

Abstract
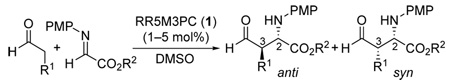
The development of catalysts for Mannich-type reactions that afford anti-products with excellent diastereo- and enantioselectivities under mild conditions and low catalyst loadings (1–5 mol%) is reported. Based on principles gained from the study of (S)-proline-catalyzed Mannich-type reactions that afford enantiomerically enriched syn-products, (3R,5R)-5-methyl-3-pyrrolidinecarboxylic acid (RR5M3PC) has been designed to catalyze the direct enantioselective anti-selective Mannich-type reactions. Computational studies of the above reaction using HF/6-31G* level of theory suggested that this design would be highly effective. The catalyst was subsequently synthesized and studied in organocatalytic Mannich-type reactions between unmodified aldehydes and N-PMP-protected α-imino esters. In accord with the design principles and in quantitative agreement with the theoretical predictions, reactions catalyzed by this catalyst afforded anti-products in good yields with excellent diastereo- and enantioselectivities (anti:syn 94:6 – 98:2, >97 – >99% ee).
Direct catalytic asymmetric Mannich reactions are highly effective carbon-carbon bond forming reactions that are used for the preparation of enantiomerically enriched amino acids, amino alcohols, and their derivatives.1–7 Because of the utility of these types of synthons, the demand for Mannich reactions that selectively afford anti- or syn-products with high enantioselectivities is high. Syn-selective direct catalytic asymmetric Mannich reactions are now common and have been performed using Zr-,1a Zn-,1b–d or Cu-derived1e catalysts, Brønsted acids,2 cinchona alkaloids,3 phase-transfer catalysts,4 and proline and related organocatalysts.5,6 Enantioselective anti-Mannich reactions are, however, considerably rarer.1a–c,7 Even a non-asymmetric anti-selective Mannich reaction would be of interest.8 Thus, the development of effective enantioselective anti-Mannich catalysts is a challenge in contemporary asymmetric synthesis. Here we present our studies regarding a solution to this problem and disclose the design, synthesis, and evaluation of amino acid catalyst 1 as a highly diastereo- and enantioselective anti-Mannich catalyst for reactions involving unmodified aldehydes (Scheme 1).
Scheme 1.

In the reaction of unmodified aldehydes with N-p-methoxyphenyl (PMP) protected imines catalyzed by the natural amino acid (S)-proline, (2S,3S)-syn-amino aldehydes are obtained with high enantioselectivities6 (Scheme 1). Although reactions involving some pyrrolidine derivatives afford anti-diastereomers as their major products, the enantioselectivities obtained with these organocatalysts are moderate.6 In order to design catalysts that provide anti-products with high levels of enantioselectivities, we revisited the key factors that control the diastereo- and enantioselectivities of (S)-proline-catalyzed reactions9 (Scheme 2 left). Four considerations are key: (1) (E)-enamine intermediates predominate. (2) The s-trans conformation of the (E)-enamine reacts in the C-C bond forming transition state. The s-cis conformation results in steric interaction between the enamine and the substituent at the 2-position of the pyrrolidine ring. (3) C-C bond formation occurs at the re-face of the enamine intermediate. This facial selection is controlled by proton-transfer from the carboxylic acid to the imine nitrogen. (4) The enamine attacks the si-face of the (E)-imine. The facial selectivity of the imine is also controlled by the proton transfer that increases the electrophilicity of the imine.
Scheme 2.

The stereoselective formation of the anti-products necessitates a reversal in the facial selectivity of either the enamine or the imine, compared to the proline-catalyzed reactions. A pyrrolidine derivative bearing substituents at 2- and 4-positions (or at 3- and 5-positions) (Scheme 2, right) was hypothesized to be an anti-Mannich catalyst. The steric features of a substituent at the 5-position of the pyrrolidine can be used to fix the conformation of the enamine (see point 2 above). This substituent can presumably be any functional group that cannot initiate proton transfer to the imine. The acid functionality was then placed at the distal 3-position of the ring, to affect control of enamine and imine face selection in the transition state (see points 3 and 4). In order to avoid steric interactions between the substituent at the 5-position of the new catalyst and the imine in the transition state, the substituents at 3- and 5-positions should be in the trans configuration.
Based on these considerations, a new catalyst (3R,5R)-5-methyl-3-pyrrolidinecarboxylic acid (RR5M3PC, 1) was designed. The major transition state of the Mannich reaction catalyzed by 1 is presented in Scheme 2 (right). Computational studies of the 1-catalyzed reaction of propionaldehyde and N-PMP-yprotected α-imino methyl glyoxylate using HF/6-31G* level of theory10 were used to test our design prior to synthesis. The catalyst was predicted to give 95:5 anti:syn diastereoselectivity and ~98% ee for the formation of the (2S,3R)-product (Table 1, entry 1).
Table 1.
RR5M3PC (1)-Catalyzed Mannich-type Reactionsa
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | R1 | R2 | time (h) | product | yield (%) | drbantisyn | eec (%) |
| 1d | Me | Me | - | - | - | 95:5 | 98 |
| 2 | Me | Et | 1 | 2 | 70 | 94:6 | >99e |
| 3 | i-Pr | Et | 3 | 3 | 85 | 98:2 | 99 |
| 4 | n-Bu | Et | 0.5 | 4 | 54 | 97:3 | 99 |
| 5f,g | n-Bu | Et | 1 | 4 | 71 | 97:3 | 99 |
| 6f,h | n-Bu | Et | 2 | 4 | 57 | 97:3 | >99 |
| 7 | n-Pent | Et | 3 | 5 | 80 | 97:3 | >99 |
| 8i | CH2CH=CH2 | Et | 3 | 6 | 72 | 96:4 | >97 |
| 9 | i-Pr | i-Pr | 1 | 7 | 92 | 97:3 | 98 |
| 10 | n-Pent | i-Pr | 1 | 8 | 85 | 96:4 | >99 |
Typical conditions: To a solution of N-PMP-protected α-imino ester (0.25 mmol, 1 equiv) and aldehyde (0.5 mmol, 2 equiv) in anhydrous DMSO (2.5 mL), catalyst RR5M3PC (1) (0.0125 mmol, 0.05 equiv, 5 mol% to the imine) was added and the mixture was stirred at room temperature.
The diastereomeric ratio (dr) was determined by 1H NMR.
The ee of the (2S,3R)-anti-product was determined by chiral-phase HPLC analysis.
Indicates computational predictions using methods described in the text.
The ee was determined by HPLC analysis of the corresponding oxime prepared with O-benzylhydroxylamine.
The reaction was performed in a doubled scale.
Catalyst 1 (2 mol%) was used.
Catalyst 1 (1 mol%) was used.
The reaction was performed with doubled concentration for each reactant and catalyst 1.
RR5M3PC (1)11 was synthesized (Scheme 3), and Mannich reactions involving a variety of unmodified aldehydes were studied (Table 1). In accord with the design principles and in quantitative agreement with the computational predictions, the reactions catalyzed by 1 afforded anti-amino aldehyde products in excellent diastereo- and enantioselectivities.12 With 5 mol% catalyst loading, the reaction rates with catalyst 1 were approximately 2- to 3-fold faster than the corresponding proline-catalyzed reactions that afford the syn-products. The high catalytic efficiency of 1 allowed the reactions to be catalyzed with only 1 or 2 mol% to afford the desired products in reasonable yields within a few hours (Table 1, entries 5 and 6).
Scheme 3.

(a) Known procedures (see supporting information); (b) (i) MsCl, Et3N, (ii) LiBHEt3, (iii) TBAF, 94% (3 steps); (c) TsCl, pyridine, 58%; (d) NH4OAc, 99%; (e) NaOH, 93%; (f) (i) MsCl, Et3N, (ii) NaCN, 58% (2steps); (g) (i) HCl, (ii) Dowex 50WX8, 90% (2 steps).
Imidazole isomerization13 of the anti-3 product obtained from the 1-catalyzed reaction and of the (2S,3S)-syn-3 product obtained from the (S)-proline-catalyzed reaction6 confirmed that the major anti-product generated from the 1-catalyzed reaction had a (2S,3R) configuration. (Scheme 4).
Scheme 4.

The relative contributions of the carboxylic acid and methyl group of 1 in directing the stereochemical outcome of the reaction were assessed. Computational studies involving the derivative lacking the 5-methyl group, (S)-3-pyrrolidinecarboxylic acid, indicate that the methyl group contributes ~1 kcal/mol towards the anti-diastereoselectivity. That is, the result in entry 1 of Table 1 changes to 82:18 anti:syn dr and 92% ee when transition states with the unmethylated catalyst are located. This unmethylated catalyst was also tested in an actual reaction, for the case where the R1 = i-Pr. This derivative afforded (2R,3S)-anti-3 in 95:5 anti:syn dr and 93% ee, which is a drop of ~0.6 kcal/mol from the 1-catalyzed reaction with the same substrate (Table 1, entry 3).
An efficient organocatalyst RR5M3PC (1) for anti-Mannich-type reactions has been developed.14 This catalyst has been demonstrated to be useful for the synthesis of amino acid derivatives with excellent anti-diastereoselective control and high enantioselectivities under mild conditions. Further studies on the full scope of this Mannich catalyst, computational studies, and other reactions catalyzed by it and its derivatives will be reported in the near future.
Supplementary Material
Experimental procedures, spectral and chromatographic data. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgment
This study was supported by The Skaggs Institute for Chemical Biology and the National Institute of General Medical Sciences, National Institutes of Health (GM36700 to KNH).
References
- 1.(a) Kobayashi S, Ishitani H, Ueno M. J. Am. Chem. Soc. 1998;120:431. [Google Scholar]; (b) Hamada T, Manabe K, Kobayashi S. J. Am. Chem. Soc. 2004;126:7768. doi: 10.1021/ja048607t. [DOI] [PubMed] [Google Scholar]; (c) Matsunaga S, Yoshida T, Morimoto H, Kumagai N, Shibasaki M. J. Am. Chem. Soc. 2004;126:8777. doi: 10.1021/ja0482435. [DOI] [PubMed] [Google Scholar]; (d) Trost B, Terrell LR. J. Am. Chem. Soc. 2003;125:338. doi: 10.1021/ja028782e. [DOI] [PubMed] [Google Scholar]; (e) Kobayashi S, Matsubara R, Nakamura Y, Kitagawa H, Sugiura M. J. Am. Chem. Soc. 2003;125:2507. doi: 10.1021/ja0281840. [DOI] [PubMed] [Google Scholar]
- 2.Akiyama T, Itoh J, Yokota K, Fuchibe K. Angew. Chem., Int. Ed. 2004;43:1566. doi: 10.1002/anie.200353240. [DOI] [PubMed] [Google Scholar]
- 3.Lou S, Taoka BM, Ting A, Scaus S. J. Am. Chem. Soc. 2005;127:11256. doi: 10.1021/ja0537373. [DOI] [PubMed] [Google Scholar]
- 4.(a) Ooi T, Kameda M, Fujii J, Maruoka K. Org. Lett. 2004;6:2397. doi: 10.1021/ol049215u. [DOI] [PubMed] [Google Scholar]; (b) Okada A, Shibuguchi T, Ohshima T, Masu H, Yamaguchi K, Shibasaki M. Angew. Chem., Int. Ed. 2005;44:4564. doi: 10.1002/anie.200500927. [DOI] [PubMed] [Google Scholar]
- 5.(a) Notz W, Watanabe S, Chowdari NS, Zhong G, Betancort JM, Tanaka F, Barbas CF., III Adv. Synth. Catal. 2004;346:1131. [Google Scholar]; (b) Wang W, Wang J, Li H. Tetrahedron Lett. 2004;45:7243. [Google Scholar]; (c) Zhuang W, Saaby S, Jorgensen KA. Angew. Chem., Int. Ed. 2004;43:4476. doi: 10.1002/anie.200460158. [DOI] [PubMed] [Google Scholar]; (d) Westermann B, Neuhaus C. Angew. Chem., Int. Ed. 2005;44:4077. doi: 10.1002/anie.200500297. [DOI] [PubMed] [Google Scholar]; (e) Enders D, Grondal C, Vrettou M, Raabe G. Angew. Chem., Int. Ed. 2005;44:4079. doi: 10.1002/anie.200500810. [DOI] [PubMed] [Google Scholar]
- 6.Notz W, Tanaka F, Watanabe S, Chowdari NS, Turner JM, Thayumanuvan R, Barbas CF., III J. Org. Chem. 2003;68:9624. doi: 10.1021/jo0347359. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 7.Yoshida T, Morimoto H, Kumagai N, Matsunaga S, Shibasaki M. Angew. Chem., Int. Ed. 2005;44:3470. doi: 10.1002/anie.200500425. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi E, Fujisawa H, Mukaiyama T. Chem. Lett. 2005;34:84. [Google Scholar]
- 9.Bahmanyar S, Houk KN. Org. Lett. 2003;5:1249. doi: 10.1021/ol034198e. [DOI] [PubMed] [Google Scholar]
- 10.HF/6-31G* was used for rapid computation of the stereoselectivity.
- 11.For racemic, cis- and trans-mixture of this compound, see: Juaristi E, Quintana D, Lamatsch B, Seebach D. J. Org. Chem. 1991 56;:2553.
- 12.DMSO provided the best anti-selectivity and enantioselectivity of the solvents tested for the RR5M3PC-catalyzed Mannich reaction to afford anti-3. Reactions in DMF (anti:syn = 97:3, 97% ee), CH3CN (96:4, 96% ee), EtOAc (94:6, 96% ee), and dioxane (97:3, 95% ee) were as efficient with respect to reaction rate as in DMSO.
- 13.Ward DE, Sales M, Sasmal P. J. Org. Chem. 2004;69:4808. doi: 10.1021/jo049626o. [DOI] [PubMed] [Google Scholar]
- 14.After submission of this paper, another anti-Mannich catalyst has been reported. Kano T, Yamaguchi Y, Tokuda O, Maruoka K. J. Am. Chem. Soc. 2005;127:16408. doi: 10.1021/ja056008w.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures, spectral and chromatographic data. This material is available free of charge via the Internet at http://pubs.acs.org.