


Abstract
ChIP-Seq, which combines chromatin immunoprecipitation (ChIP) with ultra high-throughput massively parallel sequencing, is increasingly being used for mapping protein–DNA interactions in-vivo on a genome scale. Typically, short sequence reads from ChIP-Seq are mapped to a reference genome for further analysis. Although genomic regions enriched with mapped reads could be inferred as approximate binding regions, short read lengths (∼25–50 nt) pose challenges for determining the exact binding sites within these regions. Here, we present SISSRs (Site Identification from Short Sequence Reads), a novel algorithm for precise identification of binding sites from short reads generated from ChIP-Seq experiments. The sensitivity and specificity of SISSRs are demonstrated by applying it on ChIP-Seq data for three widely studied and well-characterized human transcription factors: CTCF (CCCTC-binding factor), NRSF (neuron-restrictive silencer factor) and STAT1 (signal transducer and activator of transcription protein 1). We identified 26 814, 5813 and 73 956 binding sites for CTCF, NRSF and STAT1 proteins, respectively, which is 32, 299 and 78% more than that inferred previously for the respective proteins. Motif analysis revealed that an overwhelming majority of the identified binding sites contained the previously established consensus binding sequence for the respective proteins, thus attesting for SISSRs’ accuracy. SISSRs’ sensitivity and precision facilitated further analyses of ChIP-Seq data revealing interesting insights, which we believe will serve as guidance for designing ChIP-Seq experiments to map in vivo protein–DNA interactions. We also show that tag densities at the binding sites are a good indicator of protein–DNA binding affinity, which could be used to distinguish and characterize strong and weak binding sites. Using tag density as an indicator of DNA-binding affinity, we have identified core residues within the NRSF and CTCF binding sites that are critical for a stronger DNA binding.
INTRODUCTION
Chromatin immunoprecipitation (ChIP) is a powerful and widely used experimental technique to determine whether proteins including, but not limited to, transcription factors bind to specific regions on chromatin in vivo. ChIP requires cross-linking of living cells using formaldehyde, followed by shearing of chromatin into short fragments of desired length (usually 0.2–1 kb) using sonication. The protein-bound DNA fragments are then immunoprecipitated using an antibody specific to the protein of interest. Finally, the protein–DNA cross-links are reversed, and the DNA is purified and assayed to determine the sequence bound by that protein. Until recently, ChIP-chip (1,2), which combines ChIP with DNA microarrays, was the most widely used technique to map protein binding sites on DNA on a genome scale. ChIP-Seq (3–6), which combines ChIP with next generation massively parallel sequencing technology, is on its way to replacing ChIP-chip as the commonly used approach for genome-wide identification of protein–DNA interactions in vivo. ChIP-Seq's coverage, high resolution and cost-effectiveness, combined with its ability to sequence several million bases in a short span of time (1–2 days) allow us to map and understand protein–DNA interactions on a genome-scale.
In ChIP-Seq, the DNA fragments obtained from ChIP are directly sequenced using the next generation genome sequencers such as Illumina Genome Analyzers. Although the lengths of the input DNA could be anywhere between ∼200 bp and ∼1 kb, typically, only the first ∼25–50 nt from the DNA ends are sequenced. The resulting short reads are mapped back to a reference genome, and only those reads that map to an unique genomic locus in the reference genome are considered for further analysis. Mapped reads are commonly referred to as tags (henceforth, ‘reads’ and ‘tags’ are used interchangeably). Typically, genomic regions with high tag densities are interpreted as binding site locations (3–5). Although this approach helps identify binding ‘regions’ accurately, short read length poses challenges for determining the exact binding sites within these regions. Given that the lengths of the sequenced DNA fragments could be few hundred base pairs, such a heuristic, which uses the general framework of clustering of reads to identify binding site locations, does not take full advantage of the inherent properties of the ChIP-Seq data. Consequently, the resolution of the identified binding sites could be as much as the length of the input DNA, if not longer. However, the binding sites for transcription factors are often clustered in critical regulatory regions, and are in close proximity to each other. To understand the structure of regulatory elements and to delineate the contribution of each binding site/factor, accurate, sensitive and precise approaches for target site identification are needed. Moreover, the method needs to be robust yet flexible enough so that it allows the user to control for elements such as antibody specificity and sequencing errors, which could affect the data quality, and thus the accuracy and resolution of identified binding sites.
Here, we present SISSRs (Site Identification from Short Sequence Reads), a novel algorithm for genome-wide identification of binding sites from short reads generated from ChIP-Seq experiments. SISSRs exploits the direction of reads to first estimate the average length of DNA fragments, and then uses the fragment length, direction of reads, a background model and other user-set control parameters to narrow down the binding site resolution to within few tens of base pairs. The sensitivity and specificity of SISSRs are demonstrated by applying it on ChIP-Seq data for three widely studied and well-characterized human transcription factors: insulator protein CTCF (CCCTC-binding factor) (7–11), NRSF (neuron-restrictive silencer factor) (also known as REST, for repressor element-1 silencing transcription factor) (12–15) and transcription activator protein STAT1 (signal transducer and activator of transcription protein 1) (16–19). Using SISSRs, we identified a total of 26 814, 5813 and 73 956 binding sites for CTCF, NRSF and STAT1, respectively, which is 32, 299 and 78% more than that inferred previously for the respective proteins (3–5). Motif analysis revealed that SISSRs-inferred binding sites contained the previously established consensus binding sequence for the respective proteins, thus authenticating SISSRs accuracy.
The coverage and precision of SISSRs facilitated analyses of ChIP-Seq data revealing interesting insights, which we believe will serve as guidance for designing ChIP-Seq experiments to map in vivo protein–DNA interactions. We also show that the tag densities at the binding sites are a good indicator of protein–DNA binding affinity, which could be used to distinguish and characterize strong and weak binding sites. Using tag density as an indicator of DNA-binding affinity, we identified core residues within the NRSF and CTCF binding sites that are critical for a stable NRSF binding.
METHODS
Datasets
ChIP-Seq data for human transcription factors CTCF in CD4+ T cell (3), NRSF in Jurkat T lymphoblast cell (4) and STAT1 in interferon γ-stimulated (IFN-γ) HeLa S3 cell (5) were used in this study. The dataset and an implementation of the SISSRs algorithm are freely available at http://dir.nhlbi.nih.gov/papers/lmi/epigenomes/sissrs/.
DNA fragment length estimation
By default, SISSRs estimates the average DNA fragment length from the ChIP-Seq reads. For every tag i in the sense strand, the nearest tag k in the antisense strand, downstream of i, is identified. Let j be the tag in the sense strand immediately upstream of k. Note that i and j could be the same tag. The mean DNA fragment length F is given by 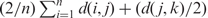 , where n is the number of sense tags for which there exists a k and a j tag, and d(i,k)⩽500 (a pictorial illustration of this approach is presented as Supplementary Figure 1). Here, d(x, y) denotes the distance (in base pairs) between tags x and y. It is assumed that sonicated DNA fragments are of length at most 500 bp. This is a reasonable assumption given that it is expected that ChIP-Seq will be used for high-resolution mapping of binding sites. SISSRs provides an option to change this setting, albeit at the cost of decreased resolution. Alternatively, if the average fragment length is known, SISSRs allows users to set the average fragment length to this value.
, where n is the number of sense tags for which there exists a k and a j tag, and d(i,k)⩽500 (a pictorial illustration of this approach is presented as Supplementary Figure 1). Here, d(x, y) denotes the distance (in base pairs) between tags x and y. It is assumed that sonicated DNA fragments are of length at most 500 bp. This is a reasonable assumption given that it is expected that ChIP-Seq will be used for high-resolution mapping of binding sites. SISSRs provides an option to change this setting, albeit at the cost of decreased resolution. Alternatively, if the average fragment length is known, SISSRs allows users to set the average fragment length to this value.
SISSRs algorithm
SISSRs uses the direction and density of reads and the average DNA fragment length to identify binding sites. The entire length of the genome spanned by mapped sequence reads/tags is scanned using a window of size w (default: 20 bp) with consecutive windows overlapping by w/2. For each window i, the net tag count ci is computed by subtracting the number of antisense tags mapped to window i from the number of sense tags mapped to the same window. As the reads are scanned by the moving window, every time the net tag count transitions from positive to negative, the transition point coordinate t is defined as the midpoint between the last seen window with positive net tag count and the current window, which has a negative net tag count. Each one of these transition points is a candidate binding site, which needs to satisfy the following conditions in order to be confirmed as a true binding site: (i) number of sense tags p in the region defined by coordinates [t − F, t] is at least E, (ii) number of antisense tags n in the region defined by coordinates [t, t+F] is at least E, and (iii) p+n is at least R, which is estimated based on the user-set false discovery rate (FDR) D. Each confirmed binding site is assigned a score, also referred to as binding site tag density, equal to p+n, which is the number of directed reads supporting the binding site. By default, parameter E is set to 2, which could be set to a desired value by the user.
Adopting a strategy similar to that used by Robertson et al. (5), for each integer value V ≥ 2E, SISSRs estimates the FDR as the ratio of the number of 2F-bp regions with V or more tags that the background model indicates should occur by chance, to the number observed in the real data. For each dataset, the number of tags R necessary to characterize a binding site is defined as the smallest V corresponding to FDR < D for binding sites defined using V. For the estimated DNA fragment length F, the expected number of tags (λ) within a window of size 2F is given by F times of the number of tags in the dataset divided by the mappable genome length M, which was estimated to be about 80% of the genome length. The probability of observing a binding site supported by at least R tags by chance is given by a sum of Poisson probabilities as
Since M could be different for different experiments depending on the reference genome being used, SISSRs gives users the option to set their own value for M. If a negative control dataset (such as IgG) is available, SISSRs provides users the option to use this dataset as background in place of the random model.
If a high sensitivity is desired, SISSRs provides an option to identify those binding sites that have tags mapped to only the sense or antisense strand (site A in Figure 6B). To identify such sites, SISSRs employs a two-pass approach (a pass each for sense and antisense tags) in which the sense and antisense tags are scanned from left-to-right and right-to-left, respectively. For instance, in the left-to-right pass, for each sense tag i SISSRs checks if (i) the distance between i and the next sense tag j is at least F, and (ii) the distance between i and the next antisense tag k is at least 2F. If tag i satisfies both the conditions, then the genomic coordinate F/2 bases upstream of i is reported as a binding site if the total number of sense tags mapped to the F-bp region upstream of i (including i) is at least R. The same strategy is applied on the right-to-left pass with the antisense tags. Since binding sites are identified using reads mapped to only one strand, it is impossible to identify the exact binding site location. Genomic coordinate F/2 bases upstream of i is a reasonable approximation of the exact binding site location, given the data.
Figure 6.

Overcoming ChIP-Seq limitations. (A) Nonspecific reads due to antibody non-specificity and/or sequencing errors could be controlled for by adjusting the window size w. (B) SISSRs provides an option to identify binding sites with tags mapped to only the sense or antisense strand, which otherwise may not have been identified (site A). Steps are taken to make sure that tags corresponding to site B are not accounted while identifying site A: the right most sense tag i for site A should be at least a fragment length F away from the nearest sense tag (j) and at least 2F away from the nearest antisense tag k. (C) False positives due to indirect DNA binding via protein–protein interaction is a cause for concern, and are difficult to avoid as it is very difficult to determine whether the binding to the DNA is direct or indirect.
Binding sites inferred in this study used default SISSRs parameters: estimated average DNA fragment length, E=2, and R as estimated using the random background model for D=10−3. The value of R was estimated to be 6, 6 and 12 tags for CTCF, NRSF and STAT1 datasets, respectively.
Genome-wide distribution of binding sites
Genome-wide distribution of identified binding sites was determined with reference to RefSeq genes downloaded from UCSC genome browser (20). The 5-kb region upstream of transcription start is defined as the gene promoter.
Motif analysis
MEME (21) with default parameters was used to identify statistically overrepresented consensus motifs within the inferred binding sites. Since the running time of motif finding algorithms are prohibitively long for large sets, we decided to use only the top X% of the binding sites. Due to the huge differences in the number of sites for the three proteins, there was not a good choice of X we could use as a compromise between being sufficiently large to include as many binding sites, and small enough for MEME to find motifs in reasonable time. For example, X=10% would have selected only 581 binding sites for NRSF but 7396 binding sites for STAT1 (few NRSF sites and too many STAT1 sites). Because of this dichotomy, we decided to use a different value for X for each of the three proteins. MEME analysis was performed only on high-scoring binding sites (sites with high tag density): top 10% of CTCF sites (2622) with 60 or more tags, top 20% of NRSF sites (1160) with 46 or more tags and top 5% of STAT1 sites (3825) with 60 or more tags. Position-specific scoring matrices (PSSMs) resulting from MEME analysis were used as input to MAST (22) to locate matching instances with P < 10−3.
Resolution of identified binding sites
The resolution of a binding site is assessed by calculating the distance (in base pairs) between the inferred binding site and the middle of the nearest canonical motif occurrence.
RESULTS
Overivew of SISSRs algorithm
A schematic overview of SISSRs algorithm is presented as Figure 1. First, the direction of mapped reads is used to estimate the average length F of the sequenced DNA fragments (see Methods section and Supplementary Figure 1 for details). Next, a w-bp window is used to scan the mapped reads, with consecutive windows overlapping by w/2. As the moving window scans the reads, the ‘net tag count’ for each window is computed by subtracting the number of antisense tags from the number of sense tags mapped to that window. At each instance, the net-tag-count profile makes a positive-to-negative transition, SISSRs identifies a candidate binding site. Candidate binding sites satisfying a set of estimated as well as user-set thresholds are confirmed as true binding sites (see Methods section for details). A background model similar to that used by Robertson et al. (5) is employed to make sure that each of the identified sites is not by chance. Alternatively, a negative control dataset, such as IgG, may be substituted for the default background model. Every inferred binding site is represented by a genomic coordinate t, and is assigned a score (referred to as ‘tag density’) equal to the sum of the number of sense tags mapped to the genomic region [t − F, t] and the number of antisense tags mapped to the genomic region [t, t+F].
Figure 1.

Schematic overview of SISSRs algorithm. (A) Sequenced short reads (typically ∼25–50 bp) from ChIP-Seq experiments are first mapped onto the reference genome. The mapped reads are then used to estimate statistical parameters, which include the estimation of the average length F of sequenced DNA fragments. (B) The entire reference genome along with mapped reads is scanned using overlapping windows of size w base pairs (overlapping not shown in the figure for clarity), and the net tag count (ci) for every window i is calculated. Every transition point (t) is a candidate binding site, and needs to satisfy a set of estimated as well as user-set thresholds in order to be classified as a true binding site.
ChIP-Seq datasets and binding sites
SISSRs was applied on recently published ChIP-Seq data for human transcription factor or insulator-binding protein CTCF (CCCTC-binding factor) in CD4+ T cell (3), NRSF in Jurkat T lymphoblast cell (4) and STAT1 in interferon γ-stimulated (IFN-γ) HeLa S3 cell (5). CTCF is an 11-zinc finger protein, which is known to play various functional roles—repressor, activator and insulator (7–11). NRSF is a zinc-finger transcriptional repressor, which binds to a DNA element called the neuron restrictive silencer elements (NRSE, also known as RE-1) to repress many neuronal genes in stem and progenitor cells, and in nonneuronal tissues) (12–15). STAT1 is a well-characterized transcription activator that is specifically activated to regulate gene transcription when cells encounter cytokines and growth factors. It shuttles between cytoplasm and nucleus, which is controlled by its phosphorylation states of key tyrosines (16–19). Upon phosphorylation by receptor-associated Janus Kinase (JAK) family of proteins, STAT1 proteins get retained in the nucleus forming homo- and hetero-dimers that bind to IFN-γ-activation site (GAS) elements and interferon-stimulated response elements (ISRE) in the DNA.
Using an FDR threshold of 10−3, we identified a total of 26 814, 5813 and 73 956 binding sites for CTCF, NRSF and STAT1 proteins, respectively. The number of sites identified in this study is 32, 299 and 78% more than what was previously identified from the same datasets for the respective proteins (3–5) (Table 1). The genome-wide distribution of the inferred binding sites relative to RefSeq genes is given as Figure 2.
Table 1.
Data and results summary
| CTCF | NRSF | STAT1 | |
|---|---|---|---|
| Number of mapped reads (million) | 2.9 | 1.7 | 15.1 |
| Estimated Fragment length (bp) | 127 | 133 | 198 |
| Binding sites identified by SISSRs | 26 814 | 5813 | 73 956 |
| Binding sites identified by previous approaches | 20 262 | 1946 | 41 582 |
Figure 2.

Distribution of binding sites across the genome. RefSeq genes were used as reference. The 5 kb region upstream of transcription start site was defined as the promoter.
Accuracy of identified binding sites
To verify whether the inferred binding sites are indeed true binding sites, we sought to examine whether these sites contained previously established consensus binding motifs for the respective proteins. First, we wanted to identify the consensus motif de novo from the inferred sites. For this, we used MEME (21) to identify statistically overrepresented motifs within the 200-bp regions centered on the genomic coordinates representing the inferred binding sites. Adopting a strategy similar to that used by Johnson et al. (4), we used only those binding sites with high tag density (signal intensity): top 10% of CTCF sites (2622) with 60 or more tags, top 20% of NRSF sites (1160) with 46 or more tags and top 5% of STAT1 sites (3825) with 60 or more tags. This analysis revealed motifs that are similar to the previously established consensus motifs for the respective proteins (9,23–25), thus authenticating the accuracy of the inferred binding sites. By ‘similar’, we mean that the conservation levels of individual residues in the motif are similar to that previously reported. While we found a single consensus motif for CTCF sites, NRSF and STAT1 sites were associated with more than one motif (Figure 3). Many NRSF sites contained partial motifs (half-sites), which is consistent with previous observations (4,26,27).
Figure 3.

Accuracy of inferred binding sites. Consensus binding motifs for CTCF, NRSF and STAT1 binding sites are shown in the left panel. The right panel shows the percentage of identified binding sites containing the consensus binding motif(s).
PSSMs of the motifs identified using MEME were then used as MAST (22) input to identify matching motif instances (P < 10−3) in the rest of the binding sites that were not used in the MEME analysis. Overall, we found that 95% of all CTCF sites contained the consensus CTCF motif. About 92% of all NRSF sites contained the full 21-bp consensus NRSF motif and/or one or both of the half-sites with variable spacing between them (4). A majority (64%) of the NRSF sites contained the 21-bp full-site, with 17% containing both the half-sites (but with variable spacing between them compared to the full site) and 17% containing just one of the half-sites. In the case of STAT1, 75% of all binding sites contained the classic GAS motif TTC(T/C)N(G/A)GAA. About 54% of all STAT1 sites contained either the M1 or the M2 motif (Figure 3), while 21% of all STAT1 sites contained both M1 and M2.
Resolution of identified binding sites
A simple but elegant strategy to infer binding sites from ChIP-Seq data is to first identify genomic regions (typically, smaller than the length of the input DNA) enriched with sequence tags, and then for each one of these regions, pick the genomic coordinate within the region that has the maximum number of overlapping tags or fragments as the binding site (3–5). Although this approach identifies the binding ‘regions’ accurately, short read length poses challenges for determining the exact binding sites within these regions. The genomic coordinate with the most number of overlapping tags or fragments may not necessarily be the exact binding site within the identified binding region. As a result, in the worst case, the inferred binding site could be as far as the length of the binding region away from the real binding site.
To assess this important attribute of binding sites identified by SISSRs, we plotted the frequency distribution of the distance between the inferred binding site and the middle of the nearest canonical motif occurrence (Figure 4A). Three-fourths of all CTCF, NRSF and STAT1 sites were within 18, 27 and 51 bp of the nearest motif, respectively (90% within 32, 52 and 73 bp, respectively). When only those high-scoring binding sites were considered, the resolution was much higher than that for all sites. Three-fourths of high-scoring CTCF, NRSF and STAT1 sites were within 13, 12 and 26 bp of the nearest motif, respectively (90% within 20, 19 and 45 bp, respectively). Given that the core CTCF and NRSF motifs are 14- and 21-bp long, respectively, and the resolution is computed with respect to the center of the motif, the precision of identified binding sites is unprecedented. Inverse correlation between the resolution and the average fragment length could explain why the resolution is different for CTCF, NRSF and STAT1, as longer the DNA fragment being sequenced, the lesser the chances of identifying the precise binding site. This suggests that if high-resolution mapping of binding sites is desired, it is important that the sequenced DNA fragments are about ∼150 bp in length.
Figure 4.

Resolution of inferred binding sites. (A) Distribution of the distance between SISSRs-inferred binding site and the middle of the nearest canonical motif occurrence. The red curve is for all binding sites, and the blue curve is for high-scoring binding sites. About 75% of all CTCF, NRSF and STAT1 sites were within 18, 27 and 51 bp of the nearest consensus motif, respectively (90% within 31, 52 and 73 bp, respectively). The resolution is much higher for high-scoring binding sites (90% within 20, 19 and 45 bp, respectively). (B) Distribution of the number of canonical motifs within the 200-bp region centered on identified binding sites. The coloring scheme is the same as that used in A. (C) A 200-bp region centered on a CTCF binding site (highlighted in black) with high tag density. Nine instances of core CTCF motif (10−8 < P < 0.0025) are highlighted in yellow.
Although SISSRs’ high sensitivity allows one to identify many more binding sites, there are extreme situations in which many potential binding sites are present within a 50–200-bp region, and SISSRs or any other approach will not be able to identify all the sites that are actually bound within this small region as it may be unreasonable to expect the individual protein–DNA complexes to immunoprecipitate independently. To assess how prevalent this situation is, we used MAST (22) to examine the distribution of the number of the consensus motifs within the 200-bp region of all sites. We were quite surprised to see that a good fraction (37–71%) of them contained more than one motif (Figure 4B). The distributions were pretty much the same even when only the high-scoring binding sites were considered, except that the fraction of sites with no canonical motif dropped to <8% in all three cases. It is not clear whether the tags mapped to such loci correspond to one or more binding events. As mentioned earlier, the only way to identify each and every binding event is to generate smaller DNA fragments during sonication, although this may not resolve the situation shown in Figure 4C as it is unreasonable to expect this 200-bp region to be sonicated into nine unique fragments containing a binding site each.
Tag density at a binding site is an indicator of protein–DNA binding affinity
DNA-binding proteins demonstrate various affinities to different DNA-binding elements. The longer the protein occupancy on the DNA, the more likely they will be pulled-down in larger numbers in ChIP experiments. This would directly translate to more ChIP-Seq tags for more stable protein–DNA complexes compared to less stable complexes. As a consequence, more tags will be detected at genomic locations corresponding to more stable protein–DNA complexes.
In an attempt to understand whether the tag density at a binding site is an indicator of strong or weak DNA binding, we first tried to determine the tag density landscapes of CTCF, NRSF and STAT1. Interestingly, we found that the distribution of the tag density exhibits a power-law behavior with many binding sites having low tag density and a small fraction of them having high tag density (Figure 5A). To understand what this means, we considered four mutually exclusive sets of 500 NRSF binding sites each, and compared the consensus 21-bp motifs from these sets. To our surprise, we noticed a positive correlation between the average tag density (of all the binding sites in the set) and the information content of the first six residues in the 21-bp NRSF consensus motif (Figure 5B). No noticeable changes were observed for the nucleotide positions 7–21. This suggested that positions 7–21 (7–9 and 12–17, in particular) are critical for DNA binding, and positions 1–6 with high information-content contribute to stable DNA binding.
Figure 5.

Tag density at a binding site is an indicator of binding affinity. (A) Distribution of the number of tags within the 200-bp region centered on binding sites exhibits a power-law behavior. (B) Motif analysis on four mutually exclusive sets of 500 NRSF binding sites each reveals a positive correlation between the information content of first 6 nt of the NRSF consensus motif and the average tag density. See Supplementary Figure 2 for an analysis of CTCF binding sites.
If positions 7–9 and 12–17 (spanning both the left and the right half-sites) are indeed as critical as we claim them to be, then it must be the case that the average tag density of those NRSF sites that contain the 21-bp full-site should be higher than that for those NRSF sites that contain only the left or right half-site. To investigate this aspect, we calculated the average tag density of sites containing full site, and those containing only the left or the right half-site. The average tag density of those NRSF sites containing the full site was 68.3, whereas the average tag density of those sites containing only the right or the left half-site was 12.7 and 14.1, respectively. Since higher tag density at a binding site is a direct result of higher protein occupancy on the DNA, a higher tag density at NRSF full-sites could only mean that these sites provides for a stable protein binding. This, in addition to the results from a recent study in which it was shown that neither the left nor the right half-site by itself is an effective repressor (27), indicates that although NRSF could bind to half-sites, half-site binding is not as strong or functional as full-site binding.
A similar analysis on CTCF binding sites revealed a 11-bp ‘core’ CTCF-binding motif (positions 4–14 in Supplementary Figure 2), whose conservation levels are similar regardless of the tag density at the binding sites. In particular, nucleotides C, G, and G at positions 5, 10, and 13, respectively, are fully conserved across all binding sites, suggesting that point mutations at these positions are highly likely to prevent CTCF binding. Indeed, a recent report used gel mobility shift assay to demonstrate that the mutation of the cytosine in position 5 strongly inhibits CTCF binding (28). Although CTCF has 11 zinc fingers, it only needs a combination of four zinc fingers for a strong binding to the DNA (28,29). In particular, it was shown that zinc finger combinations 4–7 and 5–8 show higher DNA-binding affinity, with zinc fingers 4 and 7 recognizing the 3′ and 5′ ends of the core motif, respectively (28). This finding combined with our results (Supplementary Figure 2) suggest that positions 15–19 (at the 3′ end of the CTCF consensus binding sequence) with high information content contribute to stronger DNA binding. This would mean that the DNA-binding affinity of CTCF could depend on whether or not it employs zinc finger 4 for DNA binding.
DISCUSSION
In this study, we presented SISSRs, a novel algorithm for precise identification of binding sites from short reads generated from ChIP-Seq experiments. We used SISSRs to identify 26 814, 5813 and 73 956 binding sites for CTCF, NRSF, and STAT1 proteins, respectively. Binding sites identified by SISSRs are of high resolution, i.e. the identified sites are within few tens of base pairs from the center of the canonical motif (Figure 4A). For example, >90% of CTCF sites were within 32-bp from the motif center. The resolution of SISSRs-identified binding sites is at least an order of magnitude higher than that of those sites identified by previous approaches (3–5). While SISSRs identifies the center of the binding site, previous approaches identify binding ‘regions’ ranging from a few hundred to a few thousand base pairs in length (Supplementary Figure 3), which may contain more than one binding site within them. This could be one of the reasons that the binding regions identified by these approaches are of low resolution (Supplementary Figure 4A), and the number of canonical motifs within these binding regions is relatively large (Supplementary Figure 4B).
SISSRs’ ability to identify binding sites with high resolution helped it achieve unprecedented sensitivity and specificity, as evidenced by its ability to identify 32–299% more binding sites than that by previous approaches using the same dataset (3–5). We found that 82, 68 and 92% of CTCF, NRSF and STAT1 binding regions reported by previous approaches overlap with one or more binding sites identified in this study for the respective proteins. The reason SISSRs did not recover all binding sites reported by previous studies could be one or both of the following. SISSRs was used with the option that requires at least two directional tags on either side of the binding site (Figure 1)—a stringent criteria compared to that used by previous approaches, which did not consider tag directions and just count the number of tags mapped to a region. Although SISSRs provides an option to identify binding sites with corresponding tags mapped to only one strand (Figure 6B), we did not utilize this option. Using this option and relaxing other constraints could improve the percentages listed above. It is also possible that some of the sites identified by previous approaches are false positives, which we do not expect SISSRs to identify.
SISSRs is highly accurate, which is evident from the fact that an overwhelming majority of the identified sites contained the previously established consensus binding motifs. For example, 96% and 92% of all CTCF and NRSF sites, respectively, contained the consensus binding motifs. This immediately raises questions about those sites that do not contain the consensus binding motif. Are these false-positives? While it is entirely possible that those sites without the canonical motif are false positives, one should not discount the fact that the protein of interest may have bound to the DNA indirectly via another protein (Figure 6C), which may be hard to distinguish from direct DNA binding. Also, one needs to keep in mind that the mere presence of a consensus motif in the predicted region may not necessarily imply that the protein of interest actually binds directly at this site unless it can be determined that the binding is more-or-less independent of other factors. This would mean that the accuracy of identified sites could be lower than that claimed above. This does not reflect the accuracy of SISSRs, rather it reflects the limitation of the ChIP technology, which cannot distinguish between direct and indirect DNA binding.
SISSRs is robust, yet flexible enough that it allows the user to control the elements such as antibody specificity and sequencing errors, which could affect the quality of generated data, and thus the accuracy and resolution of identified binding sites. This is a very useful attribute considering the fact that not all ChIP experiments generate high-quality data every single time, i.e. the background noise (non-specific reads) usually varies for different ChIP experiments. Non-specific reads, which may be due to antibody non-specificity and/or sequencing errors, could be controlled for by adjusting the size of the scanning window. While larger window size reduces the impact of non-specific reads and thus false positives at the cost of resolution, smaller window size provides for increased resolution but also increases the number of false positives (Figure 6A). This noteworthy feature of SISSRs is extremely useful especially when one needs to salvage information from a low-quality data. SISSRs also allows users to submit their own negative control dataset (such as IgG) to be used as a background noise, in place of the default random model.
SISSRs provides an option to identify those binding sites with tags mapped to only the sense or the antisense strand (Figure 6B). This situation arises when tags cannot be mapped to certain regions in the genome, which contain repetitive elements. Since a read aligning to a repetitive element cannot be mapped to a unique genomic location, such tags are usually left out from further consideration, and as a result certain genomic regions enriched with repetitive elements are left unmapped. SISSRs employs a simple procedure to identify those binding sites with tags mapped just to one side of the site (see Methods section). Based on our analysis with the many transcription factor binding proteins, we found that an additional ∼1–2% of binding sites could be identified by selecting this option (this option was not used to identify sites reported in this study). SISSRs also provides an option to mask out reads that fall within certain regions in the genome. This is useful especially if one needs to ignore tags that fall within, say, satellite repeat regions or regions close to centromere. Since such regions are suspected to contain disproportionately large number of mapped reads, which could be due to amplification biases or incorrect mapping of reads with one or two mismatches to regions having high sequence similarity with repetitive regions that are usually masked out during mapping, it is sometimes necessary to ignore reads mapped to these regions.
Our observation that the enrichment of tags at binding sites follows a power-law distribution raised an immediate question as to whether the tag density at the identified binding site is an indicator of the stability or affinity of protein–DNA interaction. Since stable protein–DNA interactions lead to a prolonged half-life of the protein–DNA complex, and the corresponding fragments are likely to be enriched in the ChIP sample, it is reasonable to expect high tag density at stable protein–DNA binding sites. The stability of the protein–DNA complex could depend on many factors such as how accessible the binding site is or how similar the binding site is to the canonical site. We could not assess the former possibility as it is outside the scope of this study. However, we observed a good correlation between the tag density and the information content of NRSF and CTCF binding sites indicating that tag density is a good indicator of the stability of protein–DNA binding. We have also identified the core residues within the NRSF and CTCF binding sites, which are critical for a stronger DNA binding.
In conclusion, although recent advances in sequencing technology provide us with the ability to map protein–DNA interactions on a genome-scale, development of algorithms to identify the exact binding sites from short reads generated by ultra high-throughput sequencing techniques is still in its infancy. We believe that SISSRs will serve as a useful tool for precise identification of binding sites from millions of ChIP-Seq reads. Our experimentation of SISSRs with ChIP-Seq data for three well-characterized DNA-binding proteins revealed interesting insights, which we believe will serve as a guidance for designing ChIP-Seq experiments. While a higher number of reads may increase the sensitivity (Table 1) and resolution (Figure 4), it may not necessarily translate to accuracy (Figure 3), as accuracy may be depend on other factors such as antibody specificity, and how stable the protein–DNA complex is. The length of DNA fragment, which has a direct impact on the resolution of identified binding sites, should preferably be smaller (∼120–150 bp) if high-resolution binding sites are desired.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
The authors would like to thank Dustin E Schones for suggestions and critical reading of the article, and Warren J Leonard, Tae-Young Roh, Gang Wei and Zhibin Wang for useful comments. This study was funded by Intramural Research Program of the National Heart Lung and Blood Institute, National Institutes of Health. Funding to pay the Open Access publication charges for this article was provided by the Intramural Research Program of the National Institutes of Health.
Conflict of interest statement: None declared.
REFERENCES
- 1.Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, et al. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- 2.Iyer VR, Horak CE, Scafe CS, Botstein D, Snyder M, Brown PO. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature. 2001;409:533–538. doi: 10.1038/35054095. [DOI] [PubMed] [Google Scholar]
- 3.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 4.Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 5.Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- 6.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bell AC, West AG, Felsenfeld G. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell. 1999;98:387–396. doi: 10.1016/s0092-8674(00)81967-4. [DOI] [PubMed] [Google Scholar]
- 8.Ohlsson R, Renkawitz R, Lobanenkov V. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet. 2001;17:520–527. doi: 10.1016/s0168-9525(01)02366-6. [DOI] [PubMed] [Google Scholar]
- 9.Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, Ren B. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell. 2007;128:1231–1245. doi: 10.1016/j.cell.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wallace JA, Felsenfeld G. We gather together: insulators and genome organization. Curr. Opin. Genet. Dev. 2007;17:400–407. doi: 10.1016/j.gde.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Filippova GN. Genetics and epigenetics of the multifunctional protein CTCF. Curr. Top. Dev. Biol. 2008;80:337–360. doi: 10.1016/S0070-2153(07)80009-3. [DOI] [PubMed] [Google Scholar]
- 12.Mori N, Schoenherr C, Vandenbergh DJ, Anderson DJ. A common silencer element in the SCG10 and type II Na+ channel genes binds a factor present in nonneuronal cells but not in neuronal cells. Neuron. 1992;9:45–54. doi: 10.1016/0896-6273(92)90219-4. [DOI] [PubMed] [Google Scholar]
- 13.Schoenherr CJ, Anderson DJ. The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Science. 1995;267:1360–1363. doi: 10.1126/science.7871435. [DOI] [PubMed] [Google Scholar]
- 14.Schoenherr CJ, Anderson DJ. Silencing is golden: negative regulation in the control of neuronal gene transcription. Curr. Opin. Neurobiol. 1995;5:566–571. doi: 10.1016/0959-4388(95)80060-3. [DOI] [PubMed] [Google Scholar]
- 15.Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell. 2005;121:645–657. doi: 10.1016/j.cell.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 16.Leonard WJ, O'Shea JJ. Jaks and STATs: biological implications. Annu. Rev. Immunol. 1998;16:293–322. doi: 10.1146/annurev.immunol.16.1.293. [DOI] [PubMed] [Google Scholar]
- 17.Levy DE, Darnell J.E., Jr Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell. Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 18.Ramana CV, Gil MP, Schreiber RD, Stark GR. Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol. 2002;23:96–101. doi: 10.1016/s1471-4906(01)02118-4. [DOI] [PubMed] [Google Scholar]
- 19.McBride KM, Reich NC. The ins and outs of STAT1 nuclear transport. Sci. STKE. 2003;2003:RE13. doi: 10.1126/stke.2003.195.re13. [DOI] [PubMed] [Google Scholar]
- 20.Karolchik D, Kuhn RM, Baertsch R, Barber GP, Clawson H, Diekhans M, Giardine B, Harte RA, Hinrichs AS, Hsu F, et al. The UCSC Genome Browser Database: 2008 update. Nucleic Acids Res. 2008;36:D773–D779. doi: 10.1093/nar/gkm966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bailey TL, Williams N, Misleh C, Li WW. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006;34:W369–W373. doi: 10.1093/nar/gkl198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bailey TL, Gribskov M. Combining evidence using p-values: application to sequence homology searches. Bioinformatics. 1998;14:48–54. doi: 10.1093/bioinformatics/14.1.48. [DOI] [PubMed] [Google Scholar]
- 23.Decker T, Lew DJ, Mirkovitch J, Darnell J.E., Jr Cytoplasmic activation of GAF, an IFN-gamma-regulated DNA-binding factor. EMBO J. 1991;10:927–932. doi: 10.1002/j.1460-2075.1991.tb08026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schoenherr CJ, Paquette AJ, Anderson DJ. Identification of potential target genes for the neuron-restrictive silencer factor. Proc. Natl Acad. Sci. USA. 1996;93:9881–9886. doi: 10.1073/pnas.93.18.9881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie X, Mikkelsen TS, Gnirke A, Lindblad-Toh K, Kellis M, Lander ES. Systematic discovery of regulatory motifs in conserved regions of the human genome, including thousands of CTCF insulator sites. Proc. Natl Acad. Sci. USA. 2007;104:7145–7150. doi: 10.1073/pnas.0701811104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Otto SJ, McCorkle SR, Hover J, Conaco C, Han JJ, Impey S, Yochum GS, Dunn JJ, Goodman RH, Mandel G. A new binding motif for the transcriptional repressor REST uncovers large gene networks devoted to neuronal functions. J. Neurosci. 2007;27:6729–6739. doi: 10.1523/JNEUROSCI.0091-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patel PD, Bochar DA, Turner DL, Meng F, Mueller HM, Pontrello CG. Regulation of tryptophan hydroxylase-2 gene expression by a bipartite RE-1 silencer of transcription/neuron restrictive silencing factor (REST/NRSF) binding motif. J. Biol. Chem. 2007;282:26717–26724. doi: 10.1074/jbc.M705120200. [DOI] [PubMed] [Google Scholar]
- 28.Renda M, Baglivo I, Burgess-Beusse B, Esposito S, Fattorusso R, Felsenfeld G, Pedone PV. Critical DNA binding interactions of the insulator protein CTCF: a small number of zinc fingers mediate strong binding, and a single finger-DNA interaction controls binding at imprinted loci. J. Biol. Chem. 2007;282:33336–33345. doi: 10.1074/jbc.M706213200. [DOI] [PubMed] [Google Scholar]
- 29.Filippova GN, Fagerlie S, Klenova EM, Myers C, Dehner Y, Goodwin G, Neiman PE, Collins SJ, Lobanenkov VV. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol. Cell. Biol. 1996;16:2802–2813. doi: 10.1128/mcb.16.6.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.