

Abstract
The preparation and characterization of several new cyanoligated six-coordinate low-spin iron(III) porphyrinates are reported. The synthesis and structure of the new bis(cyanide) derivative K(222)][Fe(TMP)(CN)2] (TMP= tetramesitylporphyrinate) is described. Three mixed-ligand species of the general form [Fe(Porph)(CN)(L)], where L = 1-methylimidazole or pyridine, have also been prepared and structurally characterized. All complexes have been studied with EPR spectroscopy in frozen solution and in microcrystalline form. In some cases, especially those of the bis(cyanide) derivative above and the previously reported [Fe(TPP)(CN)2]−, there are significant differences in the EPR spectra as a result of the state change. These spectral differences can be correlated with changes in the electron configuration that are the likely result of a differing environment of the coordinated cyanide ligands; core conformation and electronic structure of the porphyrin ligand are unlikely to play a role. All four new complexes and [Fe(TPP)(CN)2]− have been studied by Mössbauer spectroscopy with variable temperature and applied magnetic field measurements. The sign of the quadrupole splitting value has been established as negative. These measurements have allowed us to give estimates of the energy difference between the two close-lying dπ (dxz and dyz) orbitals. These splitting values range from ∼267 cm−1 for [Fe(TPP)(CN)2]− to ∼614 cm−1 for [Fe(TPP)(CN)(Py)].
Introduction
The cyanide ion has been an important ligand in the study of hemes and heme proteins. The strong binding of the cyanide ligand to ferric hemoproteins and porphinatoiron(III) species has been long recognized. Cyanide ion readily yields low-spin (cyano)iron(III) derivatives when there is a vacant coordination site at the heme in hemoproteins.[1] Cyanide is a frequently used inhibitor to study enzymatic hemoproteins such as oxidases and peroxidases.[2] Peroxidases such as horseradish peroxidase, lactoper-oxidase and chloroperoxidase use cyanide as a substrate and catalyze the one-electron oxidation to form the cyanyl radical.[3] The well-known acute toxicity of cyanide is the result of the inhibition of cytochrome c oxidase of the respiratory chain as a result of binding cyanide to the heme a3 oxygen site.[4, 5]
Although most work on the cyano derivatives of hemes, including much early work,[6]−[11] utilized solution measurements, these cyano species can be prepared as solid species and characterized. Thus Scheidt and co-workers prepared and determined the X-ray structures of [Fe(TPP)(CN)2]−[12] as the potassium acetone solvate salt,[13] and two mixed derivatives, [Fe(TPP)(CN)(Py)][14] and [Fe(OEP)(CN)(Py)].[15] In an important paper that closely followed this work, Strouse and co-workers[16] definitively showed that both [Fe(TPP)(CN)2]−[12] and [Fe(TPP)(CN)(Py)] possessed an unusual EPR spectral type that has been variously called HALS[17] or large “gmax.”[18] In such complexes, the largest component of the g tensor has a g-value ≥∼3.3, with the other components of the g tensor frequently unobservable in microcrystalline or frozen solution samples. Inniss et al.[16] carried out single-crystal EPR measurements on [Fe(TPP)(CN)2]− and [Fe(TPP)(CN)(Py)] and thus obtained values for all three components of the g tensor. These data, with use of the Taylor formalism,[19] provide approximate relative energies of the three lowest-lying d-orbitals.
The appearance of large gmax EPR spectra is associated with the near degeneracy of the dxz and dyz orbitals; the results of Strouse and co-workers[16] are in complete accord with this. Such degeneracy can be seen to arise from the π-accepting character of the cyanide ligand, which is isoelectronic with the well-known π-acid ligand CO. The axially symmetric π interaction appears to decrease the energy difference between the nominally Jahn-Teller split orbitals. The electron configuration of these low-spin, large gmax iron(III) states is thus best described as a subset of the usual (dxy)2(dyz, dxz)3 configuration with an unusually close energy difference between the dyz, dxz orbitals. Subsequently, Nakamura and co-workers have reported that a number of bis(cyano) derivatives of the general form [Fe(Porph)(CN)2]− displayed a different type of low-spin EPR spectrum, namely an axial EPR spectrum.[20]−[24] As had been earlier shown by Scheidt, Walker and co-workers,[25] this type of EPR spectrum can only arise from the (dyz, dxz)4(dxy)1 electron configuration. Clearly this type of spectrum can occur only if the in-plane dxy orbital is destabilized relative to the dπ orbitals. This can be effected by π-accepting axial ligands such as isocyanides[25] or 4-cyanopyridine[26] that are also only weak σ donors. Nakamura and co-workers offered stereochemical, electronic and solvent effects as explanations for the observation of axial EPR spectra. Importantly, all of the EPR measurements made by Nakamura et al. were carried out in frozen solutions and there remain significant questions about the stereochemical issues that were suggested to control electron configuration.
In this paper, we report the preparation of several new (cyano)iron(III) porphyrinate derivatives. These are a biscyanide derivative, [K(18-C-6)][Fe(TMP)(CN)2], and three mixed cyano complexes [Fe(TMP)(CN)(1-MeIm)], [Fe(TPP)(CN)(1-MeIm)], and [Fe(TPP)(CN)(Py)] (in a new crystalline solvate form). The molecular structures of these complexes have been determined. They have also been characterized by the application of multitemperature and applied magnetic field Mössbauer measurements. EPR measurements have been carried out on these and the previously reported [Fe(TPP)(CN)2]−, both in frozen solution and the microcrystalline state. These studies show that subtle environmental effects can switch the EPR spectral type and the electron configuration. No single feature is likely to uniquely define the configuration. The temperature-dependent Mössbauer measurements have provided new information into the energy gap between the dxz and dyz orbitals and have allowed us, for the first time, to make semi-quantitative determinations of the small energy difference between these nearly degenerate orbitals.
Experimental Section
General Information
THF was distilled over sodium and benzophenone ketyl; all other solvents were used as received (Fisher). 1,4,7,10,13,16-Hexaoxacyclooctadecane (18-crown-6) (Aldrich) was used as received. KCN was recrystallized and purified by a literature procedure.[27] meso-Tetraphenylporphyrin (H2TPP) was prepared according to Adler et al.[28] and meso-tetramesitylporphyrin (H2TMP) by a modified version of the procedure published by Lindsey et al.[29] The metallation of the free-base porphyrins to give [Fe(Porph)Cl] was done as previously described.[30] UV-vis spectra were recorded on a Perkin-Elmer Lambda 19 UV/vis/near-IR spectrometer and IR spectra were recorded on a Nicolet Nexus 670 FT-IR spectrometer as KBr pellets. EPR spectra were recorded on microcrystalline (powder) samples obtained by grinding a sufficient quantity of the crystalline samples. Solution measurements were made by dissolving the aforementioned samples in methylene chloride. EPR spectra were collected on a Bruker EMX EPR Spectrometer. For measurements at 4 K, a cavity with attached Oxford Continuous Flow Cryostat (ESR 900) was used. KBr (99.999%, Aldrich ), which is EPR silent at 4K, was used to embed some microcrystalline samples. Spectra were collected and analyzed using Bruker’s EPR Acquisition and WinEPR programs. Mössbauer measurements were performed on a constant acceleration spectrometer from 4.2 K to 300 K with optional small field and in a 9T superconducting magnet system (Knox College). Samples for Mössbauer spectroscopy were prepared by immobilization of the crystalline material in Apiezon M grease.
Synthesis of [K((CH3)2CO)2][Fe(TPP)(CN)2]
[Fe(TPP)(H2O)2]ClO4[31] and [K((CH3)2CO)2][Fe(TPP)(CN)2] were prepared by the methods described previously.[13] Single crystals of [K((CH3)2CO)2][Fe(TPP)(CN)2] were obtained by slow diffusion of pentane-heptane into acetone solution. IR ν(C–N): 2120 cm−1. UV-vis (CH3OH) λmax: 332, 424, 539, 560, 596 nm.
Syntheses of [Fe(TPP)(CN)(Py)]·0.65Py and [Fe(TPP)(CN)(1-MeIm)]·1-MeIm·CHCl3
[Fe(TPP)(H2O)2]ClO4 (20 mg, 0.025 mmol) was dissolved in 8 mL of methanol containing 20 mg (0.31 mmol) of KCN. The solution was stirred 30 min and the methanol was removed by rotary evaporation. 0.5 mL of pyridine or 1-methylimidazole and 3 mL of CHCl3 was then added. The solution was stirred for 24 h and then filtered through a cotton-wool plug. Crystals suitable for X-ray diffraction were grown by slow diffusion of hexanes into the CHCl3 solution. For both complexes, IR ν(C–N): 2130 cm−1. UV-vis (CH2Cl2) λmax: for [Fe(TPP)(CN)(Py)], 333, 420, 554 nm; for [Fe(TPP)(CN)(1-MeIm)], 332, 418, 555 nm.
Synthesis of [Fe(TMP)(CN)(1-MeIm)]·1.8CHCl3.
[Fe(TMP)(H2O)2]ClO4[31] (50 mg, 0.05 mmol) was stirred with KCN (50 mg, 0.77 mmol) in methanol (8 mL) for 3 h. After the evaporation of solvent, 0.6 mL of 1-methylimidazole in 3 mL of CHCl3 was added and the solution was stirred overnight. The resulting solution was filtered and hexanes were slowly diffused into the solution. Single crystals of [Fe(TMP)(CN)(1-MeIm)] (yield 30 mg) were obtained. IR ν(C–N): 2125 cm−1. UV-vis (CH2Cl2) <max: 319, 423, 558 nm.
Synthesis of [K(18-C-6)][Fe(TMP)(CN)2]
KCN (5 mg, 0.077 mmol) and 18-crown-6 (11 mg, 0.042 mmol) were added to a solution of [Fe(TMP)(H2O)2]ClO4 (20 mg, 0.021 mmol) in THF. The mixture was stirred for 5 h during which the solution turning from red to green and was then filtered through a cotton-wool plug. Hexanes were allowed to diffuse slowly into the solution. Single crystals of [K(18-C-6)][Fe(TMP)(CN)2] (as THF solvate) (yield 15 mg) was obtained. IR ν(C–N): 2111 cm−1. UV-vis (CH2Cl2) λmax: 330, 434, 579, 618 nm.
X-ray Structure Determinations
Single crystal experiments were carried out on a Bruker Apex CCD system with graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). The crystalline samples were placed in inert oil, mounted on a glass pin, and transferred to the cold gas stream of the diffractometer. Crystal data were collected at 100 K. The structures were solved by direct methods (SHELXS-97)[32] and refined against F2 using SHELXL-97.[33, 34] Subsequent difference Fourier syntheses led to the location of all remaining nonhydrogen atoms. For the structure refinement all data were used including negative intensities. All nonhydrogen atoms were refined anisotropically if not remarked upon otherwise below. Hydrogen atoms were idealized with the standard SHELXL-97 idealization methods. The program SADABS[35] was applied for the absorption correction. Complete crystallographic details, atomic coordinates, anisotropic thermal parameters, and hydrogen atom coordinates are given in the Supporting Information Tables for all structures; a brief description of crystallographic details is given in Table 1.
Table 1.
Crystallographic Details for [Fe(TPP)(CN)(Py)]0.65Py, [Fe(TPP)(CN)(1-MeIm)]1-MeIm CHCl3, 3{[K(18-C-6)][Fe(TMP)(CN)2]}·5.1THF·0.9H2O and [Fe(TMP)(CN)(1-MeIm)]·1.8CHCl3
| [Fe(TPP)(CN)(Py)] ·0.65Py |
[Fe(TPP)(CN)(1-MeIm)] ·1-MeIm CHCl3 |
3{[K(18-C-6)][Fe(TMP)(CN)2]} ·5.1THF 0.9H2O |
[Fe(TMP)(CN)(1-MeIm)] ·1.8CHCl3 |
|
|---|---|---|---|---|
| empirical formula | C53.25H36.25FeN6.65 | C54H41Cl3FeN9 | C230.40H270.60Fe3K3N18O24 | C62.80H59.80Cl5.40FeN7 |
| FW, amu | 825.09 | 978.16 | 3960.90 | 1159.86 |
| a, Å | 13.1214(2) | 20.7612(3) | 23.0103(9) | 16.4839(2) |
| b, Å | 23.5538(4) | 9.7535(1) | 20.1843(8) | 17.6960(3) |
| c, Å | 14.0324(2) | 26.5363(4) | 46.1854(18) | 20.0081(3) |
| β , deg | 102.606(1) | 121.109(1) | 98.535(2) | 99.032(1) |
| V, Å3 | 4232.29(11) | 4600.67(11) | 21213.1(14) | 5763.98(15) |
| space group | P21=n | P21=c | P21=c | P21=n |
| Z | 4 | 4 | 4 | 4 |
| crystal color | black | black | dark purple | black |
| crystal dimensions, mm | 0.55 × 0.53 × 0.40 | 0.42 × 0.23 × 0.07 | 0.49 × 0.43 × 0.22 | 0.31 × 0.24 × 0.17 |
| temp, K | 100 | 100 | 100 | 100 |
| total data collected | 48079 | 79823 | 202746 | 51896 |
| unique data | 8310 (Rint = 0.022) | 19164 (Rint = 0.031) | 46934 (Rint = 0.051) | 12045 (Rint = 0.067) |
| unique observed data [I > 2σ(I)] | 7789 | 13118 | 36729 | 6964 |
| goodness-of-fit (based on F2 | 1.044 | 1.111 | 1.100 | 0.923 |
| final R indices | R1 = 0.0515 | R1 = 0.0462 | R1 = 0.0789 | R1 = 0.0513 |
| >[I >2σ(I)] | wR2 = 0.1511 | wR2 = 0.1281 | wR2 = 0.1979 | wR2 = 0.1221 |
| final R indices | R1 = 0.0543 | R1 = 0.0765 | R1 = 0.0992 | R1 = 0.0896 |
| (all data) | ωR2= 0.1531 | ωR2 = 0.1380 | ωR2 = 0.2100 | ωR2 = 0.1292 |
[Fe(TPP)(CN)(Py)]·0.65Py
A black crystal with the dimensions 0.55 × 0.53 × 0.40 mm was used for the structure determination. The asymmetric unit contains one ordered porphyrin complex and two disordered pyridine solvate molecules. The first disordered pyridine was constrained by means of the fragment (FRAG) command, and anisotropic thermal parameters were restrained by the ‘similar Uij’ (SIMU) command. The second disordered pyridine is symmetric and N and C atoms were constrained by an EXYZ command. Finally, the occupancy parameters of pyridine groups were set to 0.4 and 0.25 respectively.
[Fe(TPP)(CN)(1-MeIm)]·1-MeIm·CHCl3
A dark purple crystal with the dimensions 0.42 × 0.23 × 0.07 mm was used for the structure determination. The asymmetric unit contains one porphyrin complex, one 1-methylimidazole and one chloroform molecule.
[Fe(TMP)(CN)(1-MeIm)]·1.8CHCl3
A black crystal with the dimensions 0.31 × 0.24 × 0.17 mm was used for the structure determination. The asymmetric unit contains one porphyrin complex, one ordered and one disordered chloroform molecule. The disordered chloroform was refined as three distinct orientations and constrained by means of a DFIX command; all atoms were anisotropically refined and constrained with an EADP instruction. The occupancy factor sum was found to be 0.8.
[K(18-C-6)][Fe(TMP)(CN)2]
A black crystal with dimensions 0.22×0.43 × 0.49 mm was used for the structure determination. The asymmetric unit contains three [K(18-C-6)][Fe(TMP)(CN)2] entities, six THF solvate sites and two water molecules. The potassium ion of each entity is coordinated to the nitrogen atom of one CN− ion. One [K(18-C-6)][Fe(TMP)(CN)2] was completely ordered, the second entity has disorder in one ethylene bridge of the [K(18-crown-6)], while the third entity has two orientations of the 18-crown-6 ring, which was constrained by means of DFIX commands. The two water molecules are each hydrogen bonded to a coordinated cyanide ion. Two hydrogen atoms of one water were located from the difference map. One of the disordered THF solvent molecules was refined as two distinct orientations and the fragment (FRAG) command was used to constrain it.
Results
The synthesis, molecular structures, IR, UV-vis, EPR, and Mössbauer spectra of several six-coordinate (cyano)iron(III) porphyrinates are reported. [K((CH3)2CO)2][Fe(TPP)(CN)2] was resynthesized[13] and further characterized by Mössbauer spectroscopy. Four new derivatives were prepared and νCN determined for all: [K(18-C-6)][Fe(TMP)(CN)2] (2111 cm−1), [Fe(TPP)(CN)(Py)] (2130 cm−1), [Fe(TPP)(CN)(1-MeIm)] (2130 cm−1) and [Fe(TMP)(CN)(1-MeIm)] (2125 cm−1). νCN for [K((CH3)2CO)2][Fe(TPP)(CN)2] (2120 cm−1) was confirmed to be identical to that reported previously. Solid-state Mössbauer spectra were measured on five samples.
The complexes are low-spin S = 1/2 species and ∼4 K EPR spectra have been measured for each complex, both as ground single crystals and as frozen solutions. Solid-state EPR spectra observed were those of typical ‘large gmax’ spectrum with the observed g = 3.3 - 3.7. For some species, a change in the EPR spectra occurred when the samples were measured as frozen solutions. These results are summarized in Table 2.
Table 2.
EPR Parameters for Low-spin (Cyano)iron(III)porphyrinates and Hemoproteins
| complex | gxx |
gyy |
gzz |
T(K) |
phase |
EPR type |
ref. |
|---|---|---|---|---|---|---|---|
| [Fe(Porph)(CN)2]− | |||||||
| [K(18-C-6)][Fe(TMP)(CN)2] | 1.20 | 1.68 | 3.47 | 4.2 | microcrystalline | large gmax | tw |
| [K(18-C-6)][Fe(TMP)(CN)2] | 2.54 | 2.54 | 4.2 | CH2Cl2 | axial | tw | |
| [K((CH3)2CO)2][Fe(TPP)(CN)2] | 0.52 | 1.05 | 3.70 | 6.0 | single crystal | large gmax | 16 |
| [K((CH3)2CO)2][Fe(TPP)(CN)2] | 3.67 | 4.2 | microcrystalline | large gmax | tw | ||
| 2.57 | 2.57 | 4.2 | microcrystalline | axial | tw | ||
| [K((CH3)2CO)2][Fe(TPP)(CN)2] | 3.54 | 4.2 | CH2Cl2-CH3OH | large gmax | tw | ||
| 1.76 | 2.21 | 2.74 | 4.2 | CH2Cl2-CH3OH | rhombic | tw | |
| [Fe(PPIX)(CN)2]− | 1.03 | 2.42 | 3.75 | – | – | large gmax | 36 |
| [Fe(THP)(CN)2]− | 3.5 | 4.2 | CH2Cl2-CH3OH | large gmax | 20 | ||
| [Fe(TPP)(CN)2]− | 3.56 | 4.2 | CH2Cl2-CH3OH | large gmax | 21 | ||
| [Fe(OMTPP)(CN)2]− | 3.48 | 4.2 | CH2Cl2 | large gmax | 37 | ||
| [Fe(OETPP)(CN)2]− | 3.31 | 4.2 | CH2Cl2 | large gmax | 37 | ||
| [Fe(OETPP)(CN)2]− | 3.47 | – | – | large gmax | 38 | ||
| [Fe(OEP)(CN)2]− | 3.73 | – | – | large gmax | 38 | ||
| [Fe(TTP)(CN)2]−a | 3.6 | 4.2 | CH2Cl2 | large gmax | 24 | ||
| [Fe(TTP)(CN)2]−a | 2.5 | 2.5 | 4.2 | CH3OH | axial | 24 | |
| [Fe(TMP)(CN)2]− | 2.47 | 2.47 | 1.5 | 4.2 | CH2Cl2-CH3OH | axial | 21 |
| [Fe(TEt3PP)(CN)2]−a,b | 2.45 | 2.45 | 1.6 | 4.2 | CD3OD | axial | 23 |
| [Fe(TEt3PP)(CN)2]−a,b | 2.45 | 2.45 | 1.5 | 4.2 | CH2Cl2-CH3OH | axial | 21 |
| [Fe(TiPr3PP)(CN)2]−a,b | 2.45 | 2.45 | 1.5 | 4.2 | CH2Cl2-CH3OH | axial | 21 |
| [Fe(TMeP)(CN)2]− | 2.43 | 2.43 | 1.69 | 4.2 | CH2Cl2-CH3OH | axial | 20 |
| [Fe(TEtP)(CN)2]− | 2.47 | 2.47 | 1.61 | 4.2 | CH2Cl2-CH3OH | axial | 20 |
| [Fe(TiPrP)(CN)2]− | 2.35 | 2.35 | 1.82 | 4.2 | CH2Cl2-CH3OH | axial | 20 |
| [Fe(TiPrP)(CN)2]− | 2.42 | 2.42 | 1.74 | 4.2 | CH2Cl2 | axial | 24 |
| [Fe(TcPrP)(CN)2]− | 2.47 | 2.47 | 4.2 | CH2Cl2 | axial | 22 | |
| [Fe(TnPrP)(CN)2]− | 2.49 | 2.49 | 4.2 | CH2Cl2 | axial | 22 | |
| [Fe(TMP)(CN)2]− | 2.56 | 2.56 | 1.70 | – | – | axial | 38 |
| [Fe(QTPP)(CN)2]− | 2.51 | 2.25 | 1.75 | 77 | CH3OH | axial | 39 |
| [Fe(MQTPP)(CN)2]− | 2.53 | 2.26 | 1.77 | 77 | CH3OH | axial | 39 |
| [Fe(PTPP)(CN)2]− |
2.46 |
2.29 |
1.7 |
77 |
CH3OH |
axial |
39 |
| [Fe(Porph)(CN)(L)] | |||||||
| [Fe(TPP)(CN)(Py)] | 0.34 | 1.76 | 3.31 | 6.0 | single crystal | large gmax | 16 |
| [Fe(TPP)(CN)(Py)] | 3.6 | 4.2 | CH2Cl2 | large gmax | tw | ||
| [Fe(TPP)(CN)(Py)] | 1.8 | 3.3 | 4.2 | microcrystalline | large gmax | tw | |
| [Fe(TPP)(CN)(Py)]c | 1.81 | 3.17 | 4.2 | microcrystalline | large gmax | tw | |
| [Fe(TPP)(CN)(1-MeIm)] | 2.04 | 3.36 | 4.2 | CH2Cl2 | large gmax | tw | |
| [Fe(TPP)(CN)(1-MeIm)] | 1.92 | 3.51 | 4.2 | microcrystalline | large gmax | tw | |
| [Fe(TMP)(CN)(1-MeIm)] | 2.04 | 3.28 | 4.2 | CH2Cl2 | large gmax | tw | |
| [Fe(TMP)(CN)(1-MeIm)] | 1.91 | 3.37 | 4.2 | microcrystalline | large gmax | tw | |
| [Fe(PPIX)(CN)(Im)]− | 1.4 | 2.2 | 3.1 | 77 | – | large gmax | 40 |
| [Fe(THP)(CN)(2-MeHIm)] | 3.3 | 4.2 | CH2Cl2-CH3OH | large gmax | 41 | ||
| [Fe(TMeP)(CN)(2-MeHIm)] | 2.5 | 2.5 | 1.6 | 4.2 | CH2Cl2-CH3OH | axial | 41 |
| [Fe(TEtP)(CN)(2-MeHIm)] | 2.5 | 2.5 | 1.6 | 4.2 | CH2Cl2-CH3OH | axial | 41 |
| [Fe(TiPrP)(CN)(2-MeHIm)] |
2.45 |
2.45 |
1.67 |
4.2 |
CH2Cl2-CH3OH |
axial |
41 |
| Cyano Hemoproteins | |||||||
| Mb, HIm, CN− | 0.93 | 1.89 | 3.45 | – | – | large gmax | 42 |
| Cytochrome c, CN− | 0.73 | 1.85 | 3.47 | – | – | large gmax | 42 |
| Sirohemin, 2CN− | 2.37 | 2.37 | 1.78 | 15-20 | – | axial | 43 |
Similar results were claimed for a number of meso-tetrakis(p-substituted-phenyl)porphyrin derivatives, p-X-TPP; X = OCH3, H, F, Cl, COOCH3, CF3 and CN, but no data shown.
the complex shows either an axial type signal or both axial and large gmax type signals in different solvents.[23]
Crystalline phase of Mössbauer sample.
The molecular structures of four six-coordinate cyano complexes, [Fe(TPP)(CN)(1-MeIm)]·1-MeIm·CHCl3, [Fe(TPP)(CN)(Py)]·0.65Py, [Fe(TMP)(CN)(1-MeIm)]·1.8CHCl3 and [K(18-C-6)][Fe(TMP)(CN)2], have been determined.
Crystalline [K(18-C-6)][Fe(TMP)(CN)2] contains three independent entities per asymmetric unit. Each entity consists of the potassium 18-crown-6 ether cation and the anionic porphyrinate. In each entity, the potassium ion is coordinated to a nitrogen atom of one of the cyanide ligands, making one cyanide an exo-bidendate ligand. A labeled ORTEP diagram of one such entity is given in Figure 1. In this and subsequent figures, and in all tables for [K(18-C-6)][Fe(TMP)(CN)2], the following atom naming convention has been used: Q(nyy), where Q is the atom type, n refers to entities 1–3 and yy are further numbers and letters needed to completely specify the atom. Similar atoms in the three entities have the same name except for the digit n. The three distinct entities have similar K—N bond lengths: entity 1, 2.848 Å; entity 2, 2.814 Å; entity 3, 2.768 Å. The major difference between the three is that entities 2 and 3 have disordered 18-C-6 macrocycles. The disorder is minor in entity 2 while somewhat more serious in entity 3. Complete ORTEP diagrams of entities 2 and 3 are given in Figures S1 and S2 of the Supporting Information. ORTEP diagrams of the potassium 18-crown-6macrocycles for each of the three entities are found in Figures S3, S4, and S5. The other difference is that only two entities have a water molecule hydrogen-bonded to the nitrogen atom of the coordinated cyanide (O···N = 2.968 or 3.073 Å). The two hydrogen atoms of one water were located in the difference map (H···N = 2.530 Å). Complete crystallographic details of the [K(18-C-6)][Fe(TMP)(CN)2] structure are given in Tables S20—S25 of the Supporting Information.
Figure 1.
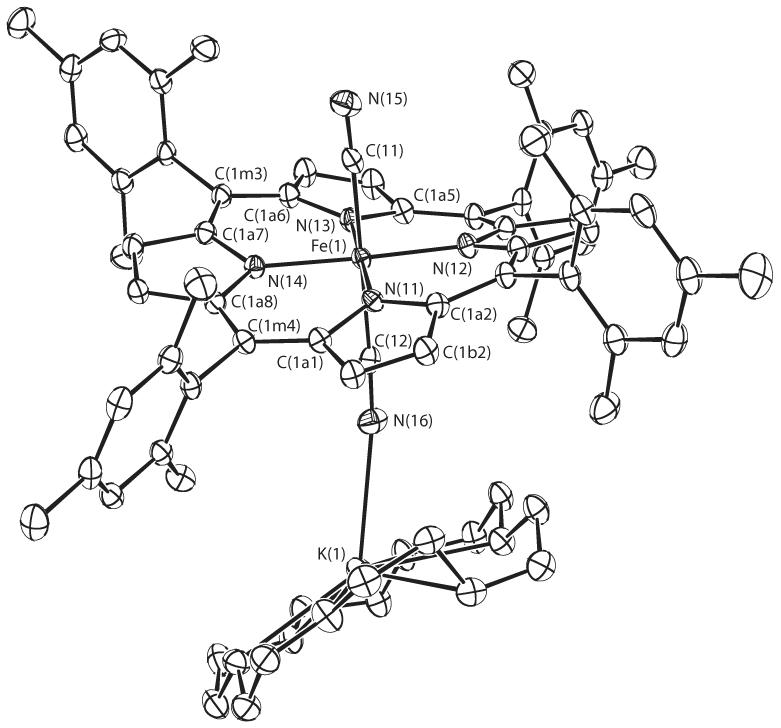
ORTEP diagram of entity 1 of [K(18-C-6)][Fe(TMP)(CN)2] displaying the atom labeling scheme. Thermal ellipsoids of all atoms are contoured at the 50% probability level. Hydrogen atoms have been omitted for clarity.
ORTEP diagrams of the molecular structures of the three new six-coordinate mixed ligand cyanide derivatives are shown in Figure 2 which displays the molecular structure of [Fe(TPP)(CN)(Py)], Figure 3 which shows that of [Fe(TPP)(CN)(1-MeIm)] and Figure 4 which displays that of [Fe(TMP)(CN)(1-MeIm)]. The projection of the axial ligand plane in the three derivatives are all between an Np –Fe–Np angle. The axial ligand plane in [Fe(TMP)(CN)(1-MeIm)] makes a dihedral angle of 20.8°; with the closest Fe–Np vector and a dihedral angle 89.6°; with the 24-atom mean plane. The corresponding dihedral angles in [Fe(TPP)(CN)(Py)] are 39.2 and 80.9°; and 26.8 and 87.1°; in [Fe(TPP)(CN)(1-MeIm)]. Complete crystallographic details of these three structure are given in Tables S2—S19.
Figure 2.
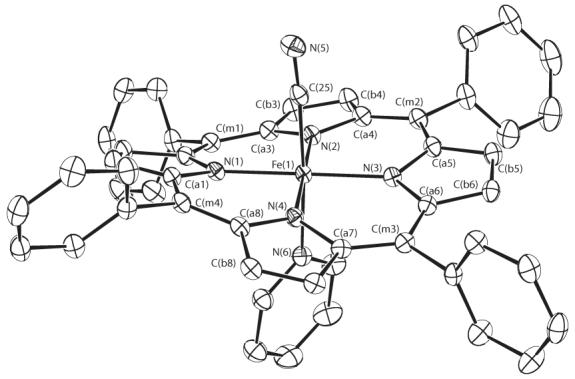
ORTEP diagram of [Fe(TPP)(CN)(Py)] displaying the atom labeling scheme. Thermal ellipsoids of all atoms are contoured at the 50% probability level. Hydrogen atoms have been omitted for clarity.
Figure 3.
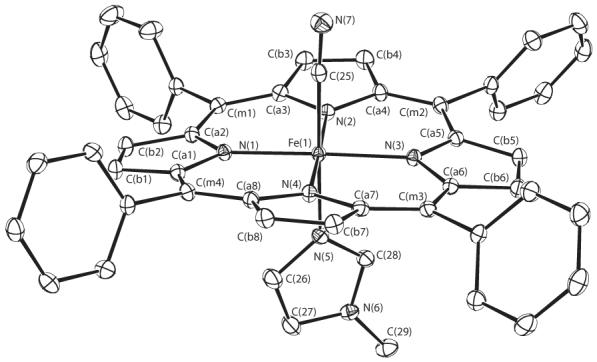
ORTEP diagram of [Fe(TPP)(CN)(1-MeIm)] displaying the atom labeling scheme. Thermal ellipsoids of all atoms are contoured at the 50% probability level. Hydrogen atoms have been omitted for clarity.
Figure 4.
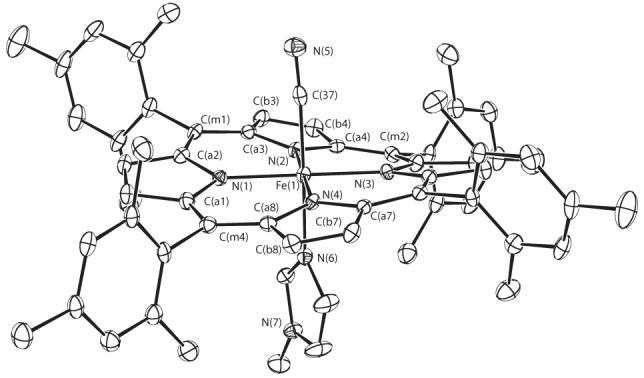
ORTEP diagram of [Fe(TMP)(CN)(1-MeIm)] displaying the atom labeling scheme. Thermal ellipsoids of all atoms are contoured at the 50% probability level. Hydrogen atoms have been omitted for clarity.
Formal diagrams showing the displacement of atoms (in units of 0.01 Å) from the 24-atom mean plane of [Fe(TPP)(CN)(1-MeIm)], [Fe(TPP)(CN)(Py)], [Fe(TMP)(CN)(1-MeIm)], and entity 1 of [K(18-C-6)][Fe(TMP)(CN)2] are given in Figure 5. As can be seen, most of the derivatives have predominantly ruffled cores, but the core in [Fe(TPP)(CN)(1-MeIm)] is primarily saddled. The diagrams also display the average values for bond lengths and bond angles. The longer average Fe–Np distance in [Fe(TPP)(CN)(1-MeIm)], compared to the other derivatives shown in Figure 5, is a result of the differing core conformation. Similar information for all entities of [K(18-C-6)][Fe(TMP)(CN)2] is given in Figures S6. A comparison of the information in these figures with that of the [K(18-C-6)][Fe(TMP)(CN)2] panel of Figure 5 shows the close correspondence in core conformation and other structural of all three entities.
Figure 5.

Formal diagrams of the porphyrin cores of [Fe(TPP)(CN)(1-MeIm)], [Fe(TPP)(CN)(Py)], [Fe(TMP)(CN)(1-MeIm)], and entity 1 of [K(18-C-6)][Fe(TMP)(CN)2]. The perpendicular displacements (in units of 0.01 Å) of the porphyrin core atoms from the 24-atom mean plane are displayed. In all diagrams, positive values of the displacement are towards the cyanide ligand. Also displayed on the formal diagrams are the averaged values of the chemically unique bond distances (in Å) and angles (in degrees). The numbers in parentheses are the esd’s calculated on the assumption that the averaged values were all drawn from the same population. The orientation of the imidazole or pyridine ligand is shown on these diagrams; the circle shows the position of the methyl group of the imidazole.
The angle between the two axial ligand donor atoms subtended at the iron are N—Fe—C = 178.88(6)°; for [Fe(TPP)(CN)(1-MeIm)], 176.05(9)°; for [Fe(TPP)(CN)(Py)], and 178.20(11)°; for [Fe(TMP)(CN)(1-MeIm)]. The equivalent C—Fe—C angle in [K(18-C-6)][Fe(TMP)(CN)2] appears to be less linear with C—Fe—C angles of 173.06(13)°; (entity 1), 171.61(14)°; (entity 2) and 172.54(14)°; (entity 3). The Fe—C—N groups are all nearly linear with observed angles ranging from 172.8 to 177.2°. A summary of selected bond distances and angles for six-coordinated (cyano)iron(III) porphyrinates and cyanide bound to hemoproteins is given in Table 3.
Table 3.
Selected Structural Parameters for (Cyano)iron Porphyrinates and Hemoproteins
| Complex | (Fe-Np)ava | Fe-Ca | Fe-NLa | C-Na | Fe-C-Nb | Tb, c | VCNd | ref. |
|---|---|---|---|---|---|---|---|---|
| Iron(III) porphyrinates | ||||||||
| [K((CH3)2CO)2][Fe(TPP)(CN)2]e | 2.000(6) | 1.975(2) | 1.147(3) | 177.8(3) | 1.6 | 2120 | 13 | |
| [K(18-crown-6)][Fe(TMP)(CN)2]f | 1.973(5) | 1.970 (4) | 1.158(1) | 175.3(12) | 3.8 | 2111 | tw | |
| 1.986(6) | 1.159(2) | 172.8(14) | 5.2 | tw | ||||
| [PPN][Fe(TMP)(CN)2]e | 2.003(4) | 1.990 (5) | 1.151(7) | 178.6(5) | 44 | |||
| [K(222)][Fe(BH(Bipy)2P)(CN)2]g | 1.949(14) | 1.949(4) | 1.152(9) | 170.8(5) | 5.2 | 45 | ||
| [Fe(OEOP)(CN)2]e | 1.966(9) | 1.969(10) | 1.128(14) | 177.4(11) | NR | 45 | ||
| [Fe(OEP)(CN)(Py)] | 1.980(4) | 1.934(4) | 2.087(3) | 1.140 | 179.1(1) | 0 | 15 | |
| [Fe(OEP)(CN)(1-MeIm)] | 1.994(7) | 1.927(8) | 2.048(6) | 1.15(1) | 175.6(5) | NR | 2115 | 47 |
| [Fe(TPP)(CN)(Py)] | 1.970(14) | 1.908(4) | 2.075(3) | 1.152(4) | 176.8(3) | 3.2 | 2130 | 14 |
| [Fe(TPP)(CN)(Py)] | 1.977(10) | 1.927(3) | 2.072(2) | 1.157(3) | 176.4(2) | 3.1 | 2130 | tw |
| [Fe(TPP)(CN)(1-MeIm)] | 1.993(3) | 1.9179(15) | 2.0149(12) | 1.1616(19) | 177.18(13) | 2.2 | 2130 | tw |
| [Fe(TMP)(CN)(1-MeIm)] | 1.982(8) | 1.929(3) | 2.026(2) | 1.155(4) | 176.2(3) | 3.2 | 2125 | tw |
| Hemoprotein Iron(III) | ||||||||
| Sperm whale Mb | 2.03 | 2.02 | 1.06 | 166 | 10 | 48 | ||
| Arthromyces ramosus peroxidase | 2.00 | 159 | 3 | 49 | ||||
Value A.
Value in degrees.
The tilt of Fe-CCN vector from the normal to the 24-atom mean plane.
Value in cm-1; KBr pellets.
These structures all have crystallographically required inversion symmetry.
Average values for the three unique [K(18-C-6)][Fe(TMP)(CN)2] entities; top line is for the simpler cyanide ligand, while the bottom line has cyanide further coordinated by [K(18-crown-6)].
Average value for the two cyanide groups.
Discussion
The diaquo complex, [Fe(Porph)(H2O)2]ClO4,[31] is a useful starting material for the preparation of all cyano-complexed derivatives reported herein. Cyanide ion is strongly basic, but the use of dried solvents and solubilizing an adequate concentration of potassium cyanide in the synthetic solvent systems is sufficient to eliminate any hydrolysis derived from the use of the diaquo complex. Although [K((CH3)2CO)2][Fe(TPP)(CN)2] had been readily synthesized from a KCN-saturated acetone solution as the cyanide ion source,[31] an attempt to prepare an analogous tetramesityl derivative [Fe(TMP)(CN)2]− using the same method failed. The use of the crown ether 18-crown-6 to solubilize potassium cyanide easily allowed the synthesis of the bis(cyano) derivative, [K(18-C-6)][Fe(TMP)(CN)2].
The mixed ligand complexes [Fe(TPP)(CN)(Py)], [Fe(TPP)(CN)(1-MeIm)], and [Fe(TMP)(CN)(1-MeIm)] were synthesized by the reaction of the appropriate diaquo complex with KCN solubilized in methanol, removal of the methanol followed by reaction of the neutral nitrogen ligand in chloroform solution. Unlike earlier syntheses of mixed ligand derivatives,[14, 15] the bis(cyano) intermediate was not isolated.
Cyanide has been extensively used as a probe ligand in hemoproteins; the properties of the resulting complexes have been studied by a variety of spectroscopic methods including infrared spectroscopy.[49, 50, 51] The IR C—N stretch provides a useful monitor of the bonding sites and ligand environment.[52, 53] As has been noted for hemoproteins, the C—N stretching band in cyano-substituted iron(III) porphyrinates is very weak. The C—N stretching frequency of the bis(cyano) derivatives are 2120 cm−1 for [Fe(TPP)(CN)2]− and 2111 cm−1 for [Fe(TMP)(CN)2]−. The C—N band shifts to higher frequency by 10–14 cm−1 when the trans-cyanide is replaced by pyridine or 1-MeIm ([Fe(TPP)(CN)(Py)], 2130; [Fe(TPP)(CN)(1-MeIm)], 2130 and [Fe(TMP)(CN)(1-MeIm)], 2125 cm−1).
Caughey and coworkers reported[52] a similar 10–13 cm−1 IR shift to higher frequency when a cyanide is replaced with pyridine in a series of 2,4-substituted deuteroporphyrin IX derivatives. They explained this “proximal or trans-effect” in terms of the substitution “rendering the iron(III) a less effective dπ-donor and a more effective σ-acceptor.” However, we note that the substitution of pyridine (a modest π-acceptor) or 1-methylimidazole (a modest π-donor) give identical IR shifts. Consequently, we conclude that the effect is clearly a result of change in the π-acceptor behavior of iron(III). Caughey and coworkers[52] also discussed possible correlations between the CN frequency and the stereochemistry of the bound cyanide. They suggested that an increasing CN represents an increased bending of the Fe—C—N bond. However, our current structural data of the cyano complexes shows no correlation of CN on the tilting of the Fe—CCN vector from the normal to the heme plane (Table 3) or on any bending of Fe—C—N. The mixed ligand porphyrinate derivatives have tilt angles (2.2 to 3.3°) both larger and smaller than the bis(cyano) species (1.6 to 5.2°) and have higher νCN frequency. With the available data, we would regard any correlation between structural distortion and C—N stretching frequency as tenuous.
We also note that the C—N stretching frequency of the tetramesitylporphyrinates are systematically lower than the corresponding TPP species. As is well-known, the CN− ion acts both as a σ-donor to the metal and a π-acceptor from the metal. It has been generally agreed that σ-donation tends to increase the value of νCN, while π back-bonding decrease the value of νCN.[53]−[57] From this and our experimental data, we deduce that the Fe(dπ)—cyanide (pπ*) interaction is enhanced for the tetramesitylporphyrinates compared to the TPPs because of the electron-donating substituents at the meso-phenyl positions and thus lead to a shift of CN to lower frequency.
A blue-shift of the Soret maxima in the absorption spectrum was observed upon replacing a trans-cyanide with a neutral nitrogen donor. For example, the Soret band of [Fe(TPP)(CN)2]− (424 nm) is blue-shifted when the trans-cyanide is replaced by pyridine ([Fe(TPP)(CN)(Py)], 420 nm) or 1-MeIm; ([Fe(TPP)(CN)(1-MeIm)], 418 nm). This is consistent with an increased ability of the iron(III) to accept electron density from the porphyrin nitrogens.[52] Interestingly, the magnitude of the blue shift is larger in the tetramesitylporphyrinate species consistent with the better σ-donating properties of this porphyrin.
The structural characterizations of four new six-coordinate cyanide derivatives [Fe(TPP)(CN)(1-MeIm)]·1-MeIm·CHCl3, [Fe(TPP)(CN)(Py)]·0.65Py, [Fe(TMP)(CN)(1-MeIm)]·1.8CHCl3 and [K(18-C-6)][Fe(TMP)(CN)2] are also presented in this paper. The average Fe–Np distances in these four new complexes range from 1.973 to 1.993 Å, distances that are all consistent with a low-spin state for the iron(III) atom.
We first consider the X-ray structure determination of the bis(cyano) complex of (meso-tetramesitylporphyrinato)iron(III). Three separate [K(18- C-6)][Fe(TMP)(CN)2] entities are found in the asymmetric unit of structure; each entity consists of an [Fe(TMP)(CN)2]− anion and a [K(18-crown-6)]+ cation. An ORTEP drawing of [K(18-C-6)][Fe(TMP)(CN)2] (entity 1) is shown in Figure 1. The diagram clearly shows that one of the cyanide ligands has the nitrogen atom coordinated to the potassium of the 18-crown-6 with a K–N distance of 2.848 (4) Å. The remaining two entities also have the same geometry and are shown in the supporting information. These tight ion-pair arrangements lead to two distinct Fe—C(CN) bond distances. The cyanide ion that is also bonded to the potassium ion has the longer Fe–C bonds (entity 1, 1.993(4); entity 2, 1.982(4); entity 3, 1.984(4) Å). The other set of Fe–C bonds are shorter (entity 1, 1.966(4); entity 2, 1.973(4); entity 3, 1.971(4) Å). The small (0.016 Å) difference in the two types of Fe–C distance is clearly a consequence of the electrostatic interaction of the one cyanide with the positively charged K(18-crown-6)] cation. The iron atom in entity 1 is displaced by 0.03 Å towards the cyanide ion complexed by the K(18-crown-6)] cation, while the iron atom in the other two entities are displaced by a small amount (0.01 Å) in the opposite direction. The conformation in all three porphyrin rings is similarly and strongly ruffled; the conformations of the three rings are compared in Supporting Information Figure S6.
The structures of three other bis(cyano) complexes have been previously determined and selected reported parameters are shown in Table 3. [K((CH3)2CO)2][Fe(TPP)(CN)2][13] and [PPN][Fe(TMP)(CN)2][44] have similar Fe–Np and Fe–C distances. The complex [PPN][Fe(TMP)(CN)2] had been reported as an iron(II) species, but in our view, the disordered I2 is more likely better described as a disordered I−3. The basket-handle porphyrin complex [K(222)][Fe(BH(Bipy)2P)(CN)2][45] has shorter distances that reflect the special steric effects that were built into the molecular structure. A somewhat different macrocyclic example, the octaethyloxoporphyrin species, [Fe(OEOP)(CN)2], also shows relatively small values for the coordination group bonds.
Table 3 also reports the same parameters for all known mixed axial ligand complexes. Three important generalizations concerning the entire group of (cyano)iron(III) complexes can be made. First, the Fe—C—N groups are all nearly linear. The two examples where the angle deviates most from linearity are the basket-handle porphyrin complex that was designed to sterically crowd the Fe—C—N group and force nonlinearity.[45] The second example are the Fe—C—N groups that are bonded to the [K(222)] cations in [K(18-C-6)][Fe(TMP)(CN)2], where the electrostatic interactions can be judged to lead to the larger deviation from linearity. Second, the Fe—C vector is always slightly tilted from the heme normal. This angle is denoted as τ in Table 3; the largest values of τ are found for the Fe—C—N groups that show the largest deviations from linearity. Finally, it can also be seen from Table 3 that all of the bis(cyano) species have longer Fe—C(CN) bonds compared to the mixed ligand species. This 0.05–0.07 Å A difference in Fe—C is readily explained by the competition of two CN− ligands on both sides of the porphyrin. There will be less π donation (per bond) by the iron to two π-accepting cyanides compared to a single cyanide acceptor in the mixed ligand species. The C—N bond distances are still seen to cluster within the narrow range of 1.15 to 1.16 Å.
The three mixed-ligand species display two different core conformations. [Fe(TPP)(CN)(1-MeIm)] has a rather strongly saddled porphyrin core while the TMP form of the same complex has a very ruffled core. The differences in core conformation probably explains the small difference in the values of the Fe–Np bond distances: 1.993(3) Å A for [Fe(TPP)(CN)(1-MeIm)] and 1.982(8) Å A for [Fe(TMP)(CN)(1-MeIm)]. The new crystalline form of [Fe(TPP)(CN)(Py)] has a ruffled porphyrin core as did the previously reported crystalline form.[14] Both have relatively short Fe–Np bond distances (1.970 and 1.977, Table 3). Similar pairs of saddled and ruffled cores are seen in a series of [Fe(TPP)(CO)(L)] complexes.[58]
Prior to the current study, the only mixed-ligand cyano complexes that had been structurally characterized were two derivatives with a pyridine ligand trans to the cyanide, [Fe(TPP)(CN)(Py)][14] and [Fe(OEP)(CN)(Py)].[15] In those two derivatives, the Fe–N(Py) bond distance was relatively long at 2.075(3) Å A or 2.087(3) Å, respectively. The new crystalline form of [Fe(TPP)(CN)(Py)] is similar with Fe–N(Py) = 2.072(2) Å; the average for the three complexes is 2.078(8) Å. The two new imidazole-ligated cyanide derivatives are seen to have significantly shorter trans Fe—N distances of 2.0149(12) Å and 2.026(2) Å or an average of 2.020(8) Å. Similar differences are seen in carbonyl or thiocarbonyl complexes with pyridines or imidazoles trans to the CO[58, 59, 60] or CS.[61] The effect of differing trans bond distances is likely the difference that imidazoles are modest π-electron-donors compared to the modest π-accepting character of pyridine.[62, 63] Clearly, one can expect that the cyanide and pyridine will compete for the same iron π density whereas the imidazole can π-donate and hence form a stronger bond.
We have also studied the electron paramagnetic spectra (EPR) of these compounds in frozen solution and especially in the solid (microcrystalline) state. The earliest EPR spectra obtained for iron(III) cyanides were all of the large gmax type and were primarily obtained on solids. The early single crystal experiments of Strouse and coworkers[16] established the value for all three components of the g tensor rather than only the largest value. The largest value is all that is commonly seen for solution or powder measurements for these complexes. Hence, Strouse et al. conclusively established the reality of the large gmax type of EPR spectrum for cyanide derivatives. This type of EPR spectrum is consistent with a (dxy)2(dxz,dyz)3 ground state with nearly degenerate energies for the two dπ orbitals. The near degeneracy of the two dπ orbitals results from the π-acceptor character of the cyanide ligand which with its axial symmetry interacts effectively equally with both orbitals. Subsequently, axial EPR spectra were observed for a number of bis(cyano)iron(III) derivatives in frozen solution.[20, 21, 23, 24] This EPR spectral type is consistent with a different ground state electron configuration, that of (dxz,dyz)4(dxy)1, as first shown by Walker, Scheidt and coworkers.[25, 26] In this state, the dxy orbital energy is higher than the energy of the dxz,dyz (dπ) orbitals. The state results from the coordination of π-accepting ligands that stabilize the dπ orbitals relative to the the dxy orbital. Nakamura has suggested that porphyrin core ruffling is the cause of many if not most of the bis(cyano) complexes that exhibit the (dxz,dyz)4(dxy)1 ground state.[20] This is consistent with the conclusion of Walker, Scheidt and coworkers[25] in that core ruffling does stabilize this state; ruffling is observed in all complexes of known structure and this (dxz,dyz)4 (dxy)1 ground state.
However, the expected structural and conformational features used by Nakamura et al. for cyano derivatives are based on analogy with observations made on related systems, they were not based directly on determined cyano derivatives structures. Our new structural and physical data clearly shows that the idea that core ruffling alone can cause the (dxz,dyz)4(dxy)1 ground state is too simple an explanation for the appearance of such a ground state for cyano derivatives. As can be seen from the EPR data given in Table 2, [K(18-C-6)][Fe(TMP)(CN)2] displays a large gmax type of EPR spectrum in the solid state with g = 3.47, 1.68, and 1.20. But, as we have noted earlier, all three distinct bis(cyano) entities in the solid state have extremely ruffled porphyrin cores. Clearly, even the substantial core ruffling found for [K(18-C-6)][Fe(TMP)(CN)2] does not lead the complex to display the (dxz,dyz)4(dxy)1 ground state as deduced by its EPR spectrum. Additional data emphasizes this point even more, when the EPR spectrum of [K(18- C-6)][Fe(TMP)(CN)2] is measured in frozen CH2Cl2 solution an axial EPR spectrum is observed. The differing EPR spectra observed in the two phases are illustrated in Figure 6. The exact nature of the core conformation in solution is unknown; but, since there are no packing considerations that suggest reasons for solid-state ruffling, a ruffled core in solution is probable. One possible effect of the solution environment is that the tight ion pairing observed for the three distinct entities in the solid state can be disrupted and an “isolated” [Fe(TMP)(CN)2]− ion, with no FeCN···K(18-C-6) interactions, will be the solution species. The crystal-field environment of the iron is thus likely to be more symmetric in solution.
Figure 6.
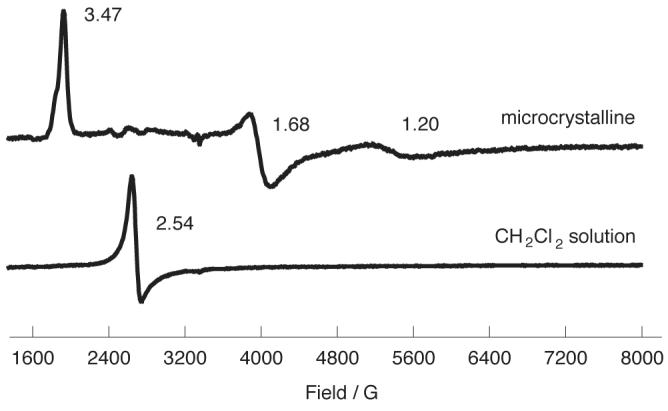
4.2 K EPR spectra for [K(18-C-6)][Fe(TMP)(CN)2] in the solid (microcrystalline) state and in a frozen CH2Cl2 solution.
Similarly, EPR spectra of [K((CH3)2CO)2][Fe(TPP)(CN)2][13] are different in the solid state and in frozen CH2Cl2/CH3OH solution. However, the differences are distinct and more complex than those of [K(18-C-6)][Fe(TMP)(CN)2]. A large gmax spectrum (g = 3.67) is observed in a microcrystalline sample of [K((CH3)2CO)2][Fe(TPP)(CN)2] along with features that are clearly the result of preferential orientations in the microcrystalline sample. As shown in Figure 7, microcrystalline [K((CH3)2CO)2][Fe(TPP)(CN)2] embedded in KBr shows 3.67 and 1.06 features associated with the complex and observed in an oriented single-crystal sample.[16] In addition, this method of sample preparation also leads to the appearance of axial spectral features with g = 2.57 and 1.45. This axial signal must also derive from a bis(cyano) derivative; replacement of cyanide with bromide is unlikely. In CH2Cl2-CH3OH solution, the large gmax feature decreases to 3.54. The solution spectrum also displays a classical rhombic EPR spectrum, the origin of which is not known. However, we believe that the spectrum is derived from a cyano-based species, not a hydrolysis species; further work on the EPR spectra of bis(cyano) derivatives and related species are in progress.
Figure 7.
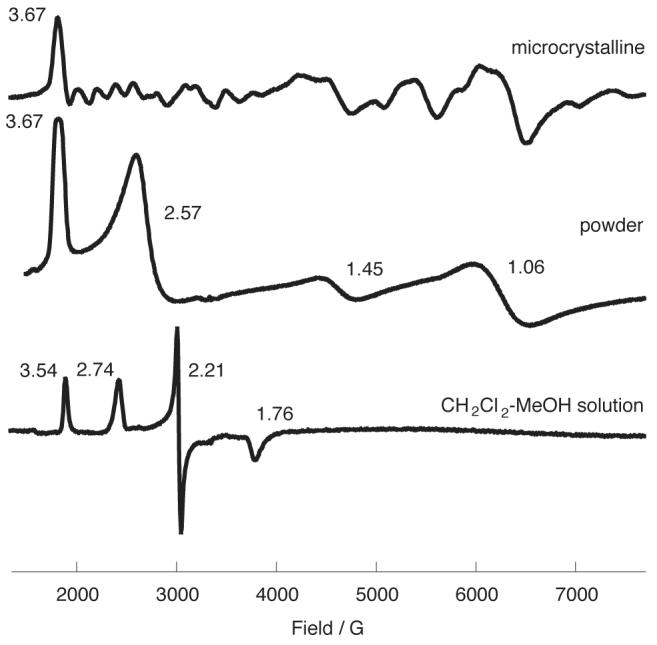
EPR spectra (6K) of [K((CH3)2CO)2][Fe(TPP)(CN)2] (top) ground microcrystalline, (middle) ground microcrystalline sample embedded in KBr, and (c) in a frozen CH2Cl2-CH3OH solution.
Environmental factors that lead to different EPR spectra, especially those caused by different solvents and conditions, have been previously reported by Nakamura et al.[23] There are clear solvent effects although we believe the origin of these effects are not necessarily those described by Nakamura et al. The appearance of two types of EPR spectra (large gmax and axial) observed in 4.2K frozen solution, such as that observed for [Fe(TEt3PP)(CN)2]− have been attributed to conformational changes between ruffled and planar porphyrin cores. However, it seems that the interconversion of two conformers on the EPR time scale is not necessary and even unlikely, given the low temperature of the measurements. Simply freezing the solution of the bis(cyanide) complex could lead to two distinct environments, one of which could be an ion-paired system; there need be no necessary change of conformers for changing EPR spectra.
The EPR spectra of the mixed axial ligand systems [Fe(Por)(CN)(L)] are simpler: in the solid state all display a large gmax type of EPR spectrum (see Table 2 and Figures S7–S9). The frozen solution spectra of these characterized solid-state species is simple, they continue to display large gmax EPR spectra, with some change in the observed value of the largest g. An unusual feature of many of these species is that they often display one additional g value. Again, ruffled porphyrin cores in cyanide complexes do not lead to the observation of EPR signals appropriate for the (dxz,dyz)4(dxy)1 ground state as two of the mixed ligand species have substantially ruffled cores.
The only mixed axial ligand species for which an axial EPR has been reported are solution spectra for complexes where the trans axial ligand is 2-methylimidazole.[41] This hindered imidazole is known to have a diminished binding constant in iron porphyrinates. It seems likely to us that the species for which an EPR spectrum has actually been obtained are those with two cyanide ligands bound. (Note the similarity in these values with those reported for the bis(cyanides) in Table 2.) No such species with a less sterically hindered imidazole has been shown to display an axial EPR spectrum, on the contrary, all show large gmax EPR spectra. It would be useful to have EPR spectra on characterized solid-state species where all questions of stoichiometry are absolutely established. At the moment, we conclude that the effect of the pyridine or imidazole ligand is to lower the symmetry of the complex sufficiently to disallow a (dxz,dyz)4(dxy)1 ground state.
Solid-state Mössbauer spectra have been obtained on two bis(cyanide) complexes, [K((CH3)2CO)2][Fe(TPP)(CN)2] and [K(18-C-6)][Fe(TMP)(CN)2], and three mixed ligand complexes, [Fe(TPP)(CN)(Py)], [Fe(TPP)(CN)(1-MeIm)], and [Fe(TMP)(CN)(1-MeIm)]. Selected attempts to obtain solution Mössbauer spectra were unsuccessful, even with57 Fe enrichment, owing to limited solubility in solvent mixtures appropriate for Mössbauer measurements. The spectra obtained over a large temperature range in the presence of a small (500 gauss) field and in high field show several common features. At room temperature the lines widths of all species are relatively sharp with normal line widths. At this temperature, the right-hand and left-hand lines of the quadrupole doublet have similar widths. As the temperature is lowered, the lines broaden and the two lines become less symmetric. In some cases, the lines become so broad that a fit for the line widths and the quadrupole splitting is impossible (at the lowest temperatures). All of the cyanide species show evidence of this magnetic broadening. The temperature-dependent spectra are illustrated for all complexes in the Supporting Information. These data have been summarized in Table 4 along with the limited amount of information already available in the literature, all of which had been measured at T ≥ 78 K. Although there is very little prior temperature-dependent data, the same pattern can be observed.
Table 4.
Mössbauer Parameters for Low-Spin (Cyano)iron(III)porphyrinatesa
| Complex | phase | ΔEqb | δb | T(L)b, c | T(R)b, c> | T(K) | ref |
|---|---|---|---|---|---|---|---|
| [Fe(Porph)(CN)2]- | |||||||
| [K((CH3)2CO)2][Fe(TPP)(CN)2] | solid | -0.40d | 0.19d | 0.4 | 4.2 | tw | |
| (microcrystalline) | no fit | no fit | very | broad | 15 | tw | |
| no fit | no fit | very | broad | 50 | tw | ||
| 0.22 | 0.20 | very | broad | 100 | tw | ||
| 0.24 | 0.17 | 0.36 | 0.43 | 150 | tw | ||
| 0.22 | 0.15 | 0.29 | 0.32 | 200 | tw | ||
| 0.23 | 0.12 | 0.27 | 0.29 | 250 | tw | ||
| 0.24 | 0.09 | 0.22 | 0.24 | 298 | tw | ||
| [K(18-C-6)][Fe(TMP)(CN)2] | solid | 1.25e | 0.14e | 0.5 | 4.2 | tw | |
| (microcrystalline) | (-1.25f) | no fit | very | broad | 15 | tw | |
| 1.27 | 0.16 | 1.24 | 0.82 | 50 | tw | ||
| 1.15 | 0.16 | 0.70 | 0.55 | 100 | tw | ||
| 1.09 | 0.16 | 0.56 | 0.47 | 200 | tw | ||
| 0.82 | 0.12 | 0.39 | 0.34 | 250 | tw | ||
| 0.68 | 0.10 | 0.34 | 0.29 | 298 | tw | ||
| K[Fe(TPP)(CN)2]·2(CH3)2CO | solid | 0.63 | 0.31 | 0.50 | 78 | 64 | |
| K[Fe(TTP)(CN)2]·2(CH3)2COg | solid | 0.69 | 0.30 | 0.40 | 78 | 64 | |
| [Fe(TTP)(CN)(OH2)]?g | 1.79 | 0.06 | 0.86 | 78 | 64 | ||
| Na[Fe(TPPS)(CN)2]·bis DMF | solid | 0.50 | 0.30 | 0.50 | 78 | 64 | |
| Na[Fe(PPIX)(CN)2]·3NaCN·6H2O | solid | 0.53 | 0.20 | 0.48 | 80 | 65 | |
| 0.40 | 0.10 | 0.28 | 298 | 65 | |||
| K[FePPIX(CN)2]g | solution | 0.73 | 0.20 | 0.50 | 80 | 66 | |
| K[FePPIX(CN)(OH)?]g | 1.51 | 0.16 | 0.62 | 80 | 66 | ||
| [Fe(TPPS)]+xsCN- | solution | ∼0 | 0.07 | 1.02 | 78 | 64 | |
| K[Fe(PPIX)(CN)2] | solution | 0.35 | 0.24 | - | 4.2 | 36 | |
| [Fe(Porph)(CN)(L)] | |||||||
| [Fe(TPP)(CN)(Py)] | solid | −1.04e | 0.19e | 0.42 | 4.2 | tw | |
| (microcrystalline) | (−1.04f) | 0.19 | 0.71 | 1.06 | 16 | tw | |
| −1.05 | 0.18 | 0.42 | 0.49 | 100 | tw | ||
| −0.99 | 0.15 | 0.30 | 0.33 | 200 | tw | ||
| −0.95 | 0.10 | 0.24 | 0.26 | 298 | tw | ||
| [Fe(TPP)(CN)(1-MeIm)] | solid | −1.35e | 0.22e | 0.4 | 4.2 | tw | |
| (microcrystalline) | (−1.35f ) | 0.29 | 0.77 | 0.87 | 15 | tw | |
| −1.35 | 0.20 | 0.56 | 0.81 | 100 | tw | ||
| −1.23 | 0.17 | 0.28 | 0.34 | 200 | tw | ||
| −1.11 | 0.11 | 0.23 | 0.25 | 298 | tw | ||
| [Fe(TMP)(CN)(1-MeIm)] | solid | −1.41e | 0.19e | 0.5 | 4.2 | tw | |
| (microcrystalline) | (−1.41f ) | no fit | very | broad | 15 | tw | |
| −1.42 | 0.23 | 1.20 | 2.02 | 50 | tw | ||
| −1.36 | 0.19 | 0.57 | 0.67 | 100 | tw | ||
| −1.25 | 0.18 | 0.36 | 0.38 | 200 | tw | ||
| −1.14 | 0.16 | 0.26 | 0.28 | 250 | tw | ||
| −1.01 | 0.12 | 0.24 | 0.25 | 295 | tw | ||
| [Fe(PPIX)(CN)(Py)] | solution | 1.24 | 0.34 | – | 4.2 | 36 | |
Unless otherwise specified, measurements in low (500 G) magnetic field.
mm/s.
Line width at FWHM.
Value from fit to high-field spectra (5 and 9 T only).
Value from fit to high-field spectra (1, 5, 9T).
Not observed, value from temperature dependent fit.
Sample is mixture of two species.
The isomer shifts for the new compounds are all smaller than those found for “classical” low-spin iron(III) porphyrinates[67] with a value of ∼0.09—0.10 mm/s at room temperature. This small isomer shift is consistent with significant covalency in the Fe–C(CN) bond. The values and the temperature dependent quadrupole splittings of these cyanide derivatives present interesting patterns.
We first consider the two bis(cyanide) derivatives. [K((CH3)2CO)2][Fe(TPP)(CN)2] and [K(18-C-6)][Fe(TMP)(CN)2] show substantially different values for the quadrupole splitting constant (ΔEq) and very different temperature dependences. [K(18-C-6)][Fe(TMP)(CN)2] shows temperature-dependent changes of the quadrupole splitting value in the usual direction: an increased magnitude of the ΔEq as the temperature decreases. The magnitude of the change in the ΔEq over temperature is extraordinarily large and varies from 1.27 at 50 K to 0.68 mm/s at RT. A large variation in the temperature dependence of the ΔEq is typically the result of a low-lying excited state. A crystal-field model fit of the temperature dependence of the ΔEq of [K(18- C-6)][Fe(TMP)(CN)2] using the experimental values between 50 and 298 K allows the prediction of the lower temperature values. The procedure gives a value for the ΔEq of −1.25 mm/s at 4.2 K, which can be checked with a fit to the spectra obtained in high field. The fit to the spectra obtained in high field also allows the determination of the sign of the ΔEq, which is negative. Importantly, the temperature-dependent fit gives an estimate of the energy gap between the two dπ orbitals of 214 cm−1.[68]
The Mössbauer spectrum of [K((CH3)2CO)2][Fe(TPP)(CN)2] at room temperature shows an extremely narrow quadrupole doublet, with ΔEq = 0.24 mm/s, and an isomer shift of 0.09 mm/s, reasonable values for a low-spin ferric system. ΔEq shows little if any temperature dependence down to 100K. Below this temperature, the magnetic hyperfine interaction is in the intermediate fluctuation limit, and magnetic broadening obscures the quadrupole doublet. This intermediate fluctuation regime persists down to 4.2K and even in an applied field of 1T. However, in larger fields of 5 and 9 T at 4.2K magnetic hyperfine lines are well-resolved, indicating the slow fluctuation limit has finally been reached (see Figure 8). When the temperature was raised to 10K in the 9T applied field, the magnetic lines were broadened and the overall magnetic splitting was slightly reduced. Both of the effects argue once again for the onset of intermediate spin fluctuations. So even in 9T, our maximum available field, we are unable to follow the temperature dependence of ΔEq above 4.2K.
Figure 8.

Figure illustrating the fits to the Mössbauer data obtained for [K((CH3)2CO)2][Fe(TPP)(CN)2] at 4.2 K and 5 and 9 T (top two panels). The bottom panel displays the fit obtained, using the 4.2 K fit parameters, for the 9 T, 10 K data, showing the onset of intermediate spin fluctuations.
The solid lines in Figure 8 are fits made using an S=1/2 spin Hamiltonian model, with final parameters given in Table S1. The large g-values for the system and the corresponding large magnetic hyperfine A-tensor results in an internal field of about 45T at 4.2K. The large magnetic splitting unfortunately makes the spectra relatively insensitive to the value of ΔEq. However, a plot of Χ2 vs. ΔEq, where all free parameters are minimized for each value of ΔEq, shows a modest minimum for ΔEq π ≈ 0.4mm/s, a value that is slightly larger than the high-temperature value, but with an uncertainty that is comparable to the difference.
Although the energies of the dxz and dyz orbitals are close, they are subject to Jahn-Teller splitting and are not degenerate. We have carried out crystal-field calculations in order to estimate ΔEq using parameters that fit the observed g-values (0.52, 1.05, 3.7),[16] various assumed values of the energy difference between dxz and dyz and a value of the spin-orbit coupling parameter of 250 cm−1. Energy splittings of 1, 20, 40, 60, 80, and 100 cm−1 between the two dπ orbitals were evaluated. After inclusion of spin-orbit coupling, the difference in the energies of the ground state and the excited state are between 264 and 282 cm−1. Moreover, for the small values of the crystal field splitting of the dπ orbitals, the spin-orbit mixing leads to electric field gradients of the ground and excited states that are very similar. Calculated values of ΔEq, as a function of temperature, have been obtained and are plotted in Figure S15. These calculation suggest, given the observed constant value of ΔEq between 100 and 298 K, that the splitting in the two dπ orbitals must be very small, near 40 cm−1 or very close to the 35 cm−1 estimate of Inniss et al.[16] based on EPR measurements. The resulting energy difference between the ground state and the excited state is 267 cm−1. Thus the observed weak temperature dependence is credible. The small magnitude of the quadrupole splitting also argues for a near cubic field around iron, viz., the axial and equatorial ligand fields are nearly equal.
The mixed ligand complexes show ΔEq values ranging from −1.04 to −1.41 mm/s. The lower value of −1.04 is observed for the [Fe(TPP)(CN)(Py)] derivative where the modest π-accepting character of the pyridine ligand could be expected to lower the ΔEq value. The values for the two [Fe(Por)(CN)(1-MeIm)] are similar (−1.35 and −1.41 mm/s); the higher values are consistent with a modest π-donating character for the imidazole ligand.
The mixed ligand complexes also show temperature-dependent changes in the magnitude of the quadrupole splitting constant. The direction of change is the usual one: a shift to increased values of the quadrupole splitting as the temperature is decreased. Although the total magnitude of the change is larger than usual, the magnitude of change observed is smaller than that of the [K(18-C-6)][Fe(TMP)(CN)2] derivative. Crystal-field model fits of the temperature dependence of the ΔEq of the three complexes using the experimental values between 100 and 298 K allows the prediction of the lower temperature values. Again the fits can be checked with a fit to the spectra obtained in high field. The fit to the spectra obtained in high field also allows the determination of the sign of the ΔEq, which is negative for all complexes. As before, the temperature-dependent fit gives an estimate of the energy gap between the singly-occupied dyz orbital and the doubly-occupied dxz orbital in all three complexes.[68] The values of this splitting are in qualitative agreement with the value of the largest g-tensor component observed in each of the three species. For the three species the values of the energy gap and the g-tensor value are: [Fe(TPP)(CN)(Py)], 556 cm−1 and 3.17; [Fe(TPP)(CN)(1-MeIm)], 396 cm−1 and 3.51; and [Fe(TMP)(CN)(1-MeIm)], 309 cm−1 and 3.37. These calculated energy differences have ignored the effects of spin orbit coupling, which has an increasingly important contribution as the crystal field splitting between the dπ orbitals becomes smaller. The energy gaps between the two dπ orbitals in the five complexes studied are shown schematically in Figure 9 and where the inclusion of spin orbit coupling effects are shown in the bottom panel of the figure.[71]
Figure 9.
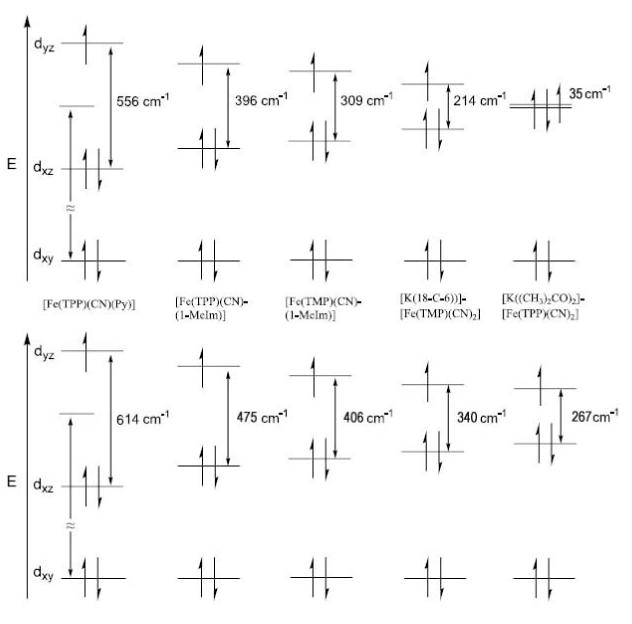
Schematic drawing showing the relative energy difference between the two near degenerate dπ orbitals of the cyanide complexes reported. The top panel shows values from a crystal field model that ignores spin orbit coupling, while the second takes into account an assumed value of 250 cm−1 for the spin orbit coupling.
Thus the Mössbauer spectra gives the ground state of all cyanide species measured thus far as (dxy)2(dxz)2(dyz)1 in the solid state. The possible exception is that of [K((CH3)2CO)2][Fe(TPP)(CN)2] where the energy may be so small as to make such distinctions unimportant and the configuration simply described as (dxy)2(dxz,dyz)3. Although other electron configurations may be accessible under different conditions, the (dxy)2(dxz)2(dyz)1 electron configuration is dominant.
Summary
The structural and spectroscopic data for the various (cyano)iron(III) porphyrinates reported herein clearly show that the ground electron configuration is always (dxy)2(dxz)2(dyz)1, with the energy gap between the dxz and dyz orbitals quite small (≤∼600 cm−1) and variable. Estimates of the gap from temperature-dependent Mössbauer are in qualitative agreement with those obtained from EPR spectra. For bis(cyanide) derivatives the two limiting low-spin states can be close in energy. It is also clear that there is not a single structural feature that assures that bis(cyano) iron(III) porphyrinates will not have the (dxy)2(dxz)2(dyz)1 ground state configuration. However, small perturbations of the bis(cyano)iron(III) porphyrinate systems may be sufficient to tip them to an alternate state. Thus far, in our experience, those circumstances always require a solution state.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health for support of this research under Grant GM-38401 to WRS. We thank the NSF for EPR support through Instrumentation Grant NSF-98-70990 and X-ray instrumentation through Grant CHE-0443233. We thank a reviewer for their helpful comments on the nature of the electronic structure of K[Fe(TPP)(CN)2].
Footnotes
Supporting Information Available: Figures S1 and S2 give OR-TEP diagrams for entities 2 and 3 of [K(18-C-6)][Fe(TMP)(CN)2]. Figures S3–S5 give ORTEP diagrams of the K(18-crown-6) rings of entities 1–3, respectively. Figure S6 gives formal diagrams of the atomic displacements from the mean plane of the porphyrin cores in entities 1–3 of [K(18-C-6)][Fe(TMP)(CN)2], Figures S7–S9 show solution and solid state EPR spectra and Figures S10–S14 show the temperature-dependent Mössbauer spectra for all five compounds. Tables S2–S25 give complete crystallographic details, atomic coordinates, anisotropic thermal parameters, fixed hydrogen atom coordinates, bond lengths, and bond angles for all four new structures. Crystallographic information as CIF files are available. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- [1].Antonini E, Brunori M. Hemoglobin and Myoglobin in their Reactions with Ligands. North-Holland; Amsterdam, London: 1971. [Google Scholar]
- [2].Solomonson LP. In: Cyanide in Biology. Vennesland B, Conn EE, Knowles CJ, Westley J, Wissing I, editors. Academic Press; New York: 1981. pp. 11–28. [Google Scholar]
- [3](a).Moreno SNJ, Stolze K, Janzen EG, Mason RP. Arch. Biochem. Biophys. 1988;265:267. doi: 10.1016/0003-9861(88)90127-0. [DOI] [PubMed] [Google Scholar]; (b) Stolze K, Moreno SNJ, Mason RP. J. Inorg. Biochem. 1989;37:45. doi: 10.1016/0162-0134(89)80028-5. [DOI] [PubMed] [Google Scholar]
- [4](a).Wilson DF, Erecinska M. In: Methods in Enzymology. Fleischer S, Leaster P, editors. Vol. 53. Academic Press; New York: 1978. pp. 191–197. [DOI] [PubMed] [Google Scholar]; (b) Erecinska M, Wilson DF, Sato N, Nicholls P. Arch. Biochem. Biophys. 1972;151:188. doi: 10.1016/0003-9861(72)90487-0. [DOI] [PubMed] [Google Scholar]
- [5].Nicholls P, Vanburen KJH, Gelder BFV. Biochem. Biophys. Acta. 1972;275:279. [Google Scholar]
- [6](a).Baumann R, Wuthrick K. Helv. Chim. Acta. 1973;56:585. doi: 10.1002/hlca.19730560204. [DOI] [PubMed] [Google Scholar]; (b) Baumann R, Wuthrick K. Helv. Chim. Acta. 1974;57:336. [Google Scholar]
- [7](a).Frye JS, LaMar GN. J. Am. Chem. Soc. 1975;97:3561. doi: 10.1021/ja00845a070. [DOI] [PubMed] [Google Scholar]; (b) LaMar GN, Viscio DB. 1974;96:7354. doi: 10.1021/ja00830a033. [DOI] [PubMed] [Google Scholar]
- [8].Goff H. J. Am. Chem. Soc. 1977;99:7723. doi: 10.1021/ja00462a022. [DOI] [PubMed] [Google Scholar]
- [9].Morishima I, Inubushi T. J. Am. Chem. Soc. 1978;100:3568. [Google Scholar]
- [10].Wang J-T, Yeh HJC, Johnson DF. J. Am. Chem. Soc. 1978;2400;100 [Google Scholar]
- [11].Goff H, Morgan LO. Inorg. Chem. 1976;15:2069. [Google Scholar]
- [12].Abbreviations: Porph, generalized porphyrin dianion; Np, porphyrinato nitrogen; NL, ligand nitrogen; Mb, Myoglobin; THP, dianion of porphine; PPIX, dianion of protoporphyrin IX; TPP, dianion of meso-tetraphenylporphyrin; TMP, meso-tetramesitylporphyrin; TTP, meso-tetra-p-tolylporphyrin; OEP, dianion of 2,3,7,8,12,13,17,18-octaethylporphyrin; OEOP, dianion of octaethyloxoporphyrin; TPPS, p-tetrasulphophenylporphyrin; QTPP, dianion of quinoxalinotetraphenylporphyrin; MQTPP, dianion of (methylquinoxalino)tetraphenylporphyrin; PTPP, dianion of pyrazinotetraphenyl-porphyrin; ORTPP (R = M or E), dianion of 2,3,7,8,12,13,17,18-octaalkyl-5,10,15,20-tetraphenylporphyrin where R is methyl (M) or ethyl (E); TEt3PP, meso-tetrakis(2,4,6-triethylphenyl)-porphyrin; TiPr3PP, meso-tetrakis(2,4,6-triisopropylphenyl)-porphyrin; TRP (R = Me, Et, iPr, cPr, or nPr), dianion of meso-tetraalkylporphyrin where R is methyl (Me) ethyl (Et), isopropyl (iPr), cyclopropyl (cPr), or propyl (nPr); p-X-TPP, meso-tetrakis(p-substituted phenyl)-porphyrin; BH(Bipy)2P, bis(6,6′-diphenylbipyridine) basket-handle porphyrin; 18-C-6, 1,4,7,10,13,16-hexaoxacyclooctadecane; PPN, bis-(triphenylphosphine)nitrogen; THF, tetrahydrofuran; Py, Pyridine; HIm, imidazole; 1-MeIm, 1-methylimidazole; 2- MeHIm, 2-methylimidazole; Kryptofix-222 or 222, 4,7,13,16,21,24- hexaoxo-1,10-diazabicyclo[8.8.8]hexacosane; 18-C-6, 1,4,7,10,13,16-hexaoxacyclooctadecane.
- [13].Scheidt WR, Haller KJ, Hatano K. J. Am. Chem. Soc. 1980;102:3017. [Google Scholar]
- [14].Scheidt WR, Lee YJ, Luangdilok W, Haller KJ, Anzai K, Hatano K. Inorg. Chem. 1983;22:1516. [Google Scholar]
- [15].Scheidt WR, Hatano K. Acta Crystallogr., Sect. C. 1991;C47:2201. doi: 10.1107/s0108270191003220. [DOI] [PubMed] [Google Scholar]
- [16].Inniss D, Soltis M, Strouse CE. J. Am. Chem. Soc. 1988;110:5644. [Google Scholar]
- [17] (a).Migita CT, Iwaizumin M. J. Am. Chem. Soc. 1981;103:4378. [Google Scholar]; (b) Tsai A, Palmer G. Biochem. Biophys. Acta. 1982;681:484. doi: 10.1016/0005-2728(82)90191-8. [DOI] [PubMed] [Google Scholar]
- [18](a).Walker FA, Huynh BH, Scheidt WR, Osvath SR. J. Am. Chem. Soc. 1986;108:5288. [Google Scholar]; (b) Walker FA, Reis D, Balke VL. J. Am. Chem. Soc. 1984;106:6888. [Google Scholar]
- [19].Taylor CPS. Biochim. Biophys. Acta. 1977;491:137. doi: 10.1016/0005-2795(77)90049-6. [DOI] [PubMed] [Google Scholar]
- [20].Nakamura M, Ikeue T, Fujii H, Yoshimura T. J. Am. Chem. Soc. 1997;119:6284. [Google Scholar]
- [21].Nakamura M, Ikeue T, Ikezaki A, Ohgo Y, Fujii H. Inorg. Chem. 1999;38:3857. doi: 10.1021/ic981184+. [DOI] [PubMed] [Google Scholar]
- [22].Ikeue T, Ohgo Y, Saitoh T, Nakamura M, Fujii H, Yokoyama M. J. Am. Chem. Soc. 2000;122:4068. [Google Scholar]
- [23].Ikezaki A, Nakamura M. Inorg. Chem. 2002;41:2761. doi: 10.1021/ic0108383. [DOI] [PubMed] [Google Scholar]
- [24].Ikezaki A, Ikeue T, Nakamura M. Inorg. Chim. Acta. 2002;335:91. [Google Scholar]
- [25].Walker FA, Nasri H, Turowska-Tyrk I, Mohanrao K, Watson CT, Shokhirev NV, Debrunner PG, Scheidt WR. J. Am. Chem. Soc. 1996;118:12109. [Google Scholar]
- [26](a).Safo MK, Gupta GP, Watson CT, Simonis U, Walker FA, Scheidt WR. J. Am. Chem. Soc. 1992;114:7066. [Google Scholar]; (b) Safo MK, Walker FA, Raitsimring AM, Walters WP, Dolata DP, Debrunner PG, Scheidt WR. J. Am. Chem. Soc. 1994;116:7760. [Google Scholar]
- [27].Armarego WLF, Perrin DD. Purification of Laboratory Chemicals. Butterworth-Heinemann; Oxford, England: 1997. p. 413. [Google Scholar]
- [28].Adler AD, Longo FR, Finarelli JD, Goldmacher J, Assour J, Korsakoff L. J. Org. Chem. 1967;32:476. [Google Scholar]
- [29](a).Wagner RW, Lawrence DS, Lindsey JS. Tetrahedron Lett. 1987;28:3069. [Google Scholar]; (b) Lindsey JS, Wagner RW. J. Org. Chem. 1988;54:828. [Google Scholar]
- [30](a).Adler AD, Longo FR, Kampus F, Kim J. J. Inorg. Nucl. Chem. 1970;32:2443. [Google Scholar]; (b) Buchler JW. Chapter 5. In: Smith KM, editor. Porphyrins and Metallo-porphyrins. Elsevier Scientific Publishing; Amsterdam, The Netherlands: 1975. [Google Scholar]
- [31].Scheidt WR, Cohen IA, Kastner ME. Biochemistry. 1979;18:3546. doi: 10.1021/bi00583a017. [DOI] [PubMed] [Google Scholar]
- [32].Sheldrick GM. Acta Crystallogr., Sect. A. 1990;A46:467. [Google Scholar]
- [33].Sheldrick GM. Program for the Refinement of Crystal Structures. Universität Göttingen; Germany: 1997. [Google Scholar]
- [34].R1 = Σ ∥ Fo| − |Fc ∥ /Σ|Fo| and ωR2 = {Σ[ω(Fo2 − Fc2)2]/Σ[ωFo4]}½. The conventional R-factors R1 are based on F , with F set to zero for negative F2. The criterion of F2 > 2σ(F2) was used only for calculating R1. R-factors based on F 2 (wR2) are statistically about twice as large as those based on F , and R-factors based on ALL data will be even larger.
- [35].Sheldrick GM. Program for Empirical Absorption Correction of Area Detector Data. Universität Göttingen; Germany: 1996. [Google Scholar]
- [36].Rhynard D, Lang G, Spartalian K, Yonetani T. J. Chem. Phys. 1979;71:3715. [Google Scholar]
- [37].Ikeue T, Ohgo Y, Saitoh T, Yamaguchi T, Nakamura M. Inorg. Chem. 2001;40:3423. doi: 10.1021/ic001412b. [DOI] [PubMed] [Google Scholar]
- [38].Walker FA, Kadish KM, Smith KM. In: The Porphyrin Handbook. Guilard R, editor. Vol. 5. Academic Press; New York: 2000. p. 132. [Google Scholar]
- [39].Wojaczynski J, Latos-Grazynski L, Tadeusz G. Inorg. Chem. 1997;36:6299. [Google Scholar]
- [40].Chacko VP, LaMar GN. J. Am. Chem. Soc. 1982;104:7002. [Google Scholar]
- [41].Nakamura M, Ikeue T, Fujii H, Yoshimura T, Tajima K. Inorg. Chem. 1998;37:2405. [Google Scholar]
- [42].Thomson AJ, Gadsby PMA. J. Chem. Soc., Dalton Trans. 1990:1921. [Google Scholar]
- [43].Kaufman J, Siegel LM, Spicer LD. Biochemistry. 1993;32:8782. doi: 10.1021/bi00085a008. [DOI] [PubMed] [Google Scholar]
- [44].Bartczak TJ, Wolowiee S, Latos-Grazynski L. Inorg. Chim. Acta. 1998;277:242. [Google Scholar]
- [45].Schappacher M, Fischer J, Weiss R. Inorg. Chem. 1989;28:390. [Google Scholar]
- [46].Balch AL, Noll BC, Safari N. Inorg. Chem. 1993;32:2901. [Google Scholar]
- [47].Scott MJ, Lee SC, Holm RH. Inorg. Chem. 1994;33:4651. [Google Scholar]
- [48].Bolognesi M, Rosano C, Losso R, Borassi A, Rizzi M, Wittenberg JB, Boffi A, Ascenzi P. Biophys. J. 1999;77:1093. doi: 10.1016/S0006-3495(99)76959-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Fukuyama K, Kunishima N, Amada F, Kubota T, Matsubara H. J. Biol. Chem. 1995;270:21884. doi: 10.1074/jbc.270.37.21884. [DOI] [PubMed] [Google Scholar]
- [50].Dalosto SD, Vanderkooi JM, Sharp KA. J. Phys. Chem. B. 2004;108:6450. doi: 10.1021/jp0310697. [DOI] [PubMed] [Google Scholar]
- [51].Milani M, Ouellet Y, Ouellet H, Guertin M, Boffi A, Antonini G, Bocedi A, Mattu M, Bolognesi M, Ascenzi P. Biochemistry. 2004;43:5213. doi: 10.1021/bi049870+. [DOI] [PubMed] [Google Scholar]
- [52].Yoshikawa S, O’Keefe DH, Caughey WS. J. Biol. Chem. 1985;260:3518. [PubMed] [Google Scholar]
- [53].Reddy KS, Yonetani T, Tsuneshige A, Chance B, Kushkuley B, Stavrov SS, Vanderkooi JM. Biochemistry. 1996;35:5562. doi: 10.1021/bi952596m. [DOI] [PubMed] [Google Scholar]
- [54].Griffith WP, Turner GT. J. Chem. Soc. A. 1970:858. [Google Scholar]
- [55].Collins TJ, Fox BG, Hu ZG, Kostka KL, Munck E, Rickard CEF, Wright LJ. J. Am. Chem. Soc. 1992;114:8724. [Google Scholar]
- [56](a).de Lange PPM, Kraakman MJA, van. Wijnkoop M, Friihauf H-W, Vrieze K, Smeets WJJ, Spek AL. Inorg. Chim. Acta. 1992;196:151. [Google Scholar]; (b) Bassett J-M, Berry DE, Barker GK, Green M, Howard JAK, Stone FGA. J. Chem. Soc., Dalton Trans. 19791003 [Google Scholar]; (c) Cotton FA, Zingales F. J. Am. Chem. Soc. 1961;83:351. [Google Scholar]; (d) Salzmann R, Mcmahon MT, Godbout N, Sanders LK, Wojdelski M, Oldfield E. J. Am. Chem. Soc. 1999;121:3818. [Google Scholar]
- [57].Carnahan EM, Rardin RL, Bott SG, Lippard SJ. Inorg. Chem. 1992;31:5193. [Google Scholar]
- [58].Silvernail NJ, Roth A, Schulz CE, Noll BC, Scheidt WR. J. Am. Chem. Soc. 2005;127:14422. doi: 10.1021/ja053148x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Salzmann R, Ziegler CJ, Godbout N, McMahon MT, Suslick KS, Oldfield E. J. Am. Chem. Soc. 1998;120:11323. [Google Scholar]
- [60].Peng SM, Ibers JA. J. Am. Chem. Soc. 1976;98:8032. doi: 10.1021/ja00441a025. [DOI] [PubMed] [Google Scholar]
- [61].Cao C, Dahal S, Shang M, Beatty AM, Hibbs W, Schulz CE, Scheidt WR. Inorg. Chem. 2003;42:5202. doi: 10.1021/ic030043r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Epstein LM, Straub DK, Maricondi C. Inorg. Chem. 1967;6:1720. [Google Scholar]
- [63].Vashi RP, Marques HM. J. Inorg. Biochem. 2004;98:1471. doi: 10.1016/j.jinorgbio.2004.05.016. [DOI] [PubMed] [Google Scholar]
- [64].Silver J, Taies JA. Inorg. Chim. Acta. 1989;159:231. [Google Scholar]
- [65].Lukas B, Silver J. Inorg. Chim. Acta. 1986;124:97. [Google Scholar]
- [66].Lukas B, Peterson J, Silver J, Wilson MT. Inorg. Chim. Acta. 1983;80:245. [Google Scholar]
- [67].Debrunner P. Chapter 2. In: Lever ABP, Gray HB, editors. Iron Porphyrins Part 3. VCH Publishers Inc.; New York: 1983. [Google Scholar]
- [68].We fit the temperature dependence of the quadrupole splitting using a simple version of the crystal field model due to Ingalls.[69] We assume the dominant contribution to the electric field gradient (EFG) is due to the unpaired dxz electron. The principal-axis components of the EFG for such an electron will be proportional to (–2/7, 4/7, –2/7).[67] The low-lying excited state for the system will be a dyz orbital, with EFG components proportional to (4/7, –2/7, –2/7). As the temperature of the sample is raised, the Boltzmann distribution results in the excited state being populated, and the average EFG at the nucleus is the Boltzmann-weighted sum of the EFGs due to the dxz and dyz orbitals. So we do a two-parameter least-squares fit to the quadrupole data, with the fit parameters being the energy of the excited state, and the constant of proportionality that relates the EFG to the quadrupole tensor. It is interesting that this model predicts that there will be a temperature-dependent rotation of the principal axes of the thermally- averaged EFG tensor, and that the asymmetry parameter η will vary with temperature as well. This analysis ignores the effects of spin orbit coupling, a point to which we subsequently return.
- [69].Ingalls R. Phys. Rev. 1964;133:A787. [Google Scholar]
- [70].Inniss.[16] have calculated this separation as 0.14 times the spin-orbit coupling constant.
- [71].The values were calculated by diagonalization of the crystal field Hamiltonian including the effects of the spin-orbit coupling term. A value of 250 cm−1 was used for λ and a value of 2000 cm−1 was used for V , the energy difference between the dxy orbital and the average energy of the dπ orbitals.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.