


Abstract
Double-stranded RNA (dsRNA) is formed in cells as intra- and intermolecular RNA interactions and is involved in a range of biological processes including RNA metabolism, RNA interference and translation control mediated by natural antisense RNA and microRNA. Despite this breadth of activities, few molecular tools are available to analyse dsRNA as native hybrids. We describe a two-step ligation method for enzymatic joining of dsRNA adaptors to any dsRNA molecule in its duplex form without a need for prior sequence or termini information. The method is specific for dsRNA and can ligate various adaptors to label, map or amplify dsRNA sequences. When combined with reverse transcription–polymerase chain reaction, the method is sensitive and can detect low nanomolar concentrations of dsRNA in total RNA. As examples, we mapped dsRNA/single-stranded RNA junctions within Escherichia coli hok mRNA and the human immunodeficiency virus TAR element using RNA from bacteria and mammalian cells.
INTRODUCTION
Double-stranded RNA (dsRNA) is widespread in living organisms and plays structural and functional roles in various biological processes and pathways (1). RNA higher order structures consist of dsRNA formed by cis and trans hybridization. Many viruses have dsRNA genomes or use dsRNA as intermediates during their life cycle. In prokaryotes, naturally occurring antisense RNAs form dsRNA complexes with mRNAs that regulate translation. In eukaryotes, dsRNA is involved in numerous processes, such as heterochromatin remodelling (2), RNA editing, interferon responses and the RNA interference/microRNA pathways (3,4). Indeed ∼5% of mammalian heterogeneous nuclear RNA (hnRNA) appears to be double stranded (1).
Although there is increasing information about the biogenesis and function of dsRNA in cells, methodologies to analyse and handle RNA in its natural duplex form remain limited. Unlike DNA and single-stranded RNA (ssRNA), dsRNA is not easily manipulated or analysed with current molecular tools. While various adaptors can be attached to DNA using restriction enzymes and ligases, there are no known dsRNA-specific enzymes with corresponding activities. Therefore, we aimed to establish a method for ligation of dsRNA adaptors to dsRNA substrates in a way that does not require prior sequence or termini information and can be applied to cellular RNA.
MATERIALS AND METHODS
Oligoribonucleotides, primers and enzymes
All oligoribonucleotides (Table 1) and primers (Table 2) used in this study were purchased from Biomers (Ulm, Germany). All enzymes were purchased from Fermentas (Ontario, Canada) unless specified otherwise.
Table 1.
Oligoribonucleotide sequences and modifications
| Oligoribonucleotide | Sequence (5′→ 3′) |
|---|---|
| dsRNA substrate | ugagguaguagguugugugguu |
| 6-FAM-aaccacacaaccuacuaccuca | |
| ssRNA substrate | ugagguaguagguugugugguu |
| Adenine hexamer | aaaaaa |
| dsRNA-adaptor | NH2-auucuagaggauccagcagcaaguuuc |
| uuuuuugaagcuugcugcuggauccucuagaau-NH2 | |
| Overhanging dsRNA | nnnugagguaguagguugugugguunnn |
| 6-FAM-aaccacacaaccuacuaccuca | |
| TxRed-adaptor | NH2-auucuagaggauccagcagcaaguuuc |
| uuuuuugaagcuugcu- Texas Red | |
| Y-adaptor | gccaccucgagucacaccguaaguuuc |
| uuuuuugaagcuugcugcuggauccucuagaau-NH2 |
‘nnn’ at the ends of ‘Overhanging dsRNA’ indicates three random nucleotides. ‘NH2’ is an amino group attached to the 3′ positions of oligonucleotides through a linker.
Table 2.
Primer sequences for RT-PCR reactions
| Primer | Sequence (5′→ 3′) |
|---|---|
| Adaptor-specific RT primer | attctagaggatcca |
| Adaptor-specific PCR primers: | |
| Y-adaptor-a | gccacctcgagtcacacc gta |
| Y-adaptor-b | attctagaggatccagcagcaag |
| Gene-specific PCR primers: | |
| O-dsRNA-p | ttggatcctgaggtagtaggttgtgtgg |
| Hok-p | ccaccacgaggcatccctatgtcta |
| TAR-p | gggtctctctggttaga |
dsRNA extraction
Escherichia coli K12 strain CSH50 containing plasmids pPR95, expressing hok and Sok RNAs and pOU82, the backbone plasmid (both received kindly from Kenn Gerdes, Newcastle University, Newcastle upon Tyne, UK) were harvested from 1.5 ml overnight cultures using centrifugation and resuspended in 100 µl of digestion buffer at final concentrations of 40 mM sodium acetate (pH 4.5 at 25°C), 300 mM NaCl, 2 mM ZnSO4 and 1 U/µl S1 nuclease. To disrupt cells, 700 mg of 0.1 mm Zirconia/Silica beads (Biospec Products Inc., Bartlesville, OK, USA) was added to the cell suspension and the mixture was vigorously vortexed in a 1.5-ml microtube for 10 min at room temperature. dsRNA was extracted from cell lysate/beads mixture using Trizol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol.
Human embryonic kidney 293 cells were transfected in 35-mm dishes using the calcium phosphate method with the plasmid pSVIII-HXB2, expressing an mRNA carrying the human immunodeficiency virus (HIV) TAR RNA (received kindly from J. Sodroski, Harvard Medical School, Boston, MA, USA) together with pcDNA3.1-Tat (5) or mock transfected and plated for 24 h. Cells were then resuspended in 300 µl of digestion buffer at final concentrations of 40 mM sodium acetate (pH 4.5 at 25°C), 300 mM NaCl, 2 mM ZnSO4, 1 U/µl S1 nuclease and 0.1% (v/v) Triton X-100 and incubated at room temperature for 30 min. Cell lysate was centrifuged to separate nuclear and cytoplasmic fractions. Nuclear fraction was frozen and thawed three times. Finally, dsRNA was extracted from both fractions using Trizol (Invitrogen).
Human lung adenocarcinoma A549 cells were incubated in normoxia and hypoxia conditions (0.5% O2 for 6 h) and dsRNA was extracted as described above.
S1 nuclease digestion (trimming)
To generate blunt ends, 40 pmol of overhanging dsRNA or 2.5 µg cellular RNA was incubated in 50 µl of the S1 digestion reaction at final concentrations of 40 mM sodium acetate (pH 4.5 at 25°C), 300 mM NaCl, 2 mM ZnSO4 and 1 U/µl S1 nuclease at 30°C for 1 h. Trimmed dsRNA was phenol/chloroform extracted, precipitated with NaOAc/Ethanol/GlycoBlue (Ambion Inc., Foster City, CA, USA) and dissolved in 10 µl H2O.
T4 RNA ligation (generating sticky-ended dsRNA in Step I)
To attach short ssRNA to blunt-ended dsRNA for generating sticky-ends, 20 pmol of synthetic dsRNA substrate or 5 µl of trimmed dsRNA was incubated along with 10 µM adenine hexamer in 50 µl of the ligation reaction at final concentrations of 50 mM HEPES-NaOH (pH 8.0 at 25°C), 10 mM MgCl2, 10 mM DTT, 1 mM ATP, 5% polyethylene glycol 6000 (w/v) and 2.5 U/µl T4 RNA ligase at 16°C for 15 h. Sticky-ended dsRNA was chloroform extracted, precipitated with NaOAc/Ethanol and dissolved in 10 µl H2O.
Phosphorylation and T4 DNA ligation (adaptor joining in Step II)
To ligate dsRNA molecules to any adaptors, 5 µl of sticky-ended RNA (from Step I) and 5 µM of adaptor were incubated in 10 µl of the coupled phosphorylation and ligation reaction at final concentrations of 50 mM Tris–HCl (pH 7.6 at 25°C), 10 mM MgCl2, 5 mM DTT, 0.1 mM spermidine and 0.1 mM EDTA, 1 mM ATP, 5% polyethylene glycol 4000 (w/v), 0.5 U/µl T4 polynucleotide kinase and 1.5 U/µl T4 DNA ligase at 30°C for 15 h. The reaction was heat inactivated at 70°C for 10 min.
Reverse transcription and PCR
Y-adaptor-ligated dsRNA was converted to cDNA using ThermoScript Reverse Transcriptase (Invitrogen) and adaptor-specific RT primer. Directly 2 µl of ligation reaction (Step II) was transferred to a 20 µl of RT reaction and incubated at 60°C for 1 h according to the manufacturer's protocol. PCR was carried out with adaptor-specific or gene-specific PCR primers and products were run in 4% agarose gels.
Polyacrylamide gel electrophoresis
Samples were applied to polyacrylamide/TBE gels (20%) for native polyacrylamide gel electrophoresis (PAGE) and (12%) containing 7 M urea (run in 60°C water bath) for denaturing PAGE. All gels were scanned for FAM or Texas Red (TxRed) dyes using Typhoon 9400 scanner and the images were analysed by ImageQuant, version 5 (Amersham Biosciences, Piscataway, NJ, USA).
RESULTS
Ligation of dsRNA adaptors to dsRNA substrate
We first tried to ligate a blunt-ended dsRNA adaptor directly to a blunt-ended dsRNA substrate. Despite repeated attempts, it was not possible to ligate adaptors to blunt-ended dsRNA using T4 DNA ligase (data not shown). However, ligation of in vitro transcribed dsRNA with defined overlapping overhangs or ‘sticky termini’ was reported using the same ligase (6). Therefore, we considered that it should be possible to ligate blunt-ended dsRNA molecules if the ends could be first converted to sticky termini. Moreover, previous reports show that T4 RNA ligase can add ssDNA oligomers to dsRNA (7,8), and also attach a short ribohomopolymers to the 5′-phosphate group of blunt-ended DNA (9). Therefore, it seemed likely that this enzyme could also catalyse ligation of short ssRNA sequences to blunt-ended dsRNA to create sticky termini. Given the above information, we designed and optimized a two-step ligation method. In the first step, sticky ends are generated from blunt-ended dsRNA. In the second step, ligation between two dsRNA molecules is completed. A schematic outline of the two-step ligation method is provided in Figure 1A.
Figure 1.

Two-step ligation of a dsRNA adaptor to blunt-ended dsRNA. (A) Illustration of the ligation method. The dsRNA substrate (red) was FAM-conjugated for easy detection in gels. In Step I, T4 RNA ligase attaches an adenine hexamer oligomer (green) to the dsRNA substrate producing an overhanging 5′ terminus (sticky terminus). In Step II, the sticky-ended dsRNA is phosphorylated by T4 polynucleotide kinase and joined to a dsRNA adaptor, which contains a complementary overhang (blue), using T4 DNA ligase. (B) Native and (C) denaturing PAGE analyses of the first and the second steps. Each band is indicated with a corresponding illustration.
To generate sticky termini from blunt-ended dsRNA in the first step, an adenine hexamer oligoribonucleotide (9) was ligated to the 5′-phosphate group of dsRNA substrate using T4 RNA ligase. The adenine hexamer was not phosphorylated to avoid concatenation. One strand of dsRNA substrate was conjugated to FAM fluorophore for detection after electrophoresis (for oligoribonucleotides information, see Table 1). PAGE analysis showed that the adenine hexamer was joined to dsRNA with high efficiency (Figure 1B). The addition of RNA oligomers to dsRNA provides a new biochemical reaction for molecular biology and in the present application provides the sticky termini needed for ligation.
In the second step, the dsRNA substrate with the added adenine hexamer overhang was incubated along with a dsRNA adaptor having an extension of six uridines at one end (dsRNA-adaptor). To simplify the analysis, the dsRNA-adaptor was blocked at the other end by adding amino groups to the 3′ positions through a six-carbon linker (www.biomers.net). These molecules were then phosphorylated using T4 polynucleotide kinase and ligated using a high concentration of T4 DNA ligase. PAGE analysis and FAM fluorophore quantification showed that adaptor ligation was moderately efficient (∼50%) (Figure 1B). To ensure that the gel shift was not due to hybridization of short complementary stretches, the reactions were rerun in a denaturing urea PAGE. Migration of the ligated product was retarded as expected, suggesting that the two molecules were joined covalently (Figure 1C). Therefore, the two-step ligation method can attach a dsRNA adaptor to a blunt-ended dsRNA substrate, without a need for prior sequence information.
Two-step ligation is specific for dsRNA
To examine the specificity of ligation to dsRNA using the two-step ligation method, an unmodified blunt-ended dsRNA and an unmodified ssRNA were used as substrates. A dsRNA adaptor conjugated to TxRed (TxRed-adaptor) (Table 1) was ligated to the substrates. The results show that the TxRed-adaptor was only attached to dsRNA but not to ssRNA (Figure 2). Therefore, the method enables specific tagging or labelling of blunt-ended dsRNA.
Figure 2.

Adaptor ligation is specific for dsRNA. A TxRed-adaptor was ligated to unmodified dsRNA and ssRNA substrates. Ligated products were fractionated using denaturing PAGE.
Amplification of dsRNA using ligated adaptors
The two-step ligation method can be combined with reverse transcription–polymerase chain reaction (RT–PCR) to amplify dsRNA sequences using the primer sites provided by ligated adaptors. However, as the same adaptors are ligated to both ends of dsRNA, their primer sites are identical. Therefore, we designed a ‘Y’ shaped adaptor (Y-adaptor) that can provide two different primer sites when attached to both ends of dsRNA (Figure 3A and Table 1).
Figure 3.

RT–PCR amplification of dsRNA. (A) Primer sites are illustrated when Y-adaptors are attached to a dsRNA. (B) RT–PCR amplification of dsRNA as well as ssRNA substrates that were ligated to Y-adaptors (expected PCR product length is 88 bp).
To test this strategy, Y-adaptors were ligated to an unmodified blunt-ended dsRNA. Next, dsRNA was reverse-transcribed to a full-length cDNA using an adaptor-specific RT primer (for primer sequences, see Table 2). We used thermo-stable reverse transcriptase to overcome secondary structure hindrance of duplex RNA. Subsequently, cDNA was amplified using adaptor-specific primers (Y-adaptor-a and -b primers). The results indicate that dsRNA ligated to adaptors can be amplified by PCR and confirmed that the dsRNA substrate and adaptors were covalently linked (Figure 3B). Therefore, the full-length dsRNA can be amplified regardless of its sequence information using primers that are specific for two arms of the Y-adaptor. Again to verify the specificity of the ligation method for dsRNA, an ssRNA substrate was included in the amplification experiment, and this reaction did not result in a product (Figure 3B). Therefore, the ligation method combined with RT–PCR amplifies only dsRNA and not ssRNA sequences.
Trimming of dsRNA to make blunt ends
In the above ligation reactions, a blunt-ended dsRNA was used as substrate; a molecule that is rarely found in total RNA derived from cells. However, certain single-strand-specific endonucleases, such as S1 nuclease, are known to remove single-stranded overhangs from DNA fragments leaving ligatable blunt ends (10–12). Therefore, we tested whether S1 nuclease is capable of reliably trimming dsRNA ends in the same way.
We designed a dsRNA substrate with heterogeneous overhangs (overhanging dsRNA) and performed trimming reactions with S1 nuclease (Figure 4A). PAGE analysis showed that dsRNA was trimmed over a range of reaction periods as well as enzyme and substrate concentrations (Figure 4B and Supplementary Figure 1). Also, two-step ligation of dsRNA adaptors followed by PAGE fractionation showed that the S1-treated dsRNA was blunt ended and ligatable (Figure 4C). Ligation of the trimmed dsRNA was less efficient relative to the defined blunt-ended molecules, possibly due to incomplete trimming activity, as reported for DNA trimming (13). However, the overall efficiency appears to be sufficient for practical applications. Therefore, the new ligation method together with S1 nuclease treatment is applicable to dsRNA with unpaired termini as typically found in cells.
Figure 4.
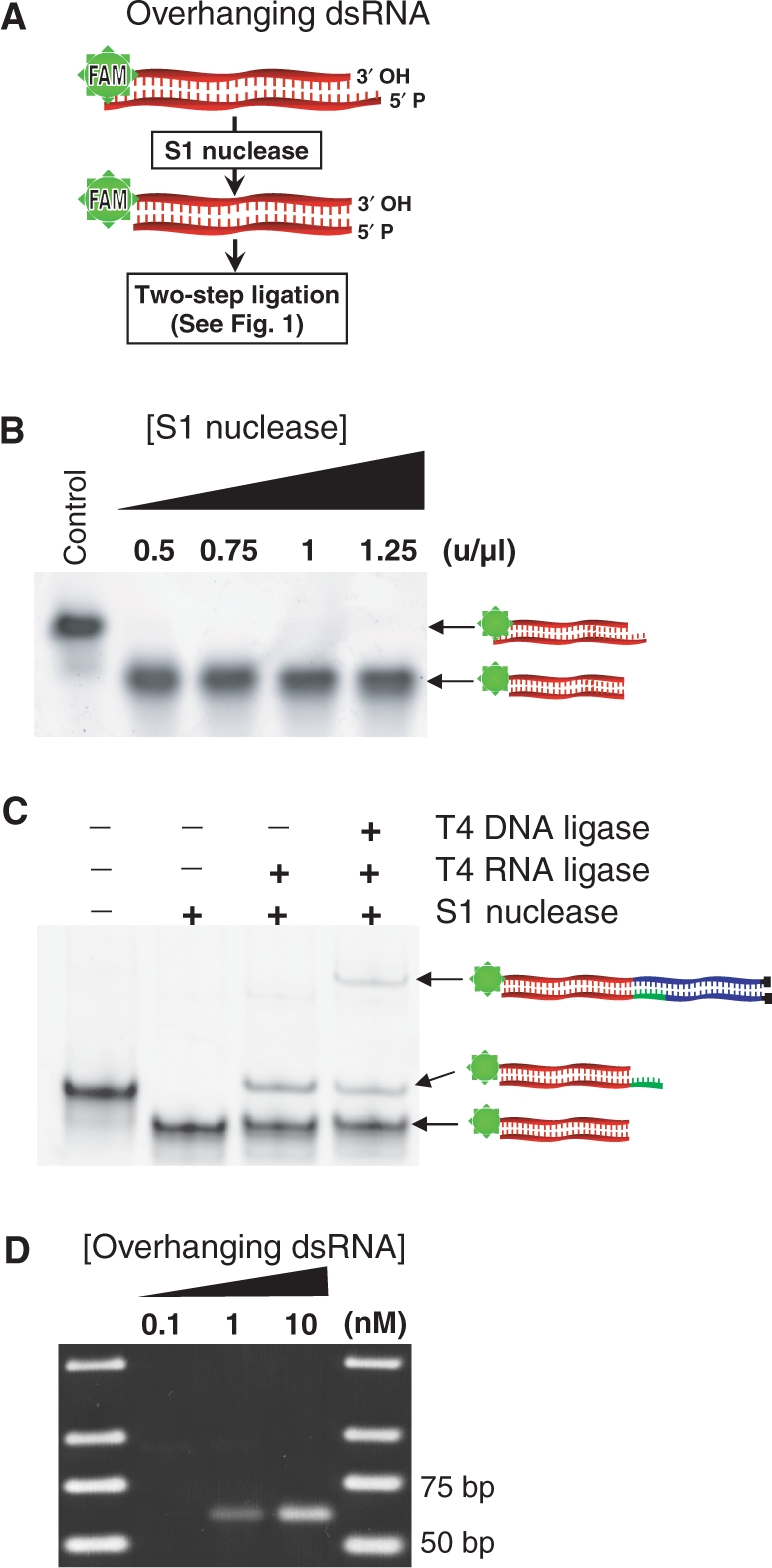
Trimming of overhanging dsRNA and adapter ligation. (A) Illustration of heterogeneous overhanging termini and their removal from dsRNA using S1 nuclease. (B) Native PAGE analysis showed that S1 nuclease cleaves overhangs but not the duplex in a range of enzyme concentrations. (C) Native PAGE analysis indicated that blunt ends generated by S1 nuclease were ligatable to dsRNA adaptors. It is notable that in the third lane from left, where only T4 DNA ligase was omitted, a faint band is observed. We cannot identify this band, which was not observed when using agarose gels. (D) RT–PCR amplification of the overhanging dsRNA that was spiked into E. coli total RNA (0.05 µg/µl) and ligated to Y-adaptors (expected PCR product length is 63 bp).
To evaluate whether the present method could be applied to cellular RNA, we aimed to detect small amounts of dsRNA in a complex pool of RNA. Low nanomolar concentrations of the unmodified overhanging dsRNA were spiked into E. coli total RNA (final concentration of 0.05 µg/µl) and subjected to S1 nuclease digestion, Y-adaptor ligation and RT–PCR using an overhanging dsRNA-specific primer (O-dsRNA-p) and an adaptor-specific primer (Y-adaptor-b) (Table 2). PCR produced an amplicon corresponding to the target dsRNA sequence, indicated by migration in an agarose gel (Figure 4D). Therefore, the sensitivity of the reaction is sufficient to detect the presence of specific dsRNA sequences at physiological concentrations in total cellular RNA.
Mapping dsRNA regions within cellular RNA
Given the specificity and sensitivity demonstrated above, we considered that it should be possible to use the two-step ligation method to map dsRNA regions in total cellular RNA preparations. Because dsRNA may arise artificially during RNA preparation (as the consequence of hybridization in concentration steps or phenol extraction), cells were disrupted using non-denaturing conditions and in the presence of S1 nuclease to remove ssRNA from native ribonucleoprotein complexes. After RNA purification, a second S1 nuclease digestion was performed to trim dsRNA, and Y-adaptors were ligated using the two-step ligation method. RT–PCR amplification using gene-specific and adaptor-specific (Y-adaptor-b) (Table 2) primers produced amplicons that correspond in size to the predicted dsRNA regions.
First, we attempted to map dsRNA regions formed by trans hybridization of two RNA molecules. Several reports show that aHIF RNA, an antisense complementary to the 3′UTR of HIF-1α mRNA is overexpressed and antagonizes HIF-1α expression in response to prolonged hypoxia (14) and in non-papillary renal cancers (15). It is hypothesized that the antisense destabilizes HIF-1α mRNA by exposing AU-rich elements located in sense 3′ UTR (14). Also, it is possible that antisense interferes with HIF-1α pre-mRNA splicing (16). However, when we treated cells with hypoxic conditions and subsequently performed the RNA ligation method we did not detect any dsRNA formed in the complementary region (data not shown). This negative evidence is not surprising as aHIF antisense RNA may function through mechanisms other than duplex formation in cells (17).
Next, we attempted to map RNAs that contain well-studied duplex regions. In bacteria, we examined the hok mRNA of E. coli R1 plasmid (Figure 5A) and in mammalian cells we examined the HIV TAR RNA element, an RNA structure involved in transcription (18) (Figure 5B). These RNAs fold to form stable cis RNA duplexes. It is notable that S1 nuclease can remove loops and bulges from secondary structures by endo-nuclease activity leaving blunt-ended dsRNA fragments (10). Sequence analysis of the amplicons confirmed ligation to ssRNA/dsRNA junctions within both hok mRNA and TAR RNA. For both RNAs, a site that is part of the loop of an extended hairpin was tagged at a point adjacent to the stem. Also, in the case of TAR RNA, a site that is part of a three-base bulge in the stem was tagged by ligation and mapped by sequencing. In addition, TAR dsRNA regions were detected only in the nuclear fraction. Therefore, the new method can be used to map dsRNA structures in cellular RNA preparations from both bacterial and mammalian cells.
Figure 5.

Mapping of dsRNA regions in cellular RNA. (A) Experimental flowchart of extraction and detection of natural dsRNA fragments from cells. Fragments of dsRNA were ligated to Y-adaptors and subjected to RT–PCR amplification. (B) Detection of a dsRNA/ssRNA junction at the loop within folded hok mRNA (PCR product is 87 bp). (C) Detection of dsRNA/ssRNA junctions at the internal bulge and the loop within the TAR RNA element (PCR products are 55 and 62 bp, respectively) in RNA from nuclear (Nuc) and cytoplasmic (Cyt) fractions. Structures of hok and TAR RNAs, the amplified dsRNA region and adaptor ligation points are indicated.
DISCUSSION
Ligation of dsRNA adaptors to dsRNA molecules independent of template sequence information provides a new reaction in molecular biology that enables further manipulation and analyses of dsRNA in its native duplex form. Here we developed a two-step ligation method that is specific for dsRNA. Indeed, the new method provides the only available dsRNA-specific labelling/tagging reaction. Previous labelling methods using T4 RNA ligase are not specific to dsRNA, where ssRNA is also tagged with even higher efficiency. Also, we demonstrate that when the ligation method is combined with S1 nuclease digestion and RT–PCR, it is possible to amplify and map dsRNA regions in structured cellular RNA.
The two-step ligation method provides a new experimental strategy to examine cellular dsRNAs. As examples, the results provide termini mapping data for the presence and structure of dsRNA within hok mRNA and HIV TAR element. On the other hand, the method can provide negative evidence for predicted RNA::RNA interactions in cases where the dsRNA may be unstable or absent in cells. There are many speculative cases of dsRNA structures in cells, including putative roles for non-coding RNAs in RNA::RNA interactions, and molecular methods are needed to probe potential roles in cells. In principle, the method reported here provides a means to evaluate the presence or abundance of any putative dsRNA.
In the future, dsRNA ligation could be used for global dsRNA amplification and library construction using cellular dsRNA. Testing putative interactions between natural antisense RNAs and their targets, which have been widely predicted in silico, is just one example of how dsRNA ligation can be used to test or discover dsRNA interactions. Furthermore, dsRNA ligation should be helpful in dsRNA tagging and pull-down experiments to investigate native dsRNA interactions with proteins, small molecules and other nucleic acids. Therefore, we anticipate that the two-step ligation of dsRNA will provide a simple and useful new tool in dsRNA studies.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
O.R.F. is supported by a scholarship from the Ministry of Science, Research and Technology I.R. Iran. This work was supported by a grant from the Swedish Research Council. Funding to pay the Open Access publication charges for this article was provided by the Swedish Research Council.
Conflict of interest statement. None declared.
REFERENCES
- 1.Nicholson AW. Structure, reactivity, and biology of double-stranded RNA. Prog. Nucleic Acid. Res. Mol. Biol. 1996;52:1–65. doi: 10.1016/s0079-6603(08)60963-0. [DOI] [PubMed] [Google Scholar]
- 2.Gullerova M, Proudfoot NJ. Cohesin complex promotes transcriptional termination between convergent genes in S. pombe. Cell. 2008;132:983–995. doi: 10.1016/j.cell.2008.02.040. [DOI] [PubMed] [Google Scholar]
- 3.Tariq M, Paszkowski J. DNA and histone methylation in plants. Trends Genet. 2004;20:244–251. doi: 10.1016/j.tig.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 4.Kumar M, Carmichael GG. Antisense RNA: function and fate of duplex RNA in cells of higher eukaryotes. Microbiol. Mol. Biol. Rev. 1998;62:1415–1434. doi: 10.1128/mmbr.62.4.1415-1434.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ott M, Emiliani S, Van Lint C, Herbein G, Lovett J, Chirmule N, McCloskey T, Pahwa S, Verdin E. Immune hyperactivation of HIV-1-infected T cells mediated by Tat and the CD28 pathway. Science. 1997;275:1481–1485. doi: 10.1126/science.275.5305.1481. [DOI] [PubMed] [Google Scholar]
- 6.Dekker NH, Abels JA, Veenhuizen PT, Bruinink MM, Dekker C. Joining of long double-stranded RNA molecules through controlled overhangs. Nucleic Acids Res. 2004;32:e140. doi: 10.1093/nar/gnh138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Imai M, Richardson MA, Ikegami N, Shatkin AJ, Furuichi Y. Molecular cloning of double-stranded RNA virus genomes. Proc. Natl Acad. Sci. USA. 1983;80:373–377. doi: 10.1073/pnas.80.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lambden PR, Cooke SJ, Caul EO, Clarke IN. Cloning of noncultivatable human rotavirus by single primer amplification. J. Virol. 1992;66:1817–1822. doi: 10.1128/jvi.66.3.1817-1822.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Higgins NP, Geballe AP, Cozzarelli NR. Addition of oligonucleotides to the 5'-terminus of DNA by T4 RNA ligase. Nucleic Acids Res. 1979;6:1013–1024. doi: 10.1093/nar/6.3.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lehman RI. Endonucleases specific for single-stranded polynucleotides. In: Boyer PD, editor. The Enzymes, Vol. 4. 3rd. New York: Academic Press Inc.; 1981. pp. 193–201. [Google Scholar]
- 11.Sakonju S, Bogenhagen DF, Brown DD. A control region in the center of the 5S RNA gene directs specific initiation of transcription: I. The 5' border of the region. Cell. 1980;19:13–25. doi: 10.1016/0092-8674(80)90384-0. [DOI] [PubMed] [Google Scholar]
- 12.Vogt VM. Purification and further properties of single-strand-specific nuclease from Aspergillus oryzae. Eur. J. Biochem. 1973;33:192–200. doi: 10.1111/j.1432-1033.1973.tb02669.x. [DOI] [PubMed] [Google Scholar]
- 13.Shishido K, Ando T. Efficiency of T4 DNA ligase-catalyzed end joining after S1 endonuclease treatment on duplex DNA containing single-stranded portions. Biochim. Biophys. Acta. 1981;656:123–127. doi: 10.1016/0005-2787(81)90035-6. [DOI] [PubMed] [Google Scholar]
- 14.Uchida T, Rossignol F, Matthay MA, Mounier R, Couette S, Clottes E, Clerici C. Prolonged hypoxia differentially regulates hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha expression in lung epithelial cells: implication of natural antisense HIF-1alpha. J. Biol. Chem. 2004;279:14871–14878. doi: 10.1074/jbc.M400461200. [DOI] [PubMed] [Google Scholar]
- 15.Thrash-Bingham CA, Tartof KD. aHIF: a natural antisense transcript overexpressed in human renal cancer and during hypoxia. J. Natl Cancer Inst. 1999;91:143–151. doi: 10.1093/jnci/91.2.143. [DOI] [PubMed] [Google Scholar]
- 16.Cayre A, Rossignol F, Clottes E, Penault-Llorca F. aHIF but not HIF-1alpha transcript is a poor prognostic marker in human breast cancer. Breast Cancer Res. 2003;5:R223–R230. doi: 10.1186/bcr652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Munroe SH, Zhu J. Overlapping transcripts, double-stranded RNA and antisense regulation: a genomic perspective. Cell. Mol. Life Sci. 2006;63:2102–2118. doi: 10.1007/s00018-006-6070-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeang KT, Xiao H, Rich EA. Multifaceted activities of the HIV-1 transactivator of transcription, Tat. J. Biol. Chem. 1999;274:28837–28840. doi: 10.1074/jbc.274.41.28837. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.