

Abstract
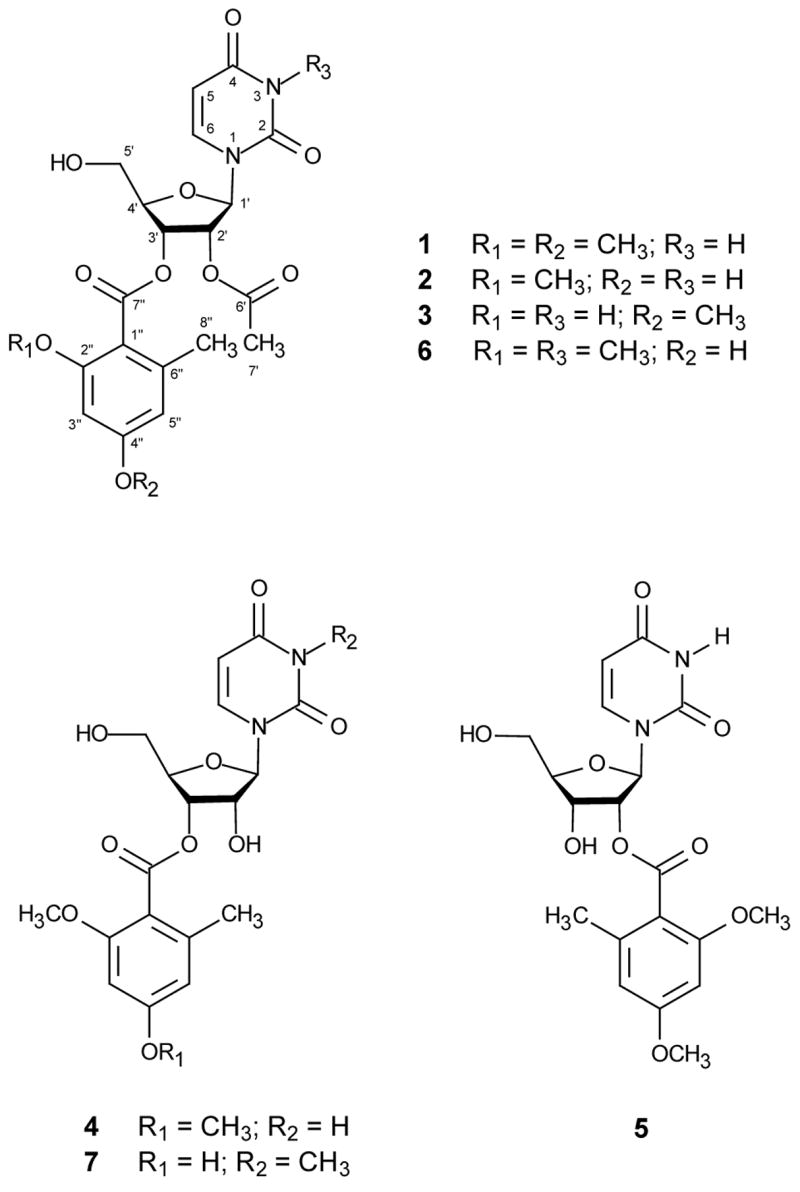
Seven new aroyl uridine derivatives (kipukasins A–G; 1–7) were isolated from solid-substrate fermentation cultures of two different Hawaiian isolates of Aspergillus versicolor. The structures of compounds 1–7 were determined by analysis of NMR and MS data. The nucleoside portion of lead compound 1 was assigned as uracil-1-β-D-ribofuranoside by spectral comparison with an authentic standard. The bioactivity of the original A. versicolor extracts was accounted for mainly by the presence of the known metabolite sterigmatocystin, but kipukasins A and B showed modest activity in assays against gram-positive bacteria.
Our continuing interest in mycoparasitic and fungicolous fungi as sources of new bioactive secondary metabolites1–3 prompted us to investigate the chemistry of an isolate of Aspergillus versicolor (Vuill.) Tiraboschi (MYC-2236 = NRRL 35600). Although A. versicolor is known as a producer of mycotoxins and other compounds,4,5 isolation of A. versicolor as a colonist of other fungi has not been previously reported to our knowledge. This isolate was obtained from a basidioma of Gandoderma australe found growing on a tree in a montane mesic forest in Hawaii, and was cultured by solid-substrate fermentation on rice. The crude extract of the resulting cultures showed significant antiinsectan activity. Sterigmatocystin5 was encountered as a major component, and was responsible for the antiinsectan activity of the original crude extract. However, initial analyses indicated the presence of a set of major constituents unrelated to sterigmatocystin. Further investigation afforded five new nucleoside derivatives, which we named kipukasins A–E (1–5). At the same time, analysis of extracts from cultures of a different fungicolous isolate of A. versicolor (also from Hawaii, but from a different location) led to recognition of the presence of a similar set of compounds. Studies of this second isolate yielded two additional related compounds (kipukasins F and G; 6 and 7). Details of the isolation, structure elucidation, and stereochemical assignment of these metabolites are described here.
Results and Discussion
The crude EtOAc extract from cultures of A. versicolor NRRL 35600 was subjected to solvent partitioning, chromatography on Sephadex LH-20, and reversed phase HPLC to afford samples of sterigmatocystin and kipukasins A–E (1–5). Sterigmatocystin was identified by comparison of NMR and MS data to those of a previously isolated sample. The molecular formula of kipukasin A (1) was determined as C21H24N2O10 (11 unsaturations) based on NMR and MS data. The 1H NMR spectrum revealed the presence of a 1,2,3,5-tetrasubstituted benzene ring, a 1,2-disubstituted olefin (J = 8.1 Hz), four oxymethine protons, one oxymethylene unit, two methoxy groups, two aryl or acetyl methyl groups, and one exchangeable proton (δH 8.65). 13C NMR data were consistent with these observations, and also indicated the presence of four carboxy or amide carbons. Aside from the data for the substituted benzene unit, the 1H and 13C NMR data were suggestive of a nucleoside derivative, more specifically, a uridine analog. Analysis of 1H NMR shifts and J-values confirmed that the four oxygenated methines and the oxygenated methylene are linked to form a furanose moiety. 13C NMR data were consistent with this conclusion. The base was identified as uracil by comparison of the relevant NMR signals to literature data.6 Observation of HMBC correlations of H-6 with C-2, C-4, and C-5, and of H-5 with C-4 and C-6, confirmed the presence of the uracil moiety. The δ-values for the anomeric proton and carbon signals (δH 6.11; d, J = 6.4 Hz; δC 87.9) were consistent with the presence of a C-N glycosidic linkage. HMBC correlations of H-1′ to C-2 and C-4 confirmed the connection of the furanose unit to the uracil moiety at the expected N-1 position.
Analysis of additional HMBC data enabled location of both methoxy groups and an aryl methyl on the benzene ring. Correlations to the remaining two carbonyl groups enabled extension of the aromatic ring to a benzoate moiety and also revealed the presence of an acetate group. Both of these carbonyls were linked to oxygen atoms of the sugar moiety to form ester linkages. The acetate CH3 singlet at δH 2.06 showed HMBC correlations to C-2′ (δC 73.4) and C-6′ (δC 170.2), enabling location of the acetate group at C-2′. A correlation between H-3′ (δH 5.67) and the second ester carbonyl at δC 167.5 established the attachment of the modified benzoate unit to C-3′. The locations of the substituents on the aromatic ring (already noted above to be 1,2,3,5-tetrasubstituted) were assigned by analysis of chemical shifts and HMBC correlations. The chemical shifts of H-3′ (δH 6.34) and C-3′ (δC 96.7) were suggestive of oxygen substitution at both ortho positions. H-3′ showed HMBC correlations to C-2′, C-4′, and C-5′. Correlations from aryl CH3-8′ to C-5′, C-6′, and C-7′ located the methyl group at C-6′. A long-range HMBC correlation between H-5′ and C-7′ established the location of the ester carbonyl at C-1′. The two methoxy singlets showed correlations to C-2′ and C-4′, thereby completing the assignment of structure 1 as shown.
Structure elucidation of kipukasins B–E (2–5) was straightforward due to their close relationships with compound 1. Compound 2 was assigned the molecular formula C20H22N2O10, having one methylene unit less than 1, on the basis of NMR and MS data. The 1H and 13C NMR spectra were consistent with the absence of one of the methoxy groups in 1. The structure was independently assigned as shown by analysis of HMQC and HMBC data. Due to the proximity of the two key carbon signals for C-2′ and C-4′ (δC 159.6 and δC 159.4), some uncertainty remained regarding the position assignment for the methoxy group. Therefore, a NOESY experiment was carried out for compound 2. A correlation between the methoxy signal and the aromatic H-3′ resonance but not the H-5′ signal suggested that the methoxy group was located at C-2′, rather than C-4′. This assignment was verified by isolation of the regioisomer 3.
The 1H and 13C NMR, and MS data for compound 3 revealed that 3 is an isomer of 2. The only significant difference in the 1H NMR spectrum was the presence of an exchangeable proton resonance at δH 11.35, which was identified as a hydrogen-bonded phenolic OH group signal. The presence of this signal in the spectrum of 3, but not in that of 2, is consistent with location of the OH group ortho to the ester carbonyl in 3. Moreover, the methoxy proton signal at δH 3.82 showed NOE correlations to both H-3′ and H-5′, unambiguously locating the methoxy group at C-4′.
Kipukasin D (4) was assigned the molecular formula C19H22N2O9 on the basis of NMR and MS data. The 1H and 13C NMR chemical shift assignments (Tables 1 and 2) matched well with those of the corresponding signals for 1, and revealed the same structural features present in 1 except for the absence of the acetate, which was consistent with the difference in formula. Correspondingly, the H-2′ resonance was shifted significantly upfield (by 0.94 ppm) relative to that of 1, leading to assignment of the structure of kipukasin D (4) as shown.
Table 1.
1H NMR data [δH (mult; JH)] for Kipukasins A–E (1–5; 300 MHz, CDCl3)
| position | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 5 | 5.79 (d, 8.1) | 5.75 (d, 8.1) | 5.80 (d, 8.1) | 5.78 (d, 8.1) | 5.76 (d, 8.1) |
| 6 | 7.76 (d, 8.1) | 7.73 (d, 8.1) | 7.71 (d, 8.1) | 7.67 (d, 8.1) | 7.59 (d, 8.1) |
| 1′ | 6.11 (d, 6.4) | 6.06 (d, 6.7) | 6.09 (d, 6.8) | 5.85 (d, 6.4) | 5.95 (d, 5.0) |
| 2′ | 5.57 (dd, 6.4, 5.6) | 5.55 (dd, 6.7, 5.5) | 5.63 (dd, 6.8, 5.7) | 4.63 (t, 6.0) | 5.70 (t, 5.3) |
| 3′ | 5.67 (dd, 5.6, 2.8) | 5.63 (dd, 5.5, 2.4) | 5.75 (dd, 5.7, 2.5) | 5.62 (dd, 5.5, 2.6) | 4.63 (t, 5.2) |
| 4′ | 4.35 (q, 2.8) | 4.35 (q, 2.3) | 4.39 (q, 2.5) | 4.35 (q, 2.5) | 4.14 (dt, 4.8, 2.4) |
| 5′ | 3.99 (br m) | 3.99 (br m) | 4.01 (br d, 2.5) | a 3.99 (dd, 12, 2.4) | a 4.03 (dd, 12, 2.4) |
| b 3.94 (dd, 12, 2.2) | b 3.88 (dd, 12, 2.5) | ||||
| 7′ | 2.06 (s) | 2.05 (s) | 2.07 (s) | -- | -- |
| 3′ | 6.34 (br s) | 6.26 (d, 2.1) | 6.35 (d, 2.3) | 6.37 (d, 2.2) | 6.34 (d, 2.2) |
| 5′ | 6.34 (br s) | 6.24 (d, 2.1) | 6.34 (d, 2.3) | 6.40 (d, 1.5) | 6.38 (d, 2.2) |
| 8′ | 2.32 (s) | 2.28 (s) | 2.58 (s) | 2.38 (s) | 2.34 (s) |
| 2′-OH | -- | 11.35 (s) | |||
| 2′-OMe | 3.82 (s)a | 3.79 (s) | -- | 3.84 (s)b | 3.84 (s)c |
| 4′-OMe | 3.81 (s)a | -- | 3.82 (s) | 3.87 (s)b | 3.82 (s)c |
| NH | 8.65 (br s) | 7.89 (br s) | 7.98 (br s) | 8.22 (br s) | 8.00 (br s) |
Assignments with identical superscripts are interchangeable. The appearance and/or presence of signals for most of the exchangeable protons was variable and sample-dependent.
Table 2.
13C NMR data (δC) for Kipukasins A–E (1–5; 100 MHz, CDCl3)
| position | 1 | 2a | 3a | 4 | 5 |
|---|---|---|---|---|---|
| 2 | 150.8 | 150.6 | 150.7 | 150.8 | 150.0 |
| 4 | 163.1 | 162.9 | 162.6 | 162.9 | 162.5 |
| 5 | 103.6 | 103.3 | 104.0 | 103.4 | 103.1 |
| 6 | 141.1 | 140.7 | 141.2 | 141.4 | 141.5 |
| 1′ | 87.9 | 87.0 | 88.6 | 91.1 | 89.9 |
| 2′ | 73.4 | 73.4 | 73.1 | 73.8 | 75.5 |
| 3′ | 72.2 | 72.3 | 72.9 | 73.8 | 70.1 |
| 4′ | 84.2 | 84.3 | 84.2 | 83.5 | 85.3 |
| 5′ | 62.5 | 62.2 | 62.3 | 62.7 | 62.2 |
| 6′ | 170.2 | 169.8 | 170.3 | -- | -- |
| 7′ | 20.9 | 20.7 | 21.0 | -- | -- |
| 1′ | 114.8 | 113.9 | 104.7 | 114.7 | 114.0 |
| 2′ | 162.4 | 159.4 | 166.7 | 162.7 | 168.8 |
| 3′ | 96.7 | 97.2 | 99.5 | 96.9 | 96.7 |
| 4′ | 159.3 | 159.6 | 165.4 | 159.2 | 162.5 |
| 5′ | 107.3 | 109.6 | 112.3 | 108.1 | 107.7 |
| 6′ | 139.4 | 139.2 | 143.5 | 141.0 | 140.9 |
| 7′ | 167.5 | 167.3 | -- | 166.7 | 166.4 |
| 8′ | 20.6 | 20.1 | 25.4 | 20.9 | 20.6 |
| 2′-OMe | 55.8b | 55.9 | -- | 55.8c | 56.4d |
| 4′-OMe | 56.3b | -- | 55.7 | 56.5c | 55.7d |
These shifts were assigned on the basis of HMBC and HMQC correlations.
Assignments with identical superscripts are interchangeable.
Kipukasin E (5) possessed the same molecular formula as that of 4, as deduced from its NMR and MS data. The 1H and 13C NMR spectra were very similar to those of 4, with only a subtle difference in the 1H NMR spectrum, consisting of a somewhat different splitting pattern for some of the ribose oxymethine proton signals. Homonuclear decoupling experiments were employed to establish proton signal assignments, and revealed that the signal for H-3′ was shifted upfield, while the H-2′ resonance was shifted downfield, thereby leading to the location of the acyl group at C-2′ in 5 as shown. This conclusion was confirmed by observation of an HMBC correlation from H-2′ to C-7′.
A second isolate of the same species (MYC-1793 = NRRL 35641) obtained from a different location showed similar biological activity and was also investigated. The known A. versicolor metabolite sterigmatocystin was again recognized after solvent partitioning, again accounting for the antiinsectan activity. Most of the sterigmatocystin was removed by trituration with MeOH, and the soluble portion was subjected to chromatography on silica gel, followed by HPLC to afford two additional new benzoylated nucleosides (kipukasins F and G; 6–7).
The elemental composition of kipukasin F (6) was determined to be C21H24N2O10 on the basis of NMR and HRESIMS data. The 1H NMR spectrum displayed resonances that were nearly identical to those observed in the spectrum of kipukasin B (2). The presence of a broad phenolic OH singlet at δ 6.50, rather than an intramolecularly hydrogen bonded OH signal supported an analogous regiochemical assignment for the substituted benzene ring. However, the NMR data for 6 also contained signals characteristic of an N-CH3 group (δH 3.31; δC 27.8) that was absent in the spectrum of 2, and lacked the exchangeable NH singlet. These data suggested that 6 differs from 2 only in the presence of an N-3 methyl group, and this conclusion was independently confirmed by analysis of HMBC, HMQC, and NOE difference data. The location of the N-CH3 group was confirmed by HMBC correlations of the corresponding proton signal to C-2 and C-4. The location of the methoxy group on the benzene ring was confirmed by analysis of NOE difference data. Irradiation of the methoxy proton signal (δ 3.75) resulted in NOE enhancement of the signal corresponding to H-3′ (δ 6.28), while no enhancement of the signal corresponding to H-5′ was observed.
The mass of kipukasin G (7) was found to be 42 daltons lower than that of 6, and the molecular formula was determined to be C19H22N2O9 by analysis of NMR and HRESIMS data. The 1H and 13C NMR spectra of 7 were very similar to those of 6, lacking only the signals corresponding to the acetyl group (δH 2.04, δC 169.8), thereby suggesting that kipukasin G (7) is the desacetyl analogue of 6. The upfield chemical shift of the H-2′ signal (δ 4.56) in the spectrum of 7 relative to its position in the spectrum of 6 (δ 5.59) supported this conclusion. Proton assignments for the furanose ring were confirmed by decoupling experiments, and independent analysis of HMBC data verified the structure of kipukasin G (7).
The sugar moiety in 1 was determined to be a β-ribose unit by analysis of coupling constants and comparison with 1H NMR data reported for modified ribose units (although ribose unit proton signals are often reported as broad).6–8 In order to establish the absolute configuration of the ribose moiety, a sample of 1 was subjected to hydrolysis with 0.1 N NaOH and the resulting free nucleoside was purified by RP-HPLC. Comparison of the CD curve with that of uridine confirmed the presence of uracil-1-β-D-ribofuranoside in 1.9 The configurations of the ribose moieties contained in 2–7 are presumed to be the same as in 1.
Novel nucleoside derivatives have been reported from a variety of sources such as sponges, red algae, mushrooms, and bacteria,10–14 and representatives of this class have been reported to show antitumor, antiviral, and antifungal, and antibiotic activity. However, such reports are relatively uncommon, despite the ubiquitous nature of nucleosides. Unlike the kipukasins, none of these previously known metabolites display acylation at positions on the sugar residue, and to our knowledge, kipukasins A–G (1–7) are the first naturally occurring aroyl nucleosides to be reported.
2′-3′-Transesterification is known to occur in some ribose derivatives,15 and such a process could be involved in the formation of some of these compounds (e.g., via interconversion of species like 4 and 5), however, warming samples of 4 and 5 in solution (acetone) did not result in observable interconversion. On a related point, compounds 1–3 and 6 each contain acetate groups, but these units seem unlikely to arise from the ethyl acetate used routinely as an extraction solvent, since the compounds were not exposed to this solvent during any of the purification steps, and none of the presumably more accessible primary alcohol positions were acetylated. However, re-fermentation and analysis of extracts made with a different solvent was not carried out.
Although the original extract showed modest antifungal activity, kipukasins A–G showed no activity in disk assays against Aspergillus flavus (NRRL 6541), Fusarium verticillioides (NRRL 25457), or Candida albicans (ATCC 14053) at 200 μg/disk. In antibacterial assays, compound 1 was active against Staphylococcus aureus (ATCC 29213), causing an 18-mm zone of inhibition at 100 μg/disk. Compound 2 showed activity against Bacillus subtilis (ATCC 6051) at the same level, affording a 12-mm zone of inhibition. Kipukasins C–G were inactive in these assays when tested at the same level.
Experimental Section
General Experimental Procedures
Optical rotations were was determined with a Rudolph automatic polarimeter, model AP III 589, and UV data were recorded with a Varian Cary III UV-visible spectrophotometer. CD data were collected using an Olis Cary-17 spectrometer (1-cm cell). 1H and 13C NMR spectra were acquired using Bruker DPX-300 and DRX-400 spectrometers, respectively. HMQC and HMBC data were obtained using a Bruker Avance-600 instrument. HPLC was carried out using a Beckman System Gold instrument with a model 166 UV detector with UV detection at 215 nm. Other general procedures and instrumentation have been described previously.16
Fungal Material
One of the cultures of A. versicolor (MYC-2236) was isolated from the surface of a basidioma of Gandoderma australe found growing on a living tree in a montane mesic forest in Kipuka Pauula (Bird Park), Volcanoes National Park, Hawaii. A subculture has been deposited in the Agricultural Research Service (ARS) collection at the USDA NCAUR with the accession number NRRL 35600. The second A. versicolor isolate (MYC-1793) was obtained from a white mycelial growth found on the undersurface of a dead hardwood branch in a subalpine dry forest, Puulaau near Highway 200 (milepost 43), Hawaii County, Hawaii. A subculture of this isolate has been deposited with the ARS Culture Collection at the NCAUR with the accession number NRRL 35641.
General fermentation procedures used have been published elsewhere.2,15 Each culture was incubated on rice (2 × 50 g for NRRL 35600 and 3× 50 g for NRRL 35641) at 25 °C for 30 days and the combined fermentation mixtures were extracted with EtOAc (3 × 500 mL in each case). The combined, filtered EtOAc solutions were evaporated to dryness, yielding 303 mg of crude extract for NRRL 35600 and 445 mg for NRRL 35641.
Extraction and Isolation
The crude extract from NRRL 35600 was partitioned between MeCN and hexanes (1:1). The MeCN fraction (133 mg) was chromatographed on a Sephadex LH-20 column eluting successfully with 4:1 CH2Cl2-hexanes; 3:2 CH2Cl2-acetone; and 1:4 CH2Cl2-acetone to give ten fractions. Fraction 1 (47 mg), eluted with 4:1 CH2Cl2-hexanes, consisted of the known compound sterigmatocystin, which was identified by spectroscopic comparison with an authentic standard. Fraction 4 (22 mg) was further separated by reversed phase HPLC (Alltech Apollo 5-μm C18 column; 10 × 250 mm; MeCN/H2O, 40–60% over 20 min, 60–100% over 5 min at a flow rate of 2 mL/min) to give compounds 1 (7.7 mg) and 3 (0.3 mg). Fraction 5 (13 mg) was processed by HPLC under the same conditions to afford compounds 1 (1.1 mg), 3 (0.5 mg), 4 (1.8 mg) and 5 (0.7 mg). Fraction 6 (17 mg), eluted with 3:2 CH2Cl2-acetone from the Sephadex column, was separated using the same HPLC column (MeCN/H2O, 30–50% over 20 min, 50–100% over 5 min) to give compounds 2 (0.9 mg), 4 (1.5 mg), and 5 (0.9 mg). Compound 2 (1.8 mg) was also obtained from fraction 7 (15 mg), eluted with 3:2 CH2Cl2-acetone from the Sephadex column, using the same HPLC conditions as above for fraction 6.
The crude NRRL 35641 extract was similarly partitioned between MeCN and hexanes (1:1). In this instance, the CH3CN-soluble portion (365 mg) was evaporated to dryness and then CH3OH (5 mL) was added. The CH3OH-insoluble solid was collected by filtration using Whatman #2 filter paper. This process was repeated two more times with decreasing volumes of CH3OH to obtain a sample of the precipitate (47 mg) which was identified as sterigmatocystin. The combined filtrate was evaporated to dryness (301 mg) and then subjected to silica gel column chromatography, eluting successively with CH2Cl2-CH3OH (49:1, 200 mL; 97:3, 75 mL; 19:1, 150 mL; 9:1, 200 mL; 87:13, 100 mL; 21:4, 100 mL; 7:3, 75 mL), and CH2Cl2-CH3OH-acetone (6:3:1, 100 mL; 2:2:1, 100 mL) to afford several fractions. HPLC separation of the seventh fraction (22 mg; eluted with 19:1 CH2Cl2-CH3OH) eluting with CH3CN and H2O (same column as above; 30% CH3CN in H2O over 15 min, 30% to 86% CH3CN in H2O over 20 min, and 86% to 100% CH3CN in H2O over 1 min) afforded kipukasin G (7; 3 mg). The sixth fraction (32 mg; eluted with 19:1 CH2Cl2-CH3OH) was subjected to HPLC (Rainin Dynamax C-18 column, 8-μm particles; 40% CH3CN in H2O over 10 min and 40% to 100% CH3CN in H2O over 5 min at 10.0 mL/min) to obtain kipukasin F (6; 12 mg).
Kipukasin A (1)
colorless glass; [α]D25 –26 (c 0.12, MeOH); UV (MeOH) λmax (log ε) 213 (4.5), 258 (4.1) nm; 1H and 13C NMR data, see Tables 1 and 2; HMBC data H-5 → C-4, 6; H-6 → C-2, 4, 5, 1′; H-1′ → C-2, 6, 2′, 4′; H-2′ → C-1′, 4′, 6′; H-3′ → C-1′, 5′, 7′; H3-7′ → C-2′, 6′; H-3′→ C-2′, 4′, 5′; H-5′ → C-1′, 3′, 7′, 8′; H3-8′ → C-1′, 5′, 6′; 2′-OMe → C-2′, 3′; 4′-OMe → C-4′, 5′; EIMS m/z 464 (M+; 34), 353 (44), 179 (100); ESIMS m/z 487 [M + Na]+, 951 [2M + Na]+; HRESIMS m/z 487.1320 (calcd for C21H24N2O10Na, 487.1329).
Kipukasin B (2)
colorless glass; [α]D25 –14 (c 0.12, MeOH); UV (MeOH) λmax (log ε) 215 (4.4), 256 (4.0) nm; 1H and 13C NMR data, see Tables 1 and 2; HMBC data H-5 → C-6; H-6 → C-2, 4, 5, 1′; H-1′ → C-2, 6, 2′; H-2′ → C-1′, 4′, 6′; H-3′ → C-1′, 7′; H-4′ → C-3′; H-5′ → C-3′; H3-7′ → C-6′; H-3′→ C-1′, 2′, 5′; H-5′ → C-1′, 3′, 8′; H3-8′ → C-1′, 5′, 6′; 2′-OMe → C-2′; ESIMS m/z 473 [M + Na]+, 923 [2M + Na]+; HREIMS m/z 450.1271 (calcd for C20H22N2O10, 450.1274).
Kipukasin C (3)
colorless glass; UV (MeOH) λmax (log ε) 214 (4.3), 265 (4.0) nm; 1H and 13C NMR data, see Tables 1 and 2; HMBC data H-5 → C-6; H-6 → H-2, 4, 5, 1′; H-1′ → C-2, 6, 2′; H-2′ → C-1′, 4′, 6′; H-3′ → C-1′; H-4′ → C-3′; H3-7′ → C-6′; H-3′→ C-1′, 4′, 5′; H-5′ → C-1′, 3′, 8′; H3-8′ → C-1′, 5′, 6′; OH-2′ → C-1′, 2′, 3′, 4′-OMe → C-4′; EIMS m/z 450 (M+; 14), 338 (15), 175 (30), 165 (100); ESIMS m/z 473 [M + Na]+. Optical rotation and HRMS data were not recorded for 3 due to sample limitations.
Kipukasin D (4)
colorless glass; [α]D25 –27 (c 0.19, MeOH); UV (MeOH) λmax (log ε) 210 (4.4), 257 (3.8) nm; 1H and 13C NMR data, see Tables 1 and 2; HMBC data H-5 → C-4, 6; H-6 → C-2, 4, 5, 1′; H-1′ → C-2, 6, 2′; H-2′ → C-4′; H-3′ → C-1′, 5′, 7′; H-4′ → C-3′; H-5′ → C-3′, 4′; H-3′→ C-1′, 2′, 4′, 5′, 7′; H-5′ → C-1′, 3′, 8′; H3-8′ → C-1′, 5′, 6′; 2′-OMe → C-2′; 4′-OMe → C-4′; EIMS m/z 422 (M+; 27), 310 (49), 195 (42), 148 (100); HREIMS m/z 422.1321 (calcd for C19H22N2O9, 422.1325).
Kipukasin E (5)
colorless glass; UV (MeOH) λmax (log ε) 216 (4.4), 258 (3.9) nm; 1H and 13C NMR data, see Tables 1 and 2; HMBC data H-5 → H-6; H-6 → C-2, 4, 5, 1′; H-1′ → C-2, 6, 2′; H-2′ → C-1′, 4′, 7′; H-3′ → C-1′, 5′; H-4′ → C-3′; H-5′ → C-3′, 4′; H-3′→ C-1′, 2′, 4′, 5′, 7′; H-5′ → C-1′, 3′, 4′, 8′; H3-8′ → C-1′, 5′, 6′; 2′-OMe → C-2′; 4′-OMe → H-4′; EIMS m/z 422 (M+; 16), 311 (49), 179 (198), 68 (100). Optical rotation and HRMS data were not recorded for 5 due to sample limitations.
Kipukasin F (6)
off-white semi-solid; [α]25D – 32 (c 0.6, acetone); UV (CH3CN) λmax (log ε) 254 (4.0) nm; IR (film on NaCl plate) vmax 3369, 3018, 2933, 1710, 1606 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.71 (d, J = 8.1 Hz, H-6), 6.50 (br s, 4′-OH), 6.28 (br d, J = 2 Hz, H-3′), 6.26 (br d, J = 2 Hz, H-5′), 6.05 (d, J = 5.8 Hz, H-1′), 5.82 (d, J = 8.1 Hz, H-5), 5.65 (dd, J = 5.8, 3.4 Hz, H-3′), 5.59 (t, J = 5.8 Hz, H-2′), 4.32 (m, H-4′), 3.98 (br d, J = 12 Hz, H-5′a), 3.94 (br d, J = 12 Hz, H-5′b), 3.75 (s, 2′-OCH3), 3.31 (s, 3-CH3), 2.24 (s, H3-8′); 2.04 (s, H3-7′); 13C NMR (CDCl3, 100 MHz) δ 169.8 (s, C-6′), 167.3 (s, C-7′), 162.9 (s, C-4), 158.7 (s, C-4′), 159.2 (s, C-2′), 151.3 (s, C-2), 139.2 (s, C-6′), 138.7 (d, C-6), 113.9 (s, C-1′), 109.4 (d, C-5′), 102.5 (d, C-5), 96.9 (d, C-3′), 89.0 (d, C-1′), 83.6 (d, C-4′), 73.1 (d, C-2′), 71.5 (d, C-3′), 61.9 (t, C-5′), 55.8 (q, 2′-OCH3), 27.8 (q, 3-CH3), 20.6 (q, C-7′), 20.0 (q, C-8′); HMBC data (CDCl3, 600 MHz) 3-CH3 → C-2, C-4; H-5 → C-4, C-6; H-6 → C-2, C-4, C-5, C-1′; 3-CH3 → C-2, C-4; H-1′ → C-2, C-6, C-2′, C-3′, C- 4′; H-2′ → C-1′, C-4′, C-6′; H-3′→ C-1′, C-2′, C-4′, C-5′, C-7′; H-4′→ C-2′, C-3′, C-5′; H2-5′→ C-3′, C-4′; H3-7′→ C-6′; H-3′→ C-1′, C-4′, C-5′, C-7′; H-5′→ C-3′, C-8′; H3-8′→ C-1′, C-5′, C-6′; 2′-OCH3 → C-2′; NOE difference data 2-OCH3 → H-3′; H3-8′→ H-5′; HRESIMS obsd m/z 465.1500 (M + H)+, calcd for C21H25N2O10, 465.1509.
Kipukasin G (7)
colorless glass; [α]25D – 24 (c 0.15, acetone); UV (CH3CN) λmax (log ε) 256 (4.1) nm; IR (film on NaCl plate) vmax 3384, 1710, 1659, 1603 cm−1; 1H NMR (CDCl3 + one drop CD3OD, 400 MHz) δ 7.71 (d, J = 8.1 Hz, H-6), 6.29 (br s, H-3′), 6.28 (m, H-5′), 5.87 (d, J = 6.3 Hz, H-1′), 5.80 (d, J = 8.1 Hz, H-5), 5.53 (dd, J = 5.4, 2.9 Hz, H-3′), 4.56 (dd, J = 6.3, 5.4 Hz, H-2′), 4.26 (m, H-4′), 3.91 (dd, J = 12, 2.2 Hz, H-5′a), 3.87 (dd, J = 12, 2.2 Hz, H-5′b), 3.81 (s, 2′-OCH3), 3.30 (s, 3-CH3), 2.29 (s, H-8′); 13C NMR (CDCl3 + one drop CD3OD, 100 MHz) δ 166.7 (s, C-7′), 162.9 (s, C-4), 159.6 (s, C-4′), 159.1 (s, C-2′), 151.5 (s, C-2), 140.6 (s, C-6′), 138.9 (d, C-6), 112.7 (s, C-1′), 110.1 (d, C-5′), 102.2 (d, C-5), 97.0 (d, C-3′), 91.1 (d, C-1′), 83.2 (d, C-4′), 73.5 (d, C-2′), 73.3 (d, C-3′), 61.9 (t, C-5′), 56.0 (q, 2′-OCH3), 27.7 (q, 3-CH3), 20.2 (q, C-8′); HMBC data (CDCl3 + one drop CD3OD, 600 MHz) 3-CH3 → C-2, C-4; H-5 → C-4, C-6; H-6 → C-2, C-4, C-5, C-1′; 3-CH3 → C-2, C-4; H-1′ → C-2, C-6, C-4′; H-2′ → C-1′, C-3′, C-4′; H-3′→ C-1′, C-4′, C-5′, C-7′; H-4′→ C-5′; H2-5′→ C-3′, C-4′; H-3′→ C-1′, C-5′, C-7′; H-5′→ C-3′, C-7′, C-8′; H3-8′→ C-1′, C-5′, C-6′; 2′-OCH3 → C-2′; HRESIMS obsd m/z 423.1399 (M + H)+, calcd for C19H23N2O9, 423.1404.
Alkaline hydrolysis of kipukasin A (1)
Compound 1 (2.4 mg) was treated with 0.1 N NaOH (0.5 mL) at room temperature for 48 h. The solution was evaporated to dryness and purified by RP-HPLC using 0.1% formic acid in H2O/MeCN (20/80) with UV detection at 250 nm to afford uridine (1.2 mg); CD (H2O). ε 267 (9.2), 240 (−4.0), 215 (−5.0) nm; 1H NMR (300 MHz, CD3OD): δ 8.44 (1H, s), 8.01 (1H, d, J = 8.1 Hz), 5.90 (1H, d, J = 4.4 Hz), 5.70 (1H, d, J = 8.1 Hz), 4.18 (1H, t, J = 5.3 Hz), 4.15 (1H, t, J = 5.3 Hz), 4.01 (1H, m), 3.84 (1H, dd, J = 12, 2.7 Hz), 3.73 (1H, dd, J = 12, 3.2 Hz).
Supplementary Material
1H NMR spectra for 1–7 and 13C NMR spectra for compounds 1 and 4–7. This material is available free of charge on the Internet at http://pubs.acs.org.
Acknowledgments
Financial support from the National Science Foundation (CHE-0315591) and the National Institutes of Health (GM 60600) is gratefully acknowledged.
References and Notes
- 1.Deyrup ST, Gloer JB, O’Donnell K, Wicklow DT. J Nat Prod. 2007;70:378–382. doi: 10.1021/np060546k. [DOI] [PubMed] [Google Scholar]
- 2.Mudur SV, Gloer JB, Wicklow DT. J Antibiot. 2006;59:500–506. doi: 10.1038/ja.2006.70. [DOI] [PubMed] [Google Scholar]
- 3.Jiao P, Swenson DC, Gloer JB, Wicklow DT. J Nat Prod. 2006;69:636–639. doi: 10.1021/np0504964. [DOI] [PubMed] [Google Scholar]
- 4.Dictionary of Natural Products, Web Version 15.2. Chapman and Hall/CRC Press; 2007. [Google Scholar]
- 5.Cole RJ, Schweikert MA. Handbook of Secondary Fungal Metabolites. Vol. 1. Academic press; 2003. pp. 597–598. [Google Scholar]
- 6.Pretsch E, Buhlmann P, Affolter C. Structure Determination of Organic Compounds: Tables of Spectral data. Springer-Verlag; Berlin: 2000. p. 421. [Google Scholar]
- 7.Ostroswki T, Maurizot J, Adeline M, Fourrey J, Clivio P. J Org Chem. 2003:6502–6510. doi: 10.1021/jo030086p. [DOI] [PubMed] [Google Scholar]
- 8.Ohigashi H, Kaji M, Sakaki M, Koshimizu K. Phytochemistry. 1989;28:1365–1368. [Google Scholar]
- 9.Miles DW, Inskeep WH, Robins MJ, Winkley MW, Robins RK, Eyring H. J Am Chem Soc. 1970;92:3872–3881. doi: 10.1021/ja00716a007. [DOI] [PubMed] [Google Scholar]
- 10.Zhao J, Ma M, Wang S, Li S, Cao P, Yang Y, Lu Y, Shi J, Xu N, Fan X, He L. J Nat Prod. 2005;68:691–694. doi: 10.1021/np040234m. [DOI] [PubMed] [Google Scholar]
- 11.Capon RJ, Trotter NS. J Nat Prod. 2005;68:1689–1691. doi: 10.1021/np0502692. [DOI] [PubMed] [Google Scholar]
- 12.Kubo I, Kim M, Wood WF, Naoki H. Tetrahedron Lett. 1986;27:4277–4280. [Google Scholar]
- 13.Sakata K, Sakurai A, Tamura S. Tetrahedron Lett. 1974;15:4327–4330. [Google Scholar]
- 14.Sakata K, Sakurai A, Tamura S. Tetrahedron Lett. 1975;16:3191–3194. [Google Scholar]
- 15.Jackson MD, Denu JM. J Biol Chem. 2002;277:18535–18544. doi: 10.1074/jbc.M200671200. [DOI] [PubMed] [Google Scholar]
- 16.Höller U, Gloer JB, Wicklow DT. J Nat Prod. 2002;65:876–882. doi: 10.1021/np020097y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1H NMR spectra for 1–7 and 13C NMR spectra for compounds 1 and 4–7. This material is available free of charge on the Internet at http://pubs.acs.org.