed by a second single electron reduction at the cobalt center to produce enzyme-bound cob(I)alamin, a step that might be catalyzed by methionine synthase reductase (26). Interactions between the ATR and the cob(II)alamin are thought to lower the reduction potential of the cobalt into the physiologically accessible range (30, 31). Finally, while bound to hATR, the four coordinate cob(I)alamin attacks the C5* atom of the bound ATP to release the triphosphate group and form the cobalt-carbon bond of adenosylcobalamin (Figure 1). The roles of specific hATR residues in the course of this reaction are unknown.
Figure 1.

Schematic outline of the reaction catalyzed by ATP:cobalamin adenosyltransferase.
Sequence analysis indicates that ATP:cobalamin adenosyltransferases fall within three unrelated families: CobA, PduO and EutT (24). Structure determinations confirm that the CobA (32) and PduO (25) families adopt quite different architectures; a structure has not yet been described for a member of the EutT family. Human ATR belongs to the PduO-like family which, as indicated by the structure of archaeal Thermoplasma acidophilium TA0546, adopts a helical bundle fold that assembles as a stable trimer (25). This structure (32% identical to hATR) was determined in the apo state, as were six other sequence-related proteins that are thought to have ATR activity. Because these structures lack bound ligands, the detailed location of the active site and the function of specific invariant residues are unknown.
In an effort to better understand the PduO-like ATR family, and the human enzyme in particular, we have determined the structure of hATR in the presence of ATP. This structure reveals previously unstructured invariant residues at the N-terminus, the functional role of the invariant residues surrounding the ATP, the putative location of the cobalamin-binding site, and residues likely to be involved in the catalytic mechanism.
Methods
A human MMAB clone, starting at the equivalent region of the sequence as previously crystallized homologs (P56), was generated from I.M.A.G.E. clone 2822202 (ATCC) and inserted into a pET3a vector (Invitrogen). Sequencing determined that the clone is the 239K variant (20). No affinity tag is present, but an initiator Met was added using the Nde1 restriction enzyme during cloning. The recombinant hATR was overexpressed in E. coli using an auto-induction method (33). Cell pellets were lysed by sonication in 20 mM HEPES, pH 7.5, 50 mM NaCl, 1 mM DTT and centrifuged to remove the particulate fraction. The soluble lysate was loaded onto a HiTrap SP HP column (Pharmacia) and eluted by a salt gradient at 300 mM NaCl. The protein was relatively pure at this stage, but was dialyzed against 20 mM HEPES, pH 7.5, 100 mM NaCl, 1 mM DTT, concentrated, and further purified by size exclusion chromatography on an SD100 column (Pharmacia). The protein was concentrated to 20 mgml−1 for crystallization trials in the presence of 1–5 mM MgATP. Lower or higher ATP concentrations did not yield crystals.
hATR-ATP crystals were grown by vapor diffusion at room temperature by mixing 2 µl of protein solution with 2 µl of reservoir; 18–20% polyethyleneglycol, MW = 3350, 0.2 M MgSO4 and 10% glycerol. The crystals belong to space group P321, with cell dimensions of a = b = 111.2 Å, c = 115.5 Å, and three molecules in the asymmetric unit. Native data were collected at the National Synchrotron Light Source (Brookhaven, NY) beamline x26c (Table 1). The structure was determined by molecular replacement (PHASER (34)) using a model of hATR based on the trimeric structure of the Bacillus subtilus YVQK (41% identical to hATR; PDBcode:1RTY). Residues of helical regions were modeled as the equivalent human sequence and flexible loops were removed prior to molecular replacement. The primary programs used for structure determination and model building were HKL (35), CCP4 (36), O (37) and Coot (38).
Table 1.
Data Collection
| Space Group | P321 |
| # Observations | 113031 |
| # Unique Reflections | 29063 |
| Resolution Range (Å) | 50 – 2.5 |
| (High res. Shell) | (2.59 – 2.5) |
| Completeness (%) | 99.8 (100) |
| I/σ(I) | 9 (2) |
| Rsym1 | 13.6 % (56%) |
| Wilson B (Å2)2 | 46.2 |
| Rcryst3 | 19.9 % (22 %) |
| Rfree3 | 25.2 % (25%) |
| RMS deviations | |
| Bonds (Å) | 0.013 |
| Angles (°) | 1.5 |
| <B> (Å2) | |
| Main chain | 33.7 |
| Side chain | 34.9 |
| ATP (2) | 25.8 |
| SO4 (3) | 60.4 |
| Mg (4) | 35.8 |
| Cl | 33.4 |
| H2O (171) | 32.7 |
Rsym = ∑|I − <I>| / ∑ I, where <I> is the average intensity from multiple observations of symmetry related reflections.
The Wilson B was calculated using TRUNCATE between 4 – 2.5Å resolution and contains an error of ± 10 Å2 (36).
Rcyst=∑||Fo| − |Fc||/∑|Fo| × 100 over 95% of the data. Rfree = Rcryst on 5% of the data not used in refinement.
The hATR construct, residues 56 to 250 (C-terminus), includes all of the highly conserved amino acids (Supplemental data). The missing upstream region includes the predicted mitochondrial targeting sequence, residues 1 – 32. The following ordered residues are included in the refined model: A79–129, A144–240, B57–135, B142–240, C58–104 and C108–240. Two of the trimer’s active sites are seen to each bind one ATP molecule and two Mg2+ ions. The final model has been refined at a resolution of 2.5 Å using REFMAC (39) to an Rfactor of 19.8 % with an Rfree value of 25.2 %, and good geometry (Table 1). The structural coordinates have been deposited at the RCSB Protein Data Bank with deposition code 2IDX.
Related cobalamin adenosyltransferase structures were identified by using the program DALI (40), the Protein Structure Comparison Service (41), and by running a BLAST search using the PDB as the amino acid sequence database. A survey of ATP-binding sites was completed by visually scanning all non-redundant PDB entries that contained ATP as a ligand (~266 structures).
Results and Discussion
Structure of the hATR monomer
The hATR monomer adopts a helical ferritin-like architecture (Figure 2) (42). Structures of this type are defined by their organization of four helices; α1, α2, α4, and α5 of hATR. These four helices form a bundle in which spatially adjacent helices are antiparallel to each other. The PduO-type cobalamin adenosyltransferase family is distinguished by an additional helix, α3 of hATR, which is located in the long loop that connects α2 to α4 (Figure 2a). Although ferritin contains a similar four-helical bundle arrangement, its long-crossover loop does not contain a helix and it wraps around the bundle in the opposite sense to that of the PduO-type cobalamin adenosyltransferases.
Figure 2.
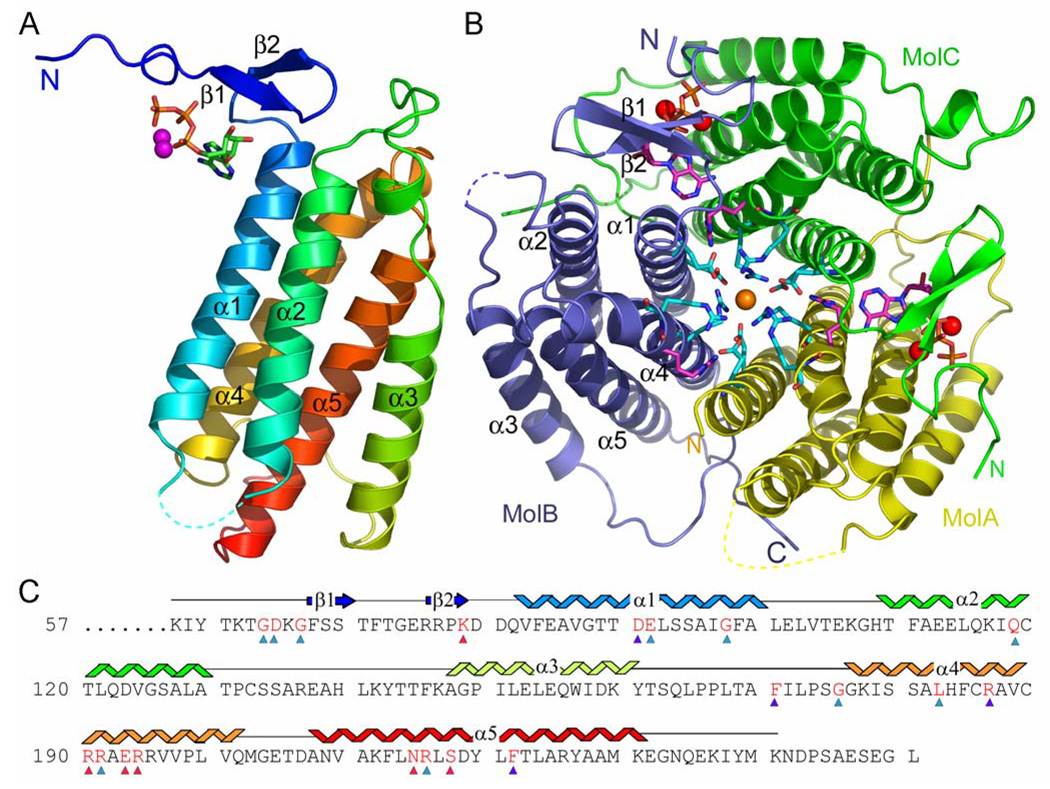
Structure of hATR. A) Side view of the hATR monomer. The trace is colored blue from the N-terminus to red at the C-terminus. The N-terminal portion is only ordered in the presence of bound ATP; molecule C is shown. B) Top view of the hATR trimer. Molecules A, B and C are colored yellow, blue and green, respectively. ATP molecules (magenta) with two Mg2+ ions each (red) are bound at the AC and CB interfaces underneath the ordered N-terminal β-strands. The apo-active site sits at the BA interface. The trimer interface formed by the supercoil of helices α1 and α4 contains alternating Glu/Arg residues (Glu84/Arg195 and Glu91/Arg191 - cyan). This charged network connects via Glu84 to the ATP binding site though a hydrogen bond to invariant Arg194 (magenta). A chloride ion has been modeled in the center of the charged ring (orange). Two rings of phenylalanine side chains lie on the trimer interface below the Arg/Glu pairs and are omitted from this figure for clarity. C) Sequence of the hATR with invariant resides highlighted in red, as determined by the extended sequence alignment included as supplemental data. Secondary structure is shown above and colored as in Figure 2a. Colored triangles below the sequence denote the functional role of the invariant residues (Table 2), ATP-binding – red, cobalamin binding pocket – blue, structural function – cyan. Figure 2a,b, Figure 3, Figure 4 and Figure 5 were made with PYMOL (45).
ATR activity has been confirmed and the structure has been described for the Thermoplasma acidophilum protein 0546 (PDBcode:1NOG) which is 32% identical to hATR (25). Several other related structures of probable ATR enzymes have also been reported by proteomics consortia, including: Bacillus subtilis YVQK (PDBcode:1RTY), Sulfolobus tokodaii ST2180 (PDBcode:1WVT), Sulfolobus Tokodaii ST1454 (PDBcode:1WOZ), Bacillus halodurans 10174212 (PDBcode:2AH6), Mycobacterium tuberculosis Mb1347c (PDBcode:2G2D) and Pyrococcus horikoshi OT3 PH0671 (PDBcode:1WY1). A sequence alignment containing a range of species and all known crystal structures is presented as Supplemental Data. The list of invariant residues as defined by this alignment is shown in Figure 2c and listed in Table 2. Of these structures, the most similar sequence (41% identical) to the human enzyme is for Bacillus subtilis YVQK, which was used for the molecular replacement solution. The r.m.s. deviation between these related structures and hATR is typically 1.2–2.0 Å over ~140 Cα residues. Interestingly, Thermoplasma acidiphilum TA1238 (PDBcode:1NIG) contains a short fifth helix indicative of the cobalamin ATR family, but does not contain the conserved amino acids that are thought to function in ATP or cobalamin binding (43). None of these structures were determined in the presence of ATP, leaving the structure and functional role of many conserved resides unknown.
Table 2.
Invariant Residues and their function within the hATR structure.
| Invariant Residue | Function |
|---|---|
| Gly63 | Forms a tight turn to allow close proximity of β-strands to active site |
| Asp64 | Forms salt bridge with Arg215 to hold N-terminal loop in place |
| Gly66 | Within a tight turn of the N-terminal loop |
| Lys78 | H-bonds directly with a beta phosphate, Glu193, water and N-terminal loop. |
| Asp90 | Potential interaction with cobalamin |
| Glu91 | Salt bridge with Arg191 at center of trimeric interface |
| Gly97* | Positioned on α1 and allows close approach of the α3- α4 loop |
| Gln118 | Sits on α2 and H-bonds the α3- α4 loop and back to α1 |
| Phe170 | Potential interaction with cobalamin |
| Gly175 | Finishes the α3- α4 loop and starts α4 |
| Leu182 | Hydrophobic core |
| Arg186* | Potential interaction with cobalamin |
| Arg190* | H-bonds with all three sections of ATP and on either side of scissile bond. |
| Arg191* | Salt bridge with Glu91 at center of trimer interface |
| Glu193* | H-bonds with one Mg-HOH and NZ of Lys78 |
| Arg194 | Forms H-bond with adenosine N1 and leads to charged trimeric interface through H-bond to Glu84. Also forms water mediated H-bond to Asp90. |
| Asn214 | A direct ligand to Mg2+ and two gamma phosphate oxygens |
| Arg215 | Salt bridge with Asp64 locking down N-terminal loop and ATP binding cleft. |
| Ser217 | Forms H-bond to Mg-HOH |
| Phe221 | Potential interaction with cobalamin |
Structure of the hATR trimer
Human ATR is a trimer in the crystal and in solution, as indicated by gel filtration chromatography. Helices α1 and α4 of each monomer associate to form the six-stranded coiled-coil core of the trimer (Figure 2b). All of the related structures listed above also crystallize in the same trimeric arrangement, with the T. acidophilum 0546 and Mycobacterium tuberculosis Mb1347c enzymes trimerizing about a crystallographic three-fold axis and the others associating via non-crystallographic symmetry. Toward the top of the structure (closer to the ATP-binding site; Figure 2b) the trimer interface contains two charged rings of Arg/Glu pairs (Glu84/Arg195 and Glu91/Arg191) that surround what appears to be a chloride ion. Glu84 from the top-most ring also forms a hydrogen bond with the guanidinium of the invariant Arg194 residue, which in turn hydrogen bonds to the N1 nitrogen of the bound ATP, thereby connecting the trimer interface to the active site. Below these two rings of charged residues lie two additional rings of phenylalanines (conserved as hydrophobic residues) that form a solid core to the interface. The hATR subunit interfaces are extensive (the apo- and substrate-bound interfaces cover 895 and 1450 Å2), and are mediated by conserved residues, thereby indicating that the formation of the trimer is required for activity.
Although the presence of differently occupied active sites could indicate allostery within the trimer, we do not favor this possibility because superposition of occupied and unoccupied subunits does not reveal an obvious pathway of conformational changes that might mediate communication between the active sites. The r.m.s. deviation between the three subunits using pairs of equivalent Cα atoms is only 0.73 – 0.78 Å (Figure 3c), although as discussed below, there are some distinctive differences at the active sites. It seems more likely that crystal contacts at each active site stabilize or prohibit conformations that are able to bind ATP. Curiously, cobalamin is also seen bound to just one of the two active sites in the crystal structure of dimeric CobA, which is structurally unrelated to ATR but catalyzes the same reaction (32). Biochemical studies indicated that, as appears be the case for hATR, the structural asymmetry of CobA is a crystallographic artifact and is probably not of mechanistic significance (44).
Figure 3.
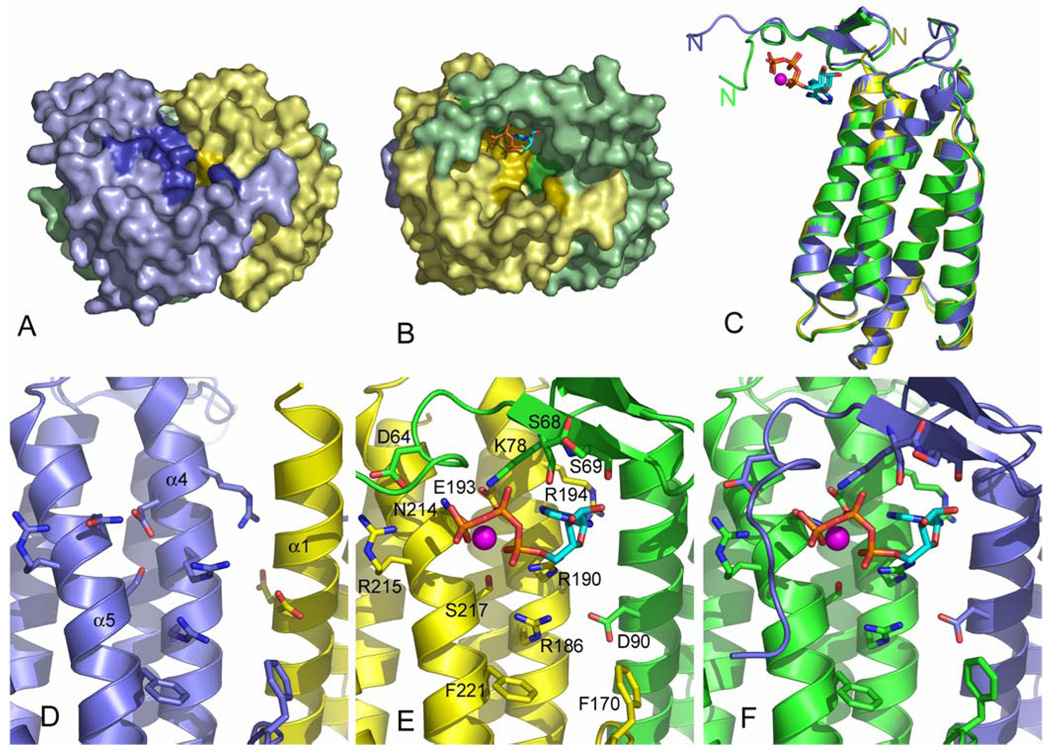
Side view of the hATR active sites. A) Surface of the apo-active site (at the BA interface) reveals several exposed invariant residues. The figure is colored as in figure 2b with invariant residues in darker hue. The right-hand molecule, molecule A (yellow), is only ordered from residue 79. B) An ATP-bound active site (the AC active site is shown in the same orientation as in panel A) reveals the ordered N-terminal residues of the right-hand molecule, molecule C (green), starting at residue 57, and wrapping over the top of the bound ATP (ball-and-stick). C) All three monomers are aligned revealing close structural similarity except at the N-terminus where binding of ATP causes ordering of ~22 N-terminal residues in molecules B and C. D) Detailed view of the apo active site (the BA interface). E) The ATP-bound active site at the AC interface with residues involved in binding ATP or positioned in the putative cobalamin binding pocket (Arg186, Phe170, Phe221, Asp90) labeled. All labeled residues are invariant except Ser68 and Ser69. F) The ATP-bound active site of the BC interface reveals slight differences in the extreme N-terminus of molecules B and C.
ATP binding site
The location of the ATP binding site at the interface between adjacent monomers further indicates that ATR is functional as a trimer. Notably, the three active sites in the asymmetric unit are distinctly different, with only two of them containing ATP and the other (between molecules A and B) being unliganded (Figure 2b, Figure 3). The active site cleft is formed between α1 and α2 of one monomer, and the α3-α4 loop and α4 and α5 of its neighbor, and is lined by many conserved residues. Comparison of bound and apo active sites reveals that binding of ATP triggers a significant local ordering of the structure. N-terminal residues surrounding the apo-active site are disordered before residue 79 (Figure 3a,d), whereas active sites occupied by ATP are ordered from residue 57 or 58 (2 or 3 residues after the N-terminus of this construct) (Figure 3b,e,f). The cleft formed by the ordered N-terminus is a deep pocket slightly bigger than twice the size of the bound ATP. This conformational change suggests that binding of ATP contributes to formation of the cobalamin binding site. The structurally unrelated CobA-type ATP:cobalamin adenosyltransferase has also been shown to increase in structural order with the binding of substrates (32).
Nucleotide binding to hATR involves the formation of numerous hydrogen bonds and Van der Waals contacts. ATP binds with the adenosine ring inserting towards the core of the enzyme and the ribose hydroxyls and several phosphate oxygens in solvent accessible positions (Figure 4a,b). The adenosine lies near strands β1, β2 and helix α1, which is a region previously proposed to be involved in nucleotide binding (21). A search of ATR-like structures and of ATP-bound protein structures did not reveal a similar mode of nucleotide binding, indicating that hATR contains a novel type of ATP-binding site. Hydrogen bonds extend from the adenosine amide group (N6) to both the side chain of Glu193 and the main chain carboxyl of Arg190 (Figure 4b, see Table 2). The adenosine N1 nitrogen hydrogen bonds with the side chain of Arg194, and this interaction connects the ATP binding pocket to the top-most charged ring of the trimer interface through a second hydrogen bond between Arg194 and Glu84. Finally, the adenosine N3 nitrogen packs against the conserved residue Gly87, and explains why PduO-like ATRs prefer ATP as the nucleotide donor over GTP. While hATR is 16% efficient at using GTP as the nucleotide donor (26), activity with GTP is below measurable levels for the T. acidophilum TA1434 and Salmonella PduO enzymes (24, 25). In contrast, CobA-like ATRs have fewer hydrogen bonds between the adenosine and the enzyme, consistent with their nucleotide promiscuity (32).
Figure 4.
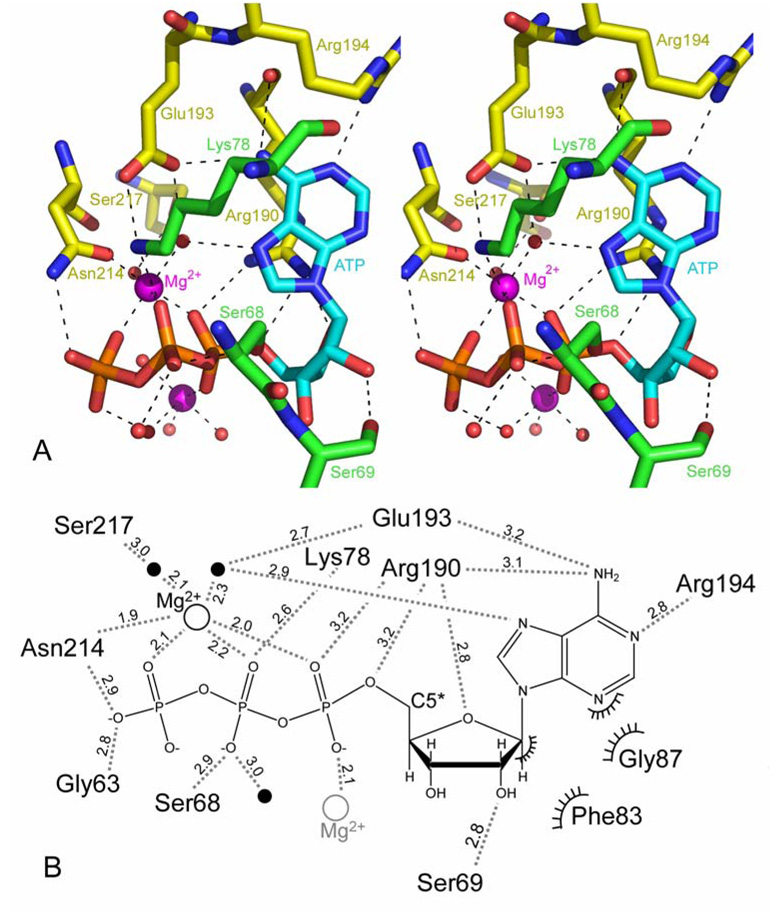
ATP binding site. A) The ATP binding site is shown in stereo. Invariant residues that are involved in forming hydrogen bonds to the bound ATP are shown explicitly. The hydrogen bond from non-conserved Ser69 extends from its main chain carbonyl not its hydroxyl side chain. Two Mg2+ ions interact with the tri-phosphate moiety. Arg194 recognizes the adenosine and connects this interaction to the trimer core. B) Schematic representation of ATP binding site and interactions with hATR residues. Hydrogen bonds are shown as dashed lines with distances shown in Ångstroms. Waters and Mg2+ are shown as closed and open circles. The secondary Mg which is likely not physiologically relevant is shown in grey. Ser68, Phe83 and Gly87 are conserved but not invariant.
The Arg190 guanidinium group packs against the five-membered ring of the nucleotide and also hydrogen bonds with the ribose oxygen. The ring oxygen (O4*) and the phosphate bridging oxygen (O5*), which are on opposite sides of the scissile C5*–O5* bond, both interact with the side chain of Arg190 (Figure 4). An additional hydrogen bond is formed between the ribose O2* hydroxyl and the main chain carbonyl of Ser69. Finally, hydrophobic contacts between Phe83 and the ribose ring, and between nearby Gly87 and a portion of the adenosine ring, further define the ATP binding conformation. Together these contacts work to orient the C5* atom towards the putative cobalamin binding site (Figure 4).
The three phosphates pack against the N-terminal loop of one subunit and helices α4 and α5 of the adjacent subunit (Figure 3e,f, 4a). Two Mg2+ ions have been modeled coordinating the phosphates; one of which binds oxygen atoms from all three phosphate groups, two water molecules, and the side chain oxygen atom of invariant Asn214. Because Asn214 also hydrogen bonds with Asp64, this interaction may be especially important for ordering of the N-terminal residues upon binding of ATP (Figure 3e,f). The other modeled Mg2+ ion is located in a much more solvent accessible environment, and may be the result of high concentrations of MgSO4 in the crystallization conditions rather than indicating a physiologically significant coordination. Phosphate oxygens also form hydrogen bonds with the side chains of the invariant residues Asn214 and Arg190, the carbonyl of Gly63, and with the hydroxyl Ser68 (conserved Ser/Thr – Supplemental Data, Figure 4a,b).
Cobalamin binding site
The ordering of hATR N-terminal residues that occurs upon binding of ATP induces formation of an active site cleft that not only holds ATP, but also contains an oval-shaped opening that measures 14 Å by 8 Å and is 7 Å deep (Figure 3b). The top of this presumed cobalamin binding cleft is bounded by the ATP molecule itself and the ATP C5* atom is oriented towards the center of the pocket as if poised for catalysis. The surface of the cleft includes four invariant residues (Asp90, Phe170, Arg186 and Phe221) that are not involved in binding ATP but could potentially interact with cobalamin (Figure 3e,f). The invariant side chain of Arg186 projects just below the ATP and is a candidate for binding the cobalamin directly, perhaps to the side chain amides. Asp90 appears well placed to interact with the bound cobalamin or may function in catalysis. Two invariant phenylalanine residues, Phe170 and Phe221, project into the active site cleft and are good candidates for binding the hydrophobic surfaces of cobalamin, or the dimethylbenzimidazole (DMB) tail.
Modeling cobalamin binding
A key question to understanding the mechanism of hATR is the conformation of cobalamin when bound to the enzyme, because this can dramatically alter reactivity at the Co center. UV-VIS, EPR and MCD spectra all indicate changes in the cobalt coordination suggestive of a four-coordinate "base-off" conformation when cobalamin binds to an ATP:hATR complex (28, 29). Because the DMB tail is inherently very flexible, there is a wide range of potential base-off conformations. The conformation of the DMB tail shown for our model is especially speculative and is only included in order to provide a sense of scale.
Our model of docked cobalamin is approximate, but illustrates that the cleft formed upon ATP binding is an appropriate size and shape to accommodate the cobalamin ring and that invariant residues are suitably positioned to make important interactions (Figure 5). The only constraints on the docking are placing the cobalt atom ~3–4 Å away from the target carbon (C5*) of the ATP and avoidance of obvious steric clashes. In our model, the upper axial position of the cobalt points towards the ATP and the DMB tail, extending off ring D, points towards solvent. The plane of the macrocycle is positioned such that rings A and B are inserted into the cleft. In this conformation, the invariant phenylalanine residues below the active site are in a position to interact with the lower corrin ring structure and Phe170 in particular may assist with exclusion of solvent below the cobalt. No polar amino acid residue appears to be available to interact with the cobalt ion from the lower face as a replacement for the benzimidazole, consistent with the earlier conclusion that the DMB is not replaced by a protein residue (28, 29).
Figure 5.
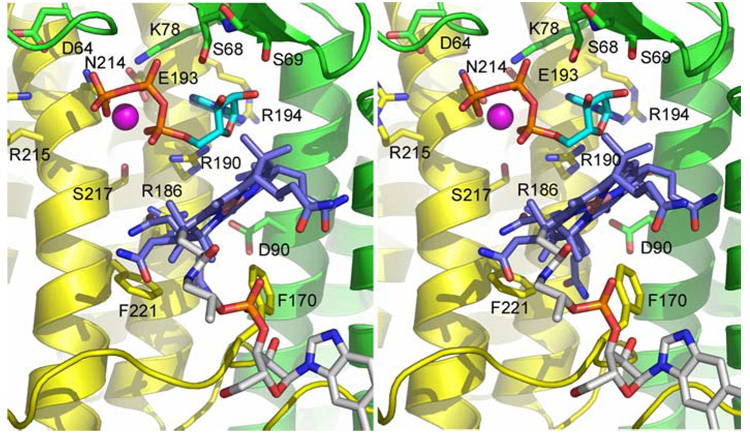
Model of base-off cobalamin bound to the active site, shown in stereo in the same orientation as figure 3e. The model is not intended to imply specific residue interactions, but rather to provide a sense of scale and demonstrate the plausibility of the apparent cobalamin binding pocket. The corrin ring (blue) is positioned ~4Å away from the ATP (cyan) and the amide side chains of the A- and B-rings point towards the base of the binding cleft. The benzimidazole tail (white) is extended away from the structure in a position that is highly speculative in the absence of additional structural information.
Known methylmalonic aciduria mutations
Mutations in hMMAB that contribute to disease have been identified in methylmalonic aciduria patients at positions Gly97*, Ser174, Arg186*, Arg190*, Arg191*, Glu193* and Gln234 (* indicates invariant residues listed in Table 2) (20, 22, 23). Mutations in MMA patients have also been observed at Arg19 in the mitochondrial targeting sequence and at nonconserved positions Ala135, Ser180 and Tyr219. The functional significance of these mutations is unclear, however, because they have only been found in patients that are heterozygous for mutations in an invariant residue. Amino acid substitutions at Gly97 or Ser174, or a stop codon at position Gln234 would disrupt the orientation of the α3–α4 loop at the bottom of the active site. The most common mutation is a substitution of Arg186 for Trp or Gln. This arginine does not contact ATP in the structure, but rather lies below the ATP in the cobalamin binding site and appears well positioned to interact with the cobalamin directly (Figure 3e,f, 5). As described above, Arg190 forms hydrogen bonds with the ATP and orients the C5* atom for catalysis. Arg191 is an invariant residue involved in the charged trimer interface. Mutation to Trp or Ala does not eliminate trimer formation, but does knock out or reduce catalytic activity, while increasing the Km for ATP three-fold over wild-type levels (20, 22, 25). Clearly, the trimeric interface is crucial for adenosyltransferase activity. Finally, Glu193 is involved in coordinating the physiological Mg2+ ion, and mutation to Lys would inhibit ATP binding.
In summary, our structure of the hATR-ATP complex explains the importance of residues found mutated in methylmalonic aciduria patients and suggests a role for each of the invariant residues in stabilizing the structure, binding ATP, binding cobalamin, or in catalysis (Table 2). This advance will facilitate future studies aimed at understanding the mechanism of the ATR enzymes.
Supplementary Material
ACKNOWLEDGMENT
We thank Anne Heroux for data collection at the National Synchrotron Light Source Brookhaven National Laboratory, beamline x26c.
Abbreviations
- ATP
Adenosine triphosphate
- Cbl
cobalamin
- CNCbl
cyanocobalamin
- MeCbl
methylcobalamin
- AdoCbl
adenosylcobalamin
- OHCbl
hydroxycobalamin
- MMCA
methylmalonyl CoA Mutase
- MMA
methylmalonic aciduria
- MMAB
MMA gene for complementation group CblB
- ATR
adeonsyltransferase
- PDBcode
Protein Data Bank code
- r.m.s.
root mean square
- EPR
electron paramagnetic resonance
- MCD
magnetic circular dichroism
Footnotes
This work was supported by the NIH (Grants GM56775 and DK072437). Data collection at NSLS was supported by the Offices of Biological and Environmental Research and of Basic Energy Sciences of the U.S. Department of Energy/National Center for Research Resources of the NIH.
REFERENCES
- 1.Banerjee R, editor. Chemistry and Biochemistry of B12. New York: John Wiley and Sons; 1999. [Google Scholar]
- 2.Roth JR, Lawrence JG, Bobik TA. Cobalamin (coenzyme B12): synthesis and biological significance. Annu Rev Microbiol. 1996;50:137–181. doi: 10.1146/annurev.micro.50.1.137. [DOI] [PubMed] [Google Scholar]
- 3.Kräutler B. B12 Coenzymes, the Central Theme. In: Kräutler B, Arigoni D, Golding BT, editors. Vitamin B12 and B12-Proteins. New York: Wiley-VCH; 1997. pp. 3–43. [Google Scholar]
- 4.Cauthen SE, Foster MA, Woods DD. Methionine synthesis by extracts of Salmonella typhimurium. Biochem J. 1966;98:630–635. doi: 10.1042/bj0980630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drummond JT, Matthews RG. Cobalamin-dependent and cobalamin-independent methionine synthases in Escherichia coli: two solutions to the same chemical problem. Adv Exp Med Biol. 1993;338:687–692. doi: 10.1007/978-1-4615-2960-6_142. [DOI] [PubMed] [Google Scholar]
- 6.Banerjee R, Chowdhurry S. Methylmalonyl-CoA Mutase. In: Banerjee R, editor. Chemistry and Biochemistry of B12. New York: John Wiley and Sons; 1999. pp. 707–730. [Google Scholar]
- 7.Pezacka EH. Identification and characterization of two enzymes involved in the intracellular metabolism of cobalamin. Cyanocobalamin beta-ligand transferase and microsomal cob(III)alamin reductase. Biochim Biophys Acta. 1993;1157:167–177. doi: 10.1016/0304-4165(93)90061-c. [DOI] [PubMed] [Google Scholar]
- 8.Watanabe F, Saido H, Yamaji R, Miyatake K, Isegawa Y, Ito A, Yubisui T, Rosenblatt DS, Nakano Y. Mitochondrial NADH- or NADPH-linked aquacobalamin reductase activity is low in human skin fibroblasts with defects in synthesis of cobalamin coenzymes. J Nutr. 1996;126:2947–2951. doi: 10.1093/jn/126.12.2947. [DOI] [PubMed] [Google Scholar]
- 9.Seetharam B. Receptor-mediated endocytosis of cobalamin (vitamin B12) Annu Rev Nutr. 1999;19:173–195. doi: 10.1146/annurev.nutr.19.1.173. [DOI] [PubMed] [Google Scholar]
- 10.Banerjee R. B12 Trafficking in Mammals: A Case for Coenzyme Escort Service. ACS Chem. Biol. 2006;1:149–159. doi: 10.1021/cb6001174. [DOI] [PubMed] [Google Scholar]
- 11.Rosenberg LE. The Metabolic Basis of Inherited Disease. 5th ed. New York: McGraw-Hill; 1983. pp. 474–497. [Google Scholar]
- 12.Rosenblatt DS, Fenton WA. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver CR, Beaudet AL, Valle D, Sly WS, editors. New York: McGraw-Hill; 2001. pp. 3897–3933. [Google Scholar]
- 13.Chandler RJ, Venditti CP. Genetic and genomic systems to study methylmalonic acidemia. Mol Genet Metab. 2005;86:34–43. doi: 10.1016/j.ymgme.2005.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kapadia CR. Vitamin B12 in Health and Disease; Part-1 Inherited Disorders of Function, Absorption and Transport. The Gastroenterologist. 1995;3:329–344. [PubMed] [Google Scholar]
- 15.Shevell MI, Rosenblatt DS. The neurology of cobalamin. Can J Neurol Sci. 1992;19:472–486. [PubMed] [Google Scholar]
- 16.Watkins D, Rosenblatt DS. Cobalamin and Inborn Errors of Cobalamin Absorption and Metabolism. The Endocrinologist. 2001;11:98–104. [Google Scholar]
- 17.Ledley FD, Lumetta M, Nguyen PN, Kolhouse JF, Allen RH. Molecular cloning of L-methylmalonyl-CoA mutase: gene transfer and analysis of mut cell lines. Proc Natl Acad Sci U S A. 1988;85:3518–3521. doi: 10.1073/pnas.85.10.3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dobson CM, Wai T, Leclerc D, Wilson A, Wu X, Dore C, Hudson T, Rosenblatt DS, Gravel RA. Identification of the gene responsible for the cblA complementation group of vitamin B12-responsive methylmalonic acidemia based on analysis of prokaryotic gene arrangements. Proc Natl Acad Sci U S A. 2002;99:15554–15559. doi: 10.1073/pnas.242614799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fenton WA, Rosenberg LE. The defect in the cblB class of human methylmalonic acidemia: deficiency of cob(I)alamin adenosyltransferase activity in extracts of cultured fibroblasts. Biochem Biophys Res Commun. 1981;98:283–289. doi: 10.1016/0006-291x(81)91900-8. [DOI] [PubMed] [Google Scholar]
- 20.Dobson CM, Wai T, Leclerc D, Kadir H, Narang M, Lerner-Ellis JP, Hudson TJ, Rosenblatt DS, Gravel RA. Identification of the gene responsible for the cblB complementation group of vitamin B12-dependent methylmalonic aciduria. Hum Mol Genet. 2002;11:3361–3369. doi: 10.1093/hmg/11.26.3361. [DOI] [PubMed] [Google Scholar]
- 21.Leal NA, Park SD, Kima PE, Bobik TA. Identification of the human and bovine ATP:Cob(I)alamin adenosyltransferase cDNAs based on complementation of a bacterial mutant. J Biol Chem. 2003;278:9227–9234. doi: 10.1074/jbc.M212739200. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J, Dobson CM, Wu X, Lerner-Ellis J, Rosenblatt DS, Gravel RA. Impact of cblB mutations on the function of ATP:cob(I)alamin adenosyltransferase in disorders of vitamin B12 metabolism. Mol Genet Metab. 2006;87:315–322. doi: 10.1016/j.ymgme.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 23.Lerner-Ellis JP, Gradinger AB, Watkins D, Tirone JC, Villeneuve A, Dobson CM, Montpetit A, Lepage P, Gravel RA, Rosenblatt DS. Mutation and biochemical analysis of patients belonging to the cblB complementation class of vitamin B12-dependent methylmalonic aciduria. Mol Genet Metab. 2006;87:219–225. doi: 10.1016/j.ymgme.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 24.Johnson CL, Pechonick E, Park SD, Havemann GD, Leal NA, Bobik TA. Functional genomic, biochemical, and genetic characterization of the Salmonella pduO gene, an ATP:cob(I)alamin adenosyltransferase gene. J Bacteriol. 2001;183:1577–1584. doi: 10.1128/JB.183.5.1577-1584.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saridakis V, Yakunin A, Xu X, Anandakumar P, Pennycooke M, Gu J, Cheung F, Lew JM, Sanishvili R, Joachimiak A, Arrowsmith CH, Christendat D, Edwards AM. The Structural Basis for Methylmalonic Aciduria. J Biol Chem. 2004;279:23646–23653. doi: 10.1074/jbc.M401395200. [DOI] [PubMed] [Google Scholar]
- 26.Leal NA, Olteanu H, Banerjee R, Bobik TA. Human ATP:Cob(I)alamin adenosyltransferase and its interaction with methionine synthase reductase. J Biol Chem. 2004;279:47536–47542. doi: 10.1074/jbc.M405449200. [DOI] [PubMed] [Google Scholar]
- 27.Fonseca MV, Escalante-Semerena JC. An in vitro reducing system for the enzymic conversion of cobalamin to adenosylcobalamin. J Biol Chem. 2001;276:32101–32108. doi: 10.1074/jbc.M102510200. [DOI] [PubMed] [Google Scholar]
- 28.Stich TA, Yamanishi M, Banerjee R, Brunold TC. Spectroscopic evidence for the formation of a four-coordinate Co2+ cobalamin species upon binding to the human ATP:cobalamin adenosyltransferase. J Am Chem Soc. 2005;127:7660–7661. doi: 10.1021/ja050546r. [DOI] [PubMed] [Google Scholar]
- 29.Yamanishi M, Labunska T, Banerjee R. Mirror "base-of" conformation of coenzyme B12 in human adenosyltransferase and its downstream target, methylmalonyl-CoA mutase. J Am Chem Soc. 2005;127:526–527. doi: 10.1021/ja044365l. [DOI] [PubMed] [Google Scholar]
- 30.Lexa D, Seveant J-M. The electrochemistry of vitamin B12. Acc. Chem. Res. 1983;16:235–243. [Google Scholar]
- 31.Stich TA, Buan NR, Escalante-Semerena JC, Brunold TC. Spectroscopic and computational studies of the ATP:corrinoid adenosyltransferase (CobA) from Salmonella enterica: insights into the mechanism of adenosylcobalamin biosynthesis. J Am Chem Soc. 2005;127:8710–8719. doi: 10.1021/ja042142p. [DOI] [PubMed] [Google Scholar]
- 32.Bauer CB, Fonseca MV, Holden HM, Thoden JB, Thompson TB, Escalante-Semerena JC, Rayment I. Three-dimensional structure of ATP:corrinoid adenosyltransferase from Salmonella typhimurium in its free state, complexed with MgATP, or complexed with hydroxycobalamin and MgATP. Biochemistry. 2001;40:361–374. doi: 10.1021/bi002145o. [DOI] [PubMed] [Google Scholar]
- 33.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 34.McCoy A, Grosse-Kunstleve R, Storoni L, Read R. Likelihood-enhanced fast translation functions. Acta Cryst. 2005;D61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 35.Otwinowski Z. Oscillation data reduction program. In: Sawyer L, Isaacs N, Bailey S, editors. Data Collection and Processing. Warrington, UK: SERC Daresbury Laboratory; 1993. pp. 56–62. [Google Scholar]
- 36.CCP4. The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 37.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 38.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 39.Murshudov GN, Vagin AA, Dodson EJ. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 40.Holm L, Sander C. Touring protein fold space with Dali/FSSP. Nucl. Acid Res. 1998;26:316–319. doi: 10.1093/nar/26.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Cryst. 2004;D60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 42.Andreeva A, Howorth D, Brenner SE, Hubbard TJP, Chothia C, Murzin AG. SCOP database in 2004: refinements integrate structure and sequence family data. Nucl. Acid Res. 2004;32:D226–D229. doi: 10.1093/nar/gkh039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanishvili R, Pennycooke M, Gu J, Xu X, Joachimiak A, Edwards AM, Christendat D. Crystal structure of the hypothetical protein TA1238 from Thermoplasma acidophilum: A new type of helical super-bundle. J.STRUCT.FUNCT.GENOM. 2004;5:231. doi: 10.1007/s10969-005-3789-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suh S, Escalante-Semerena JC. Purification and initial characterization of the ATP:corrinoid adenosyltransferase encoded by the cobA gene of Salmonella typhimurium. J Bacteriol. 1995;177:921–925. doi: 10.1128/jb.177.4.921-925.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeLano WL. DeLano Scientific. San Carlos, CA, USA: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.