

Abstract
Alkylating agents, including environmental and endogenous carcinogens, and DNA targeting antineoplastic agents, that adduct DNA via intermediates with significant cationic charge show a sequence selectively in their covalent bonding to nucleobases. The resulting patterns of alkylation eventually contribute to the agent-dependent distributions and types of mutations. The origin of the regioselective modification of DNA by electrophiles has been attributed to steric and/or electronic factors, but attempts to mechanistically model and predict alkylation patterns have had limited success. In this review, we present data consistent with the role of the intrinsic sequence-dependent electrostatic landscape (SDEL) in DNA that modulates the equilibrium binding of cations and the bonding of reactive charged alkylating agents to atoms that line the floor of the major groove of DNA.
1. Introduction
Genomic DNA is considered thermodynamically and chemically stable. However, because of its enormous size (> 3 × 109 base pairs) and the sensitivity of cells to even very low levels of DNA damage, the relatively inefficient chemical reactions between DNA and endogenous and exogenous compounds has significant biological relevance. Our comments here are directed toward alkylation reactions in which DNA serves as a nucleophile, a common theme for many mutagens, carcinogens and/or antineoplastic agents (1, 2). In most cases, electrophilic attack on DNA affords a complex mixture of lesions derived from reactions at different positions on the four nucleobases and the phosphate backbone (3, 4). If not correctly repaired before DNA replication, many of the lesions can induce cellular toxicity and/or mutagenicity. The former is associated with chemotherapeutic drugs and the latter with agents, including the same chemotherapeutic drugs, involved in the initiation of cancer.
Mutation frequencies vary depending on DNA sequence, and often concentrate at sites referred to as hotspots (5). The hotspots reflect a combination of factors starting with the level of modification at a particular site, i.e., the lower the level of adduct, the lower the mutation frequency. This phenomenon is the basis for the dose-response between mutagen exposure and mutation levels, and presumably cancer incidence and latency (6, 7). If a base is modified, then the generation of a mutation will depend on the rate and accuracy of repair, and the way the lesion is processed by DNA polymerase(s). Both repair and replication may also be sequence-dependent (8–10). These factors (level of damage, rate of accurate repair and error rate in polymerization) provide the real mutation frequency at an individual site, but the apparent mutation frequency is dependent on the selection factor(s) used to detect mutations in a particular assay. The bottom line is that the mutation frequency at an individual base follows a dose-dependent response to exposure to mutagen using non-saturating doses. This is the case for alkylating agents, e.g., N-methyl-N-nitrosourea (MNU1), that target 3′-G′s in 5′-GG sequences (see below), and induce mutations with high frequency at 3′-G′s in 5′-GG sequences in all species. More complex DNA damaging molecules often show unique alkylation profiles (11) and there is also strong correlation between sites of covalent reaction of these complex electrophiles, e.g., B[a]P-diol epoxide, and sites of mutations (12). The goal of our research is to understand the origin of sequence-dependent alkylation patterns.
2. Reactions of DNA with simple alkylating agents
After it became apparent that covalent modification of DNA is an early critical step in mutagenesis and carcinogenesis (13, 14), elegant work from many laboratories provided a detailed qualitative and quantitative picture of the lesions formed by a variety of DNA alkylating agents, including some that are, or were, used in the treatment of cancer. Some of these agents directly react with DNA while others undergo either chemical or enzymatic changes to reveal the reactive electrophilic intermediate. For reasons that will become clear later, we divide the agents into those that react via intermediates that are neutral and those that react via intermediates with significant positive charge. Examples of the former class are alkyl halides, alkane sulfonates, epoxides, etc., which directly add to DNA via an SN2 pathway. The latter group includes N-nitroso compounds (nitrosamines and nitrosoureas), triazenes, nitrogen mustards, etc. It is thought that the reactive intermediates generated from these compounds also react with DNA via a concerted bimolecular pathway (15, 16).
The types of lesions formed by methylating agents from the two classes, neutral and cationic, are shown in Figure 1 (3, 4). The yields are qualitatively and quantitatively similar, but there are some important differences. For both classes of alkylating agents, the major product forms at N7-G in the major groove. Alkylation levels at other ring nitrogens are comparable. The main difference is the level of attack at the exocyclic oxygens, e.g., O6-G, O4-T, and the non-bridging phosphate oxygens (3, 4). From a chemical yield standpoint the differences are not large, but the biological consequences are significant as a result of the mispairing opposite some O-alkylated nucleobases by replicative or translesion polymerases, and the resulting increase in the mutation frequency (17, 18). It is important to note that the yields shown in Figure 1 are averages for genomic DNA and do not reflect yields at individual sites.
Figure 1.

Methyl adducts produced from alkylating agents that react via cationic (blue) and neutral (red) intermediates: yields are normalized relative to the predominant 7-mG lesion (based upon 3, 4).
There are few reactions better understood than those of nucleophiles with alkyl sulfonate esters and halides. For reactions with ds-DNA, the issues of accessibility and nucleophilicity of individual DNA sites need to be considered (19). Based upon a combination of these two factors, the major product of ds-DNA alkylation occurs at N7-G. Obviously sites that are reactive in nucleosides and ss-DNA become refractory to alkylation if they happen to be inaccessible upon formation of the Watson-Crick duplex. In general, yields at all positions are lower in ds-DNA than ss-DNA with the agents that react via non-charged intermediates due to steric factors.
A well-studied class of charged alkylating agents are the N-nitroso compounds, including nitrosamides, nitrosoureas and nitrosamines. Through enzymatic or base-catalyzed hydrolysis, or enzymatic α-oxidation (20, 21), these compounds are converted into transient alkanediazonium ions (Figure 2) that provide the array of adducts shown in Figure 1 (3,4). For larger alkyl groups, e.g., propyl or butyl, rearrangements via carbonium ion type intermediates are observed (22, 23). Therefore, the array of products can become even more complicated due to the presence of structural isomers (24). Unsymmetrical N,N-dialkylnitrosamines, which have two sets of α-H’s, can be oxidized to give two different alkanediazonium ions that in turn can yield two sets of DNA adduct products. The formation of structural adduct isomers has been well-documented for the tobacco specific nitrosamines that can methylate and 4-oxo-4-(3-pyridyl)butylate DNA (25).
Figure 2.
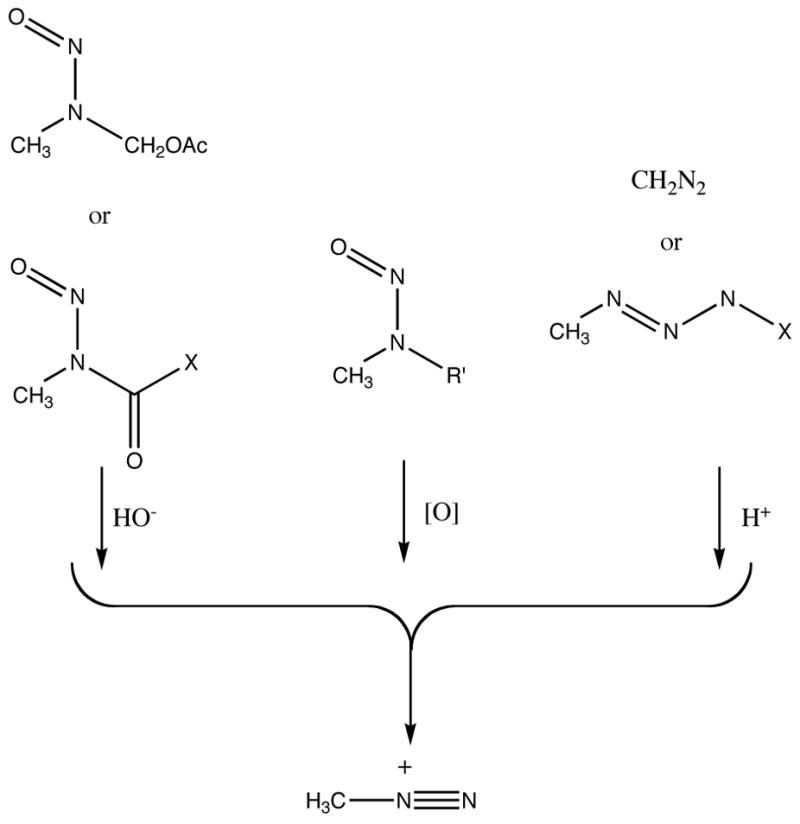
Conversion of α-acyl-N-nitrosamines, N,N-dialkylnitrosoamines and triazenes to a common methanediazonium ion.
3. Sequence selective reactions of alkylating agents with DNA
In general, the quantitation of adducts involves digestion of the DNA to the nucleotide or nucleoside level, and this obliterates information concerning reactivity at specific sequences. In the first studies that provided insight into this question, it was shown that dimethyl sulfate (DMS) treated ds-DNA is uniformly methylated at all N7-G’s (26) (Figure 3). The procedure involves treatment of the methylated DNA with hot piperidine to: (a) selectively open the N-methylimidazolium ring of 7-mG by specific base-catalyzed hydrolysis; (b) remove the aminoformamidiopyrimidine from the DNA backbone by nucleophilic displacement; and (c) specific base-catalyzed hydrolysis of the resulting abasic site to a single strand break (27). The breaks are observable using polyacrylamide gel electrophoresis (PAGE) if the DNA is end-labeled. This process is often referred to as Maxam-Gilbert G-lane chemistry (26, 28). For sequencing proposes, the use of DMS is ideal because it alkylates all G’s to a similar extent and provides a uniform G ladder.
Figure 3.

Methylation patterns at N7-G in 85 bp restriction fragment produced from dimethyl sulfate (DMS) and N-methyl-N-nitrosourea (MNU) in 10 mM Tris-HCl buffer (pH 8.0) in the absence and presence of NaCl, distamycin, spermine or ethidium (32).
The lack of sequence selectivity turns out not to be the case for all methylating agents. It was first noticed by Briscoe and Cotter that the yield of methylated guanines in MNU-treated DNA was sequence-dependent with poly(dG)·poly(dC) being methylated 2-fold more than poly(dG-dC), poly(dA-dC)·poly(dG-dT) and poly(dA-dG)·poly(dC-dT) (29). We showed that MNU, which reacts with DNA via a methanediazonium ion (CH3N2+) intermediate (30), produces a clear sequence dependent methylation pattern (Figures 3, 4) (31). The regiospecificity approaches 10-fold between the most and least reactive G’s within restriction fragment size DNA. To demonstrate that the specificity was due to an interaction of the CH3N2+ with DNA, rather than some intermediate precursor, we substituted in place of MNU a number of compounds that generate CH3N2+, and studied the reaction products. Diazomethane, acetoxymethylmethylnitrosamine and N-methyl-N′-nitro-N-nitrosoguanidine all gave identical 7-G methylation patterns and the same yield of 7-mG (Figure 4) (32). Therefore, the sequence selectivity is due in part to some characteristic of the CH3N2+. The gel fragmentation pattern indicates that a G that is on the 3′-side of another G is generally the most reactive site for CH3N2+ and the order of reactivity is 5′-GG > 5′-AG > 5′-CG > 5′-TG. The 3′-base has a smaller effect and follows the order of GG-3′ > GA-3′ > GT-3′ > GC-3′.
Figure 4.

Methylation patterns at N7-G in the same sequence shown in Figure 3 produced from different concentrations of N-methyl-N-nitrosourea (MNU), N-nitroso(1-acetoxyethyl)methyl-amine (NAEMA), N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) and diazomethane (CH2N2) (32).
Virtually the same alkylation pattern seen with CH3N2+ was also observed for a number of 2-chloroethylating compounds: nitrogen mustards (33), N-nitrosoureas (34, 35) and triazenes (36). In the case of the nitrogen mustards, it was reported that attaching an N-substituted uracil or quinacrine group altered the specificity of the alkylation pattern, presumably due to weak equilibrium binding interactions of the heterocyclic ring of the mustard to selective DNA sequences prior to the alkylation event (37). There are more striking examples of changes in the methylation pattern when an N-methyl-N-nitrosourea functionality is linked to DNA equilibrium binders. Attaching the nitrosourea to a methidium intercalator causes the methylation pattern at N7-G to become uniform (38). In contrast, using a minor groove binding peptide to deliver the alkanediazonium afforded a very significant increase in the amount of N3-alkyladenine relative to free nitrosoureas (39, 40).
There have been numerous efforts to explain and predict DNA alkylation patterns by nitrosoureas. The Pullman’s calculated the electrostatic potential at the different bases and the influence of flanking bases on the potential (19, 41). While their calculations provided the initial insight into the electrostatic environment in the grooves of ds-DNA, the approach was limited in being able to predict alkylation patterns. Klopman et al. attempted, with limited success, to relate the alkylation patterns to effects of base stacking on reaction potentials (42). Another approach to the question was proposed by LeBreton who tried to relate reactivity at different G’s to an increase in p polarizability in the highest HOMO of G that reduces the transition state activation involving charge transfer (43–45). It was calculated that the ionization potential of G, which has the lowest ionization potential of the nucleobases, is decreased when it is in the interior of a hyper-reactive G3–4 stretch. Integration of all of these theoretical studies and other experimental work, including data presented below, are consistent with an electrostatic association of the charged alkylating intermediate that is sequence selective.
The above alkylation studies described above were performed in vitro so an obvious question is whether the same adduct patterns would be observed in vivo since in the latter case the DNA is associated with proteins, including those involved in the formation of nucleosomes. It was demonstrated using ligation-mediated PCR that both MNU and N-nitroso(acetoxymethyl)methylamine afford the same methylation pattern at N7-G in human cell lines as they do in solution (46). This result indicates that in the millions of cells exposed in vivo to alkylating agent, protein occupancy varies at any particular DNA sequence, i.e., in some cells a DNA site may be bound by protein and in others the DNA sequence is unbound. At low doses of alkylating agent, i.e, low levels of DNA modification, the availability of unbound sequences in DNA is in large excess. This does not mean that methylation in a single cell and at a specific sequence will not be affected by the presence of a DNA binding protein. It simply means that within the environment of the nucleus of a cell the factors responsible for the alkylation patterns are the same as in “naked” DNA.
4. Effect of DNA structure and conformation on alkylation reaction
To determine if the sequence-dependent reaction of MNU stemmed from steric influences in the major groove, we studied the alkylation pattern of MNU in single-strand (ss) DNA. The pattern observed in ds-DNA is lost in ss-DNA, and most importantly, the alkylation yield was significantly reduced (Figure 5) (47). In contrast, DMS, which shows no sequence selectivity with either ss- or ds-DNA, reacts more efficiently with the former. The greater reactivity of ds-DNA relative to ss-DNA suggests that steric factors are not important determinants in the sequence selectivity. This indicates that the formation of the double helix from ss-DNA creates a sequence-dependent electronic environment to which some alkylating agents, i.e., those intermediates with cationic character, are sensitive.
Figure 5.

Methylation patterns of N-methyl-N-nitrosourea (MNU) in ss-DNA and ds-DNA, and in ds-DNA with mismatches and bulge sites (47): (a) sequences of oligomers; (b) gel pattern; lane a, G-lane; lane b, G + A; lane c, control; lane d, 1 + 500 μM MNU; lane e, 1 + 2 + 500 μM MNU; 1 + 3 + 500 μM MNU; 1 + 4 + 500 μM MNU; 1 + 5 + 500 μM MNU; 1 + 6 + 500 μM MNU; 1 + 7 + 500 μM MNU; (c) densitometry analysis of gel in (b).
To test whether more subtle disruptions of the canonical Watson-Crick helix would affect the methylation pattern, we reacted MNU with duplex oligomers without and with mismatches at the central G of a G3 target sequence. Consistent with the requirement for a canonical double helix, a G that is mispaired with G, T or A becomes less reactive than a G paired with C (Figure 5) (47). Similarly, the creation of G’s in a single or double bulge show a decrease in alkylation (Figure 5). In all cases the DNA retains the B-conformation based upon the CD spectra, although the base stacking, measured using the negative band at 240 nm, is somewhat reduced in the non-canonical structures. The results show that a non-canonical structure significantly reduces the methylation at the G that is at a mismatch or bulge, while the affect at flanking G’s is relatively small. To obtain further verification that steric factors were not in play in the sequence selectivity of MNU, the DNA was treated with 2,12-dimethyl-3,7,11,17-tetraazobicyclo-[11.3.1]heptadeca-1-[17],2,11,13,15 pentaene-Ni(II) in the presence of KHSO5. This bulky agent is quite sensitive to steric accessibility and has been used to mark non-canonical DNA base pairing motifs (48). The data from these studies confirm that there is no correlation between steric accessibility of the Ni(II)-complex and electrophilic reactivity of MNU. A final set of experiments that show the preference for a duplex is the MNU-methylation of DNA over a temperature range from 0 to 80°C. As the temperature of the reaction is raised and the DNA starts to denature, the pattern disappears and the alkylation intensity decreases.
It has also been reported that the reaction of calf thymus DNA, supercoiled plasmid DNA and synthetic oligomers show similar alkylation pattern when treated in vitro with CH3N2+ (49). Therefore, the topology of the DNA target does not affect reactivity assuming that the DNA retains a B-conformation. There is one study on the methylation of putative Z- and H-DNA by CH3N2+ that reports that the difference in methylation was minimal (50). However, in these studies the Z- and H-conformations of oligomeric inserts in plasmid DNA were induced by topoisomerase I catalyzed relaxation of the DNA in the presence of different concentrations of ethidium bromide. Therefore, it is likely that a mixture of B- and Z- or H-conformations was present in the methylation reactions. Since only a fraction of the DNA is alkylated in the sequence studies, the more reactive conformation, possibly B-DNA, will be preferentially methylated. In the same studies, DMS showed no change in its reactions with putative H-DNA. Clearly, in H-DNA the steric accessibility to major groove atoms should have been significantly impeded since the reduced accessibility to alkylating agents is used in “footprinting” experiments as proof of H-DNA structure (51). Therefore, the effect of conformation on the DNA methylation pattern by charged alkylating intermediates remains unresolved.
5. Effect of salt on DNA alkylation
DNA methylation reactions were carried out in the presence of various inorganic and organic salts and at various salt concentrations to determine the nature of electrostatic interactions between atoms in the major groove and the CH3N2+ intermediate (31, 32). The level of DNA methylation was inversely dependent on salt concentration and the binding affinity of the cation; however, the methylation pattern itself remained unperturbed (Figure 3). Other laboratories observed an inhibitory effect of distamycin A on adduct yields, but attributed the inhibition to its minor groove binding properties (52). However, we showed that the effect was on both major and minor groove products, and that the former inhibition was independent of sequence while the latter occurred at distamycin A equilibrium binding sites (53). The degree of inhibition followed what would be predicted from the equilibrium binding constants of the cations (32). Accordingly, it requires approximately 10 mM MgCl2 to inhibit N7-G methylation by MNU to the same extent as 200 mM NaCl (32). The addition of salt does not affect DNA methylation by DMS (Figure 3) (31).
These studies confirmed that there is an intrinsic sequence dependent electrostatic field in the major groove, irrespective of ionic strength of the buffer or the nature of the diffusible cations present. We have termed this the Sequence-Dependent Electrostatic Landscape (SDEL) of DNA (54). We propose that the SDEL is to a large measure responsible for the sequence-dependent alkylation of DNA by charged intermediates, as well as the equilibrium binding of small cationic molecules to DNA.
6. Alkylation patterns at other positions
Are sequence selective events that occur at N7-G unique? Another DNA lesion generated by MNU is O6-methylguanine (6-mG). Although it is formed at 10% of the level of 7-mG, its biological role in toxicity and mutagenicity is well-documented (17, 55–57). Because 6-mG is relatively stable to chemical approaches to selectively cleave it off DNA, and because it is formed along with the more labile 7-mG adduct, it is not possible to directly and unambiguously sequence 6-mG using standard chemical approaches. By isotopically labeling the individual G’s in a short duplex, two groups have been able to determine the sequence selectivity for 6-mG formation (58, 59). Both studies show that the patterns observed at 7-G and 6-G are similar. Thus, the same electrostatic factors are at work in determining the electrophilic reactivity of the two major groove G atoms that are just under 3 Å apart. The mechanistic factors responsible for the partitioning of the alkanediazonium ion between reaction at N7-G versus O6-G remain unclear, although recently a model involving electrostatics has been suggested (60)
In contrast to the sequence dependency observed in the major groove, N3-methylation of A in the minor groove of restriction fragment length DNA is virtually uniform (49). Only in the presence of minor groove binding ligands does the alkylation pattern become non-uniform, and this coincides with the steric inhibition effect of the bound ligand at specific minor groove sequences.
7. Electrostatic binding of cations to DNA
The factors governing the sequence-selective methylation of DNA at N7-G, and at O6-G, suggest that there is an electrostatic code associated with ds-DNA (the SDEL) that is sensed and covalently marked by CH3N2+ and related alkylating agents. If so, then inorganic cations might be expected to preferentially associate with the same regions. Is there any evidence for this? The B-form helix is a polyanion with characteristic axial charge density. In the theory developed by Manning and others, this results in the ‘condensation’ of diffusible cations in a region proximal to the DNA, with an effective neutralization of approximately 75% of the formal anionic charge (61–63). The theory predicts that this condensation effect is a function of the axial charge density, and is constant over a broad range of bulk cation concentration. This type of electrostatic cation association is not predicted by the theory to exhibit any sequence dependency; the sequence does not change the axial charge density, as confirmed in high resolution NMR and x-ray crystallography studies (61). To date, these cations associated with the phosphate backbone have been infrequently observed using either high resolution structural method.
Monovalent cations are sporadically observed in high resolution x-ray crystal structures of DNA, primarily in the major groove near G:C pairs and in the minor groove near A:T pairs (Figure 6) (54, 64). Cations in the major groove cluster within 3.5 Å of the N7- and O6-positions of G, and in some cases they appear as bifurcated bridges between the two electronegative atoms on the same G or between the atoms on neighboring G’s. The use of Tl+ as a K+ substitute, because of its anomalous scattering, has facilitated the accurate mapping of monovalent cation locations (65). Monovalent Na+, K+, Rb+, Cs+, NH4+ ions and H2O molecules, and even polyamines and divalent cations, compete for similar sites on DNA or RNA (65–67). Various species bind with partial and mixed occupancies in x-ray structures. Na+ and NH4+ scatter x-rays with nearly the same power as water or partially occupied K+. Each of these species has irregular and variable coordination. Therefore the solvent/ion environment can be difficult to fit unambiguously during structure refinement. A well-developed K+ substitute with a distinctive x-ray scattering fingerprint that obviates interpretation of subtle differences in coordination geometry and scattering power is provided by Tl+. Tl+ and K+ have similar ionic radii [K+ = 1.33 Å, Tl+ = 1.49 Å] (68 and enthalpies of hydration [K+ = −77 kcal mol−1, Tl+ = −78 kcal mol−1] (69). Tl+ can substitute for K+ in the catalytic mechanisms of sodium-potassium pumps (70), fructose-1-6-bisphosphatase (71), and pyruvate kinase (72). Tl+ has been shown by NMR to stabilize guanine tetraplexes in a manner analogous to K+ and NH4+ (73). We have used Tl+ as a marker for sites of K+ localization adjacent to B-DNA (65) and DNA–drug complexes (54). Doudna and coworkers used Tl+ as a marker for sites of K+ localization tetrahymena ribozyme P4-P6 domain (74). Tl+ was used by Caspar and coworkers to determine counterion positions adjacent to insulin (75, 76) and by Gill and Eisenberg to determine the location of NH4+ ions in the binding pocket of glutamine synthetase (77).
Figure 6.
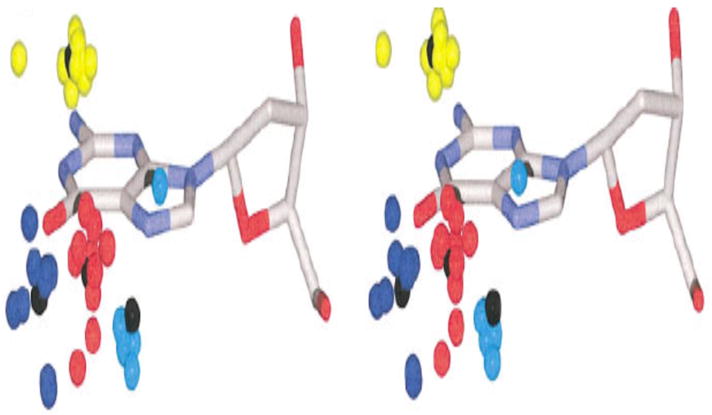
Relaxed stereoview of cation localization at consensus sites obtained from superimposition of guanine residues in the Protein Database that are adjacent to monovalent cations (54). This composite shows that G’s are high occupancy cation binding sites, even for monovalent cations: blue, near O6-G; red, near both O6- and N7-G; cyan, near N7-G; yellow, near both bases in dinucleotide step (generally O6- and N7-G).
The self-complementary Drew-Dickerson dodecamer (DDD) (Figure 9), has been crystallized in the presence of Tl+ (65). The locations of monovalent and divalent cations are depicted in Figure 7. In addition to the location, the x-ray structure also provides information on occupancy of cations at the different sites. The highest occupancy is calculated to be at the C9:G4 pair. One significant caveat of these crystallographic studies is that the DNA sequences studied using high resolution crystallography are quite limited. Structures of the sequences that are hot spots for alkylation (and mutagenesis) have not been determined. Despite this limitation, it is reassuring that the relative occupancies of inorganic cations in the major groove of DDD correlates well with the methylation intensities (Figure 8, bottom right). The methylation data show that the yield of 7-mG at G4 is 25% higher than at G2 and G10, and G4 is associated with the highest Tl+ occupancy derived from x-ray crystallography.
Figure 9.
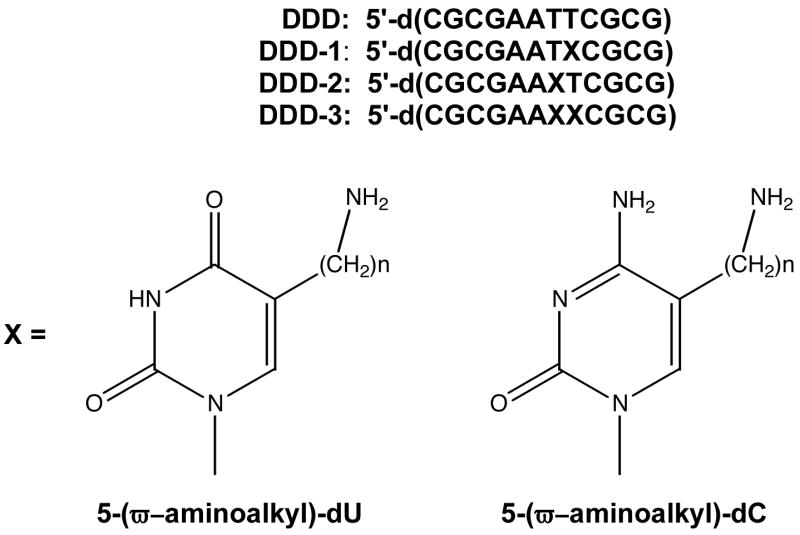
Structures of dodecamer sequences and modified bases used in study (n = 3 or 6).
Figure 7.
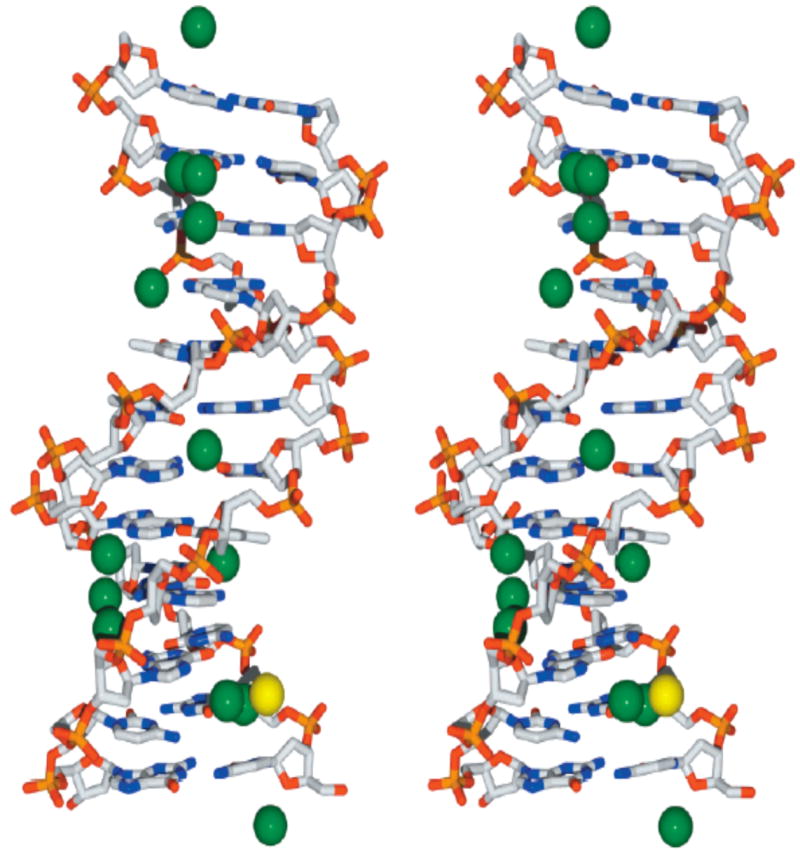
Relaxed stereoview of 5′-d(CGCGAATTCGCG)2 (DDD) and associated Tl+ ions based upon 1.2 Å resolution x-ray data(65). Tl+ ions are depicted by green spheres; the sole Mg2+ ion is depicted by a yellow sphere. The DNA is shown in stick representation with standard CPK color coding of atoms. H2O molecules have been omitted for clarity. For reference the minor groove is in the vertical center of the DNA molecule.
Figure 8.

(a) Methylation pattern observed in unmodified DDD and 3-aminopropyl-dU modified DDD-I and DDD-II. (b) Depiction of position of sidechains based upon the methylation inhibition pattern.
In a series of line broadening NMR studies, Braunlin’s group (78–80) showed that di- and tri- valent ions, such as Mg2+, Ca2+ and Co3+, associate with DNA with distinct sequence preferences related to G/C content. As with the crystallographic results described above, these NMR results are not predicted by the counterion condensation theory of Manning (61) or by the cylindrical Poisson-Boltzmann cell model (81). Braunlin proposed that neighboring G’s might provide favorable divalent cation binding sites via interaction with the major groove N7 and O6-atoms (79). These same G/C sequences have also been associated with cation mediated bending in crystal structures (66, 70, 82–84). It is important to point out that using the same NMR line-broadening technique and 23Na+, the Braunlin group found no evidence for a G/C association preference (85). As mentioned above, monovalent cations are also seldom observed in high resolution crystal structures of DNA, but when they are it is at many of the same regions that divalent cations bind, i.e., at G/C sequences. This is not to say that hydrated divalent cations, and weakly hydrated divalent and monovalent cations uniformly have the same DNA association characteristics.
8. Probing and manipulating the electrostatic potential of DNA
The SDEL appears to modulate or even drive the major groove alkylation pattern, as well as influence cation association. Therefore, experiments were designed to manipulate the electrostatic environment to determine if, and how, DNA alkylation would be affected. This was done by covalently tethering cations at a specific sites using an ω-aminoalkyl sidechain at the 5-position of a pyrimidine base (Figure 9).
The 5-(ω-aminoalkyl)pyrimidines, which can conveniently be introduced into DNA oligomers at specific positions using standard phosphoramidite chemistry, have been previously prepared and used to provide a primary amino group for post-column modification with a number of functionalities, including anisotropy probes (86). Originally, it was assumed that in ds-DNA the terminal ammonium ion in ω-aminoalkylpyrimidines would form a salt bridge with the 5′-non-bridging phosphate oxygen (87–89). However, this conformation appeared to be unlikely based upon molecular simulations and grid search computational approaches that predicted that the alkyl tethered ammonium ion would associate with major groove N7- and O-G sites that were on the 3′-side of the modification (90). Replacement of the ω-amino functionality with an ω-hydroxyl in the simulations reduced the interaction between the atoms on G in the major groove and the sidechain but the sidechain still retained the 3′-orientation. The reaction of MNU with oligomers modified with the 5-(ω-aminoalkyl)-2′-deoxypyrimidines revealed that there was selective inhibition of methylation directed toward the 3′-direction, which is what was predicted from molecular modeling (Figure 8) (91). The site of inhibition of 7-mG formation could be controlled by shortening or lengthening the aliphatic tether between the pyrimidine and the ammonium ion (90). Significantly, the cationic sidechain has virtually no effect on DNA methylation by DMS, which is consistent with electrostatic repulsion of the tethered cation with the attacking CH3N2+ but not with DMS. It is not clear whether the tethered ammonium ion prevents alkylation by electrostatically repulsing the CH3N2+ and/or whether it tightly binds to the N7- and O6-positions on the G’s and sterically blocks the covalent attack. Since DMS is not affected by the presence of the sidechain, the former electrostatic repulsion scenario appears more likely since methylation by DMS would also be sensitive to the presence of the tethered cation stably interacting with N7-G.
9. Localization of major groove charge and DNA structure
In analyzing the alkylation data, it was observed that inhibition of methylation occurs at sites that are too remote from the cationic adduct to be affected by steric or short–range electrostatic interactions in canonical B-DNA (90, 91). The 3-aminopropyl side chain can physically extend approximately 4 Å through space, but inhibition is seen 2–3 bases pairs away, which is equivalent to >8 Å in B-DNA. The 6-aminohexyl sidechain, with its six-atom bridge, can extend ~8 Å above the plane of the pyrimidine ring, yet it influences methylation 3 base pairs away, or >11 Å. The unanticipated electrostatic “reach” of the tethered cations suggested that they deform DNA from the classical B-conformation. There is precedence for the ability of cations to induce changes in DNA structure. The same 5-(ω-aminoalkyl)-2′-deoxypyrimidines, when appropriately phased so that they are on the same face of the double helix with an inherently bent A-tract, cause increased aberrant gel mobility (88, 89). This aberrant mobility is generally interpreted to indicate that the DNA has a static bend or anisotropic flexibility (92). The bending was reduced when the 5-(ω-aminoalkyl)-2′-deoxypyrimidines were orthogonal to the A-tract, and when trans, the bending disappeared. Increasing the salt concentration also reduces bending, which implies an electrostatic role in the bending (88, 89). More recently the incorporation of the same 3-aminopropyl-dU residues were shown to shorten the 5′-3′ length of DNA using fluorescence resonance energy transfer (93). In these studies, two adjacent cationic sidechains were required to observe measurable bending and it was proposed that the cations had a cooperative effect. As mentioned above, divalent cations, which preferentially bind at G rich sequences (77–80), appear to bend DNA at these sequences (82).
There are additional cases where the introduction of charge in the major groove causes dramatic bending. The interstrand crosslinks formed by nitrogen mustards, e.g., mechlorethamine, were predicted to form between the N7-positions of G’s in 5′-GC sequences since the G’s are appropriately spaced to accommodate the bridging -(CH2)2-N(CH3)-(CH2)2- linkage without distorting the B-DNA structure (94). However, it was independently shown by the Loechler and Hopkins labs that the major site for interstrand crosslinking was at N7-G in 5′-GNC sequences despite the fact that the crosslinked DNA is highly distorted (95–98). Distortion is required because the covalent linkage between the two strands is at least 1.4 Å too “short” to accommodate a classical B-DNA conformation (Figure 10). A priori there are two explanations for the unanticipated crosslink sequence specificity: (i) DNA normally adopts an array of non-classical structures and the 5′-GNC crosslink is the kinetically favored product; or (ii) the initial mono-functional mustard adduct stabilizes a conformational perturbation in B-DNA that allows the 5′-GNC crosslink to efficiently form. The first explanation is not in accord with the time-averaged structures on the μs time scale obtained by NMR nor do the static crystal structures, which also do not reveal the presence of highly distorted structures (64). In addition, other techniques such as fluorescence resonance energy transfer (99) and electron paramagnetic resonance (100) also do not indicate the degree of conformational diversity in DNA required for the 5′-GNC crosslink.
Figure 10.
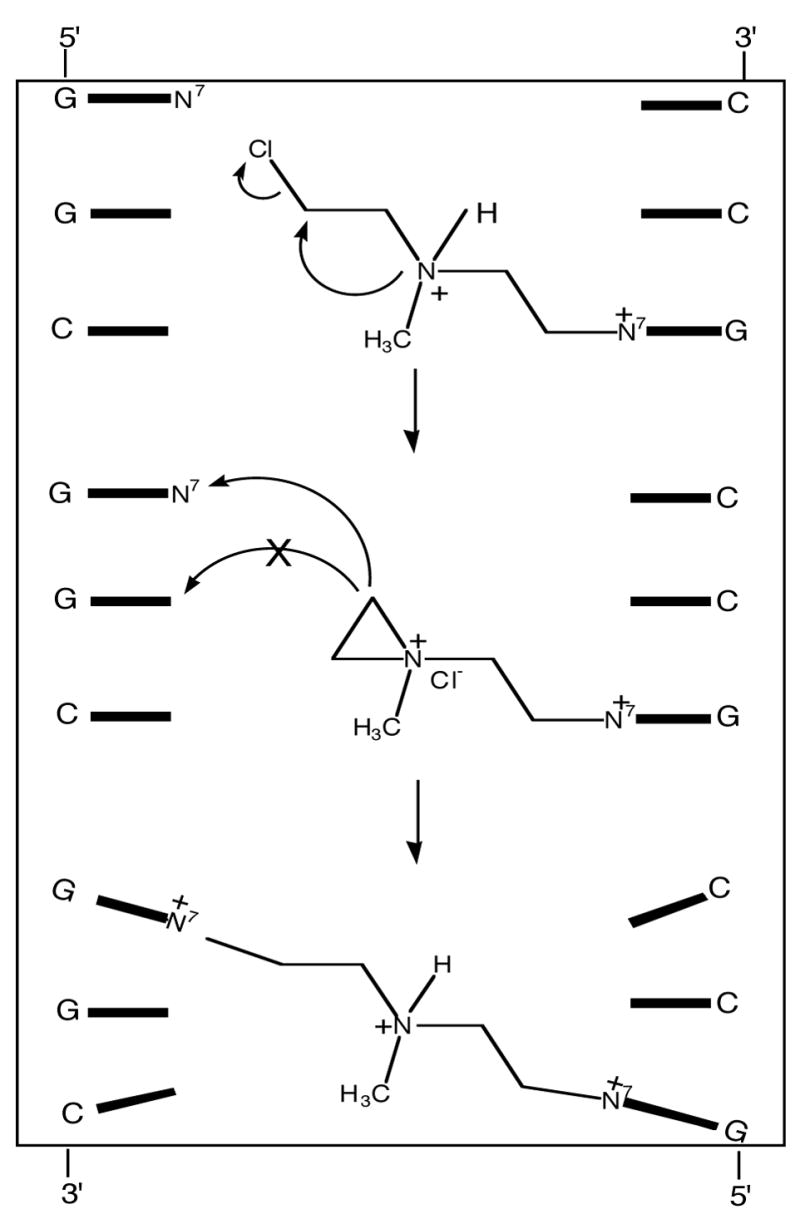
Interstrand crosslinking by mechlorethamine. The monofunctional lesion (top) which has a cationic purine and a cationic sidechain distorts the DNA so the 5′-GNC crosslink forms in favor of the predicted 5′-GC lesion (middle). The distance of the 5′-GNC crosslink is ~ 1.5 A longer than the covalent bridge between the strands; therefore the DNA must be distorted (bottom).
The initial reaction of nitrogen mustards with DNA results in a mono-functional N7-G adduct that has both a cationic purine ring and a ionized amino group on the sidechain appendage (Figure 10). To dissect out the role of the cationic purine on the sequence specificity of DNA interstrand crosslinking, we studied a series of α, ω-alkanediol dimethylsulfonate esters, CH3SO2O-(CH2)n-OSO2CH3 (n = 4–6 or 8) (Figure 11) (101). These compounds, some of which have been clinically used to treat acute myeloid leukemia (102), crosslink DNA via monofunctional cationic N7-alkylG adducts, which do not have cationic sidechains (103). The sequences shown in Figure 11 were treated with one of the sulfonate esters or mechlorethamine. The crosslinked DNA and non-crosslinked DNA were separated on non-denaturing gels. Quantification of the bands gives the relative crosslinking efficiency of sulfonate esters with 6, 5, 8 and 4-carbon linkers as 22 : 6 : 1 : 0, respectively (101). To determine if the preferred crosslink site reflects sequence selective formation of the mono-functional precursor, the locations of the monofunctional lesions were also quantified. The results indicate that the very weak sequence selectivity for monofunctional adduction by the sulfonate esters and the strong specificity for crosslinking do not overlap. Since the linker is too short to span the modification sites of the reactant B-form DNA, the DNA must be distorted in an intermediate state, prior to the closure of the second linkage, so that the two atoms involved in bond formation are close enough to efficiently form a transition state on the crosslinking pathway. The presence of a cationic nucleobase (i.e., N7-alkyl-dG) is sufficient to induce distortion in the DNA so that the GNC crosslink is efficiently formed. This conclusion is consistent with the similar sequence specificity of diepoxybutane crosslinking, which also occurs at N7-G’s in 5′-GNC (104). Therefore, the deformation of the crosslink is independent of the alkylating agent; an initial charged N7-G monofunctional intermediate is sufficient.
Figure 11.
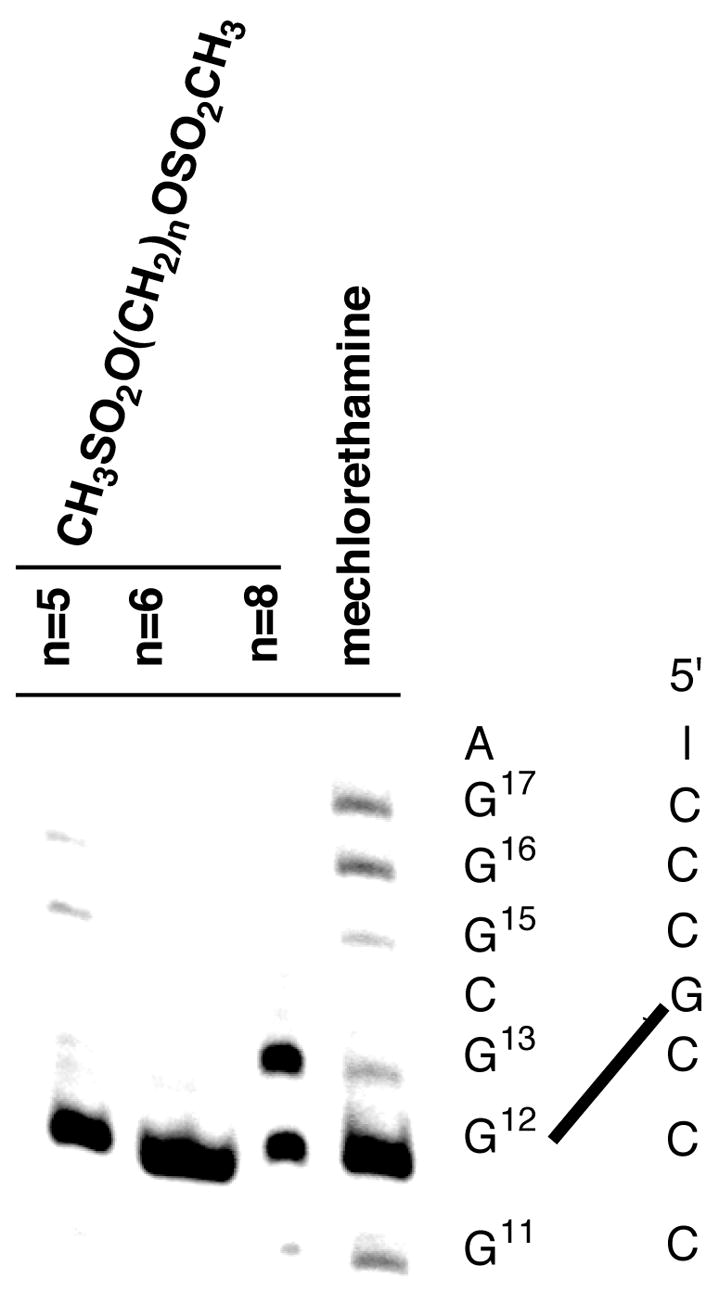
Crosslinking specificity of bis-sulfonates in 5′-[32P]-A2C2TATAT2G3CG3AT2A2 (101). The gel isolated crosslink products are treated with piperidine to generate strand breaks at N7-alkyl guanine lesions. The results indicate the preference for the 5′-GNC crosslink (G-12 band) for n = 5 and 6, but an equal amount of crosslink at 5′-GNC and 5′-GC for n = 8.
10. Structural studies on DNA with tethered cationic sidechains
To understand the affects of tethered cations, such as 5-(ω-aminoalkyl)-2′-deoxypyrimidine, on DNA methylation and conformation, structural studies using both NMR and x-ray crystallography were initiated. We chose the DDD sequence mentioned above since even when chemically modified it frequently affords crystals that provide high resolution information. The first structure analyzed was 5′-d(CGCGAATXCGCG) (DDD-1) where X is 5-(3-aminopropyl)dU. NMR data indicate that the amino moiety is oriented in the 3′-direction from the site of modification (105), which is the same preference seen in the MNU methylation and molecular modeling studies (90, 91). Structures emergent from molecular dynamics calculations in which the amino group was constrained to be proximate to O6-position at G10, on the floor of the major groove, were consistent with experimental NOE’s, and indicated the possibility of axial bending (Figure 12). The interpretation of the data that emerged from electrostatic footprinting (90, 91) and NMR (105) studies posited that, as a consequence of favorable electrostatic interactions, the tethered amine occupied conformational space in the vicinity of O6 and N7 of G10, inducing DNA bending, and enabling the release of a diffusible cation(s) from a high-affinity major groove cation binding site associated with the CGCG tracts of the DDD (65). Indeed, as mentioned above, the preferential binding of monovalent cations within the major groove in CGCG has been substantiated by crystallographic analysis in the presence of Tl+ (Figure 7) (65).
Figure 12.
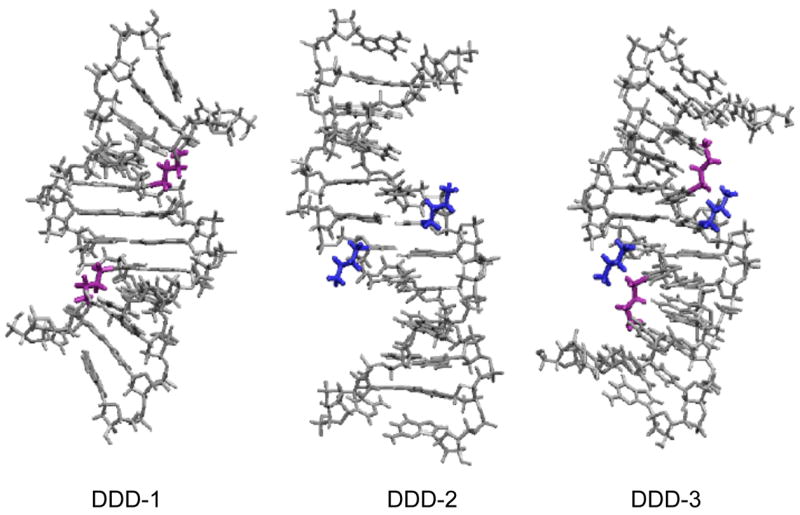
NMR derived structures of DDD-1 (105), DDD-2 and DDD-3: purple sidechains are attached to position X-8; blue sidechains are attached to X-7 (Figure 9).
The model of the tethered cation interacting with a site near G10 is also supported by molecular dynamics simulations of solvated oligodeoxynucleotides with counterions (106–109). Auffinger & Westhof (110) suggested preferential cation binding near the floor of the major groove adjacent to the edges of G·C base pairs close to the O6- and N7-G. These cations exhibited significant residence life-times (ca. 500 ps). Additional NMR studies on DDD-1 involving the measurement of residual H3′-31P dipolar couplings were performed and the data confirm the conformation preference of the 3-aminopropyl sidechain.
11. Effect of cation localization in the major groove on DNA stability and conformation
To fully understand how the sequence specific introduction of cationic charge in the major groove affects local DNA structure, including the organization of cations and water, a detailed thermodynamic characterization of DNA containing the tethered cations was initiated. It was equally important to correlate the thermodynamic effects of the incorporation of charged sidechains with the structural characteristics of similar DNA complexes. For this reason, we used a combination of optical and calorimetry techniques to investigate the helix-coil transition of DDD and DDD-1, and to characterize the counterion and water releases accompanying the unfolding of these duplexes (111). The comparison of the resulting thermodynamic profiles yields the specific energetic, ion and hydration contributions for placing a cationic chain at specific positions in the major groove of the DDD. A comparison of the melting characteristics of DDD and DDD-1 at 260 (unstacking of A:T and G:C pairs) and 275 nm (most sensitive to G:C unstacking) show that the denaturation behavior corresponds to sequential duplex → hairpin, and hairpin → random coil transitions. This is in agreement with earlier unfolding measurements of DDD (112). Placing a 3-aminopropyl-dU residue at X8 drops the TM by ~3 °C relative to DDD. The thermodynamic parameters (Table 1) provide additional insights into the effect that the tethered cation has on the structure. There is an unfavorable ΔΔG° of 3.6 kcal/mol for DDD-1 relative to DDD resulting from a large compensation of an unfavorable ΔΔHcal of 48 kcal/mol with a favorable (ΔTΔScal) term of 44 kcal/mol. The differential enthalpy term indicates differences in base stacking and hydration between the two duplexes, while the differential entropy term is consistent with both a net counterion release of 0.8 mol Na+/mol duplex and a release of 31 mol water/mol duplex. The overall effects are consistent with the tethered amino group directly or indirectly neutralizing charge, perhaps causing some increase in the exposure of aromatic nucleobases to the solvent, as would be anticipated if the DNA was bent. The indirect charge neutralization could occur by the proposed electrostatic mechanism of Rouzina and Bloomfield (113). In this mechanism, the local presence of the cationic aminopropyl chain (rather than divalent cations used in the calculations of Rouzina and Bloomfield) located in the major groove of DNA regiospecifically repels phosphate screening counterions, and the unscreened phosphates collapse toward the propyl tethered ammonium ion (Figure 13). This scenario is consistent with our thermodynamic results and nicely explains how the incorporation of -aminoalkyl side chains can induce DNA bending.
Table 1.
Thermodynamic Profiles for the Formation of DDD, DDD-1, DDD-2 and DDD-3 at 20 °C. (111)
| Duplex | TM (°C) | ΔHcal (kcal/mol) | ΔG°cal (kcal/mol) | TΔScal (kcal/mol) | ΔnNa+ (per/mol) | ΔnW (per/mol) |
|---|---|---|---|---|---|---|
| Duplex → Single Strands | ||||||
| DDD | 33.3 | −116 | −6.9 | −109 | −2.3 | −38 |
| DDD-1 | 29.8 | −68 | −3.3 | −65 | −1.5 | −7 |
| DDD-2 | 37.3 | −53 | −3.7 | −49 | −1.7 | −15 |
| DDD-3 | 25.3 | −50 | −1.8 | −49 | −1.0 | - |
|
| ||||||
| Hairpin → Single Strands | ||||||
| DDD | 62.8 | −37 | −5.3 | −32 | −0.2 | −9 |
| DDD-1 | 64.5 | −27 | −4.0 | −23 | −0.2 | −6 |
All parameters are measured from DSC and UV melting curves in 10 mM sodium phosphate buffer at pH 7.0. The experimental uncertainties are as follow: TM (±0.5 °C), ΔHcal (±3%), ΔG°cal (±5%), TΔScal (±3%), ΔnNa+ (±5%) and ΔnW (±8%).
Figure 13.
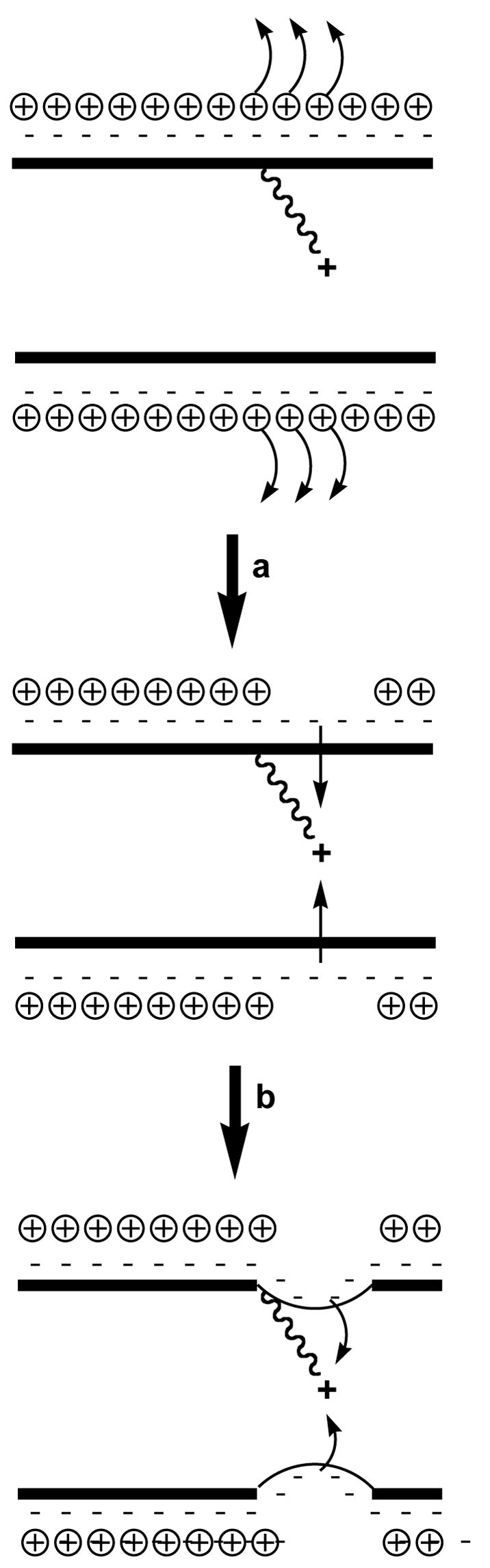
Proposed pathway to explain the bending induced by major groove localized cations: (a) repulsion between tethered NH3+ ion and diffusible cations that screen the non-bridging phosphate; (b) collapse of partially unscreened backbone onto tethered cation (based upon 113).
If the apparent aminopropyl-induced bending of the DNA, to which is attributed the slower gel mobility (88, 89) and methylation inhibition pattern (90, 91), is indeed driven by the tethered ammonium ion moving into a high-affinity binding site in the major groove, then altering the position of the modified base in the dodecamer should modulate the effect. Specifically, relocating the modification from position X8 to position X7 (i.e., 5′-CGCGAAX7TCGCG, DDD-2) should prevent the tethered amine from reaching and displacing a bound cation in the major groove adjacent to N7 and O6 at G10 because of the increased energetic penalty for the distortion of the DNA required for the 3-carbon tether to reach as far G10. It should be noted that this additional one base pair offset is roughly equivalent to a (CH2)3 extension of the aliphatic tether between the base and the amine.
We synthesized DDD-2, and DDD-3 with 5-(3-aminopropyl)dU modifications at X7, and at both X7 and X8, respectively. In contrast to what was observed in the NMR spectra of DDD-1, when located at X8 the cationic moiety in DDD-2 is not positioned toward the floor of the major groove (Figure 12). Furthermore, restrained molecular dynamics calculations yield ensembles of structures suggesting that DDD-2 is not bent. In DDD-3, the moiety located at X8 behaves similar to the X8 sidechain in DDD-1, whereas the aminopropyl moiety located at X7 behaves similar to the X7 sidechain in DDD-2. In DDD-3, bending is proposed based upon the NMR data, which is in agreement with that observed for DDD-1 (Figure 12). Overall, comparison of modified DDD-I, 2 and -3 indicates that only the cation attached via X8 induces bending while placing the cationic moiety at X7 does not.
While the NMR and thermodynamic experiments were being performed, we were able to get diffracting crystals for DDD-3. The results of this work are generally consistent with the NMR picture in that one of the tethered ammonium ions at X8 points down into the groove and approaches G10 (114). The X8 on the complementary strand, which should be identical, points out into solution. In contrast, both of the ammonium ions tethered to X7 point out into solvent (Figure 14). The reason that the structure displays asymmetry is unclear. It may result from forces exerted by the crystal lattice or from an equilibrium between the two predominant DNA conformations: bent with sidechain partially occupying the major groove cation site or linear with the sidechain pointing out into the solvent. Using a combination of modifications that precisely positions cation charge in the major groove, e.g., 7-aminomethyl-7-deazaguanine, or deletes the potential cation binding site near N7 and O6-G, e.g., 7-deazaguanine, we are trying to resolve this question.
Figure 14.
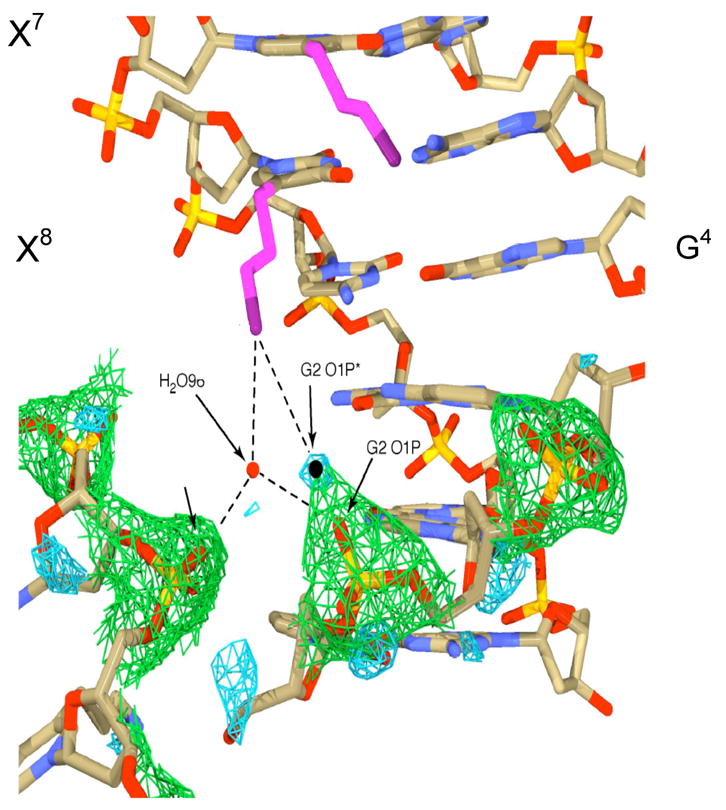
Close-up view of contact between one of the amino propyl groups (in purple) attached to position X8 and the backbone on the same DDD-3 molecule (Figure 9) and with a symmetry related molecule (lower left): this sidechain is near the floor of the major groove while the other three point out into solution (114). The aminopropyl sidechain attached to X7 (upper center) can be seen pointing out from the major groove into solvent near G4.
Using the MNU (CH3N2+) based electrostatic footprinting approach (91), we characterized the position of the tethered ammonium ion in DDD-2 (Figure 8). In comparison to DDD, the location of the modification at X8 (DDD-1) results in a 45% decrease in methylation at G4 and G10 with no significant difference at G2. The data are interpreted to mean that the aminopropyl sidechain in DDD-1 is conformationally flexible and moves between G10 and G4. Attaching the sidechain to X7 (DDD-2) induced a > 50% decrease at G4 with no effect at the other G’s. This is what would be expected if the sidechain in DDD-2 predominantly pointed out into solution, which would situate it in the vicinity of G4. Therefore, the electrostatic footprinting results are consistent with a dynamic sidechain in DDD-1 where the cation moves down into the groove near G2 or extends out into solution near G4. In DDD-2, only the latter occurs.
12. Affect of sequence and cation position on DNA stability
Comparative thermodynamic analysis of DDD, DDD-1, DDD-2 and DDD-3 provided an insight into how the movement of the aminopropyl sidechains was affecting the stability and organization of cations and water in the duplexes. When the aminopropyl group is located at X8 (DDD-1), the TM drops by 3 °C relative to DDD (Table 1) (111), while placing it at X7 (DDD-2) increases the TM by 4 °C relative to DDD. Therefore, moving the 3-aminopropyl-dU:A pair by one position (~3.5 Å) causes a > 7 °C change in TM. The introduction of two modified residues on each strand (DDD-3) causes the TM to decrease by ~8 and 11.5 °C relative to DDD and DDD-2, respectively. DDD-2 yields a similar free energy destabilization as DDD-1 (ΔΔG° = 3.2 kcal/mol), but there is larger enthalpy-entropy compensation (ΔΔHcal = 63 kcal/mol and Δ(TΔScal) = 60), and lower ion (ΔΔnNa+ = 0.6) and H2O (ΔΔnW = 23) release. In DDD-3, where a total of 4 cationic sidechains are present, the differential release of counterions is the only parameter that is additive (1.3 mol Na+/mol duplex versus 1.4 mol Na+/mol duplex); all others parameters have small changes relative to DDD-2. The thermodynamic data demonstrate that the location of the cationic sidechain has a pronounced impact on how it alters DNA stability, and cation and water organization. The thermodynamic observations are consistent with the structural differences seen using NMR and crystallography.
In sequences other than DDD, the thermodynamic effect of the aminopropyl sidechain has also been unpredictable: in some cases stabilizing and in others destabilizing. For example, in the non-complementary duplex, 5′-d(CGTAGXCGTGC)·3′-d(GCACGACTACG), the aminopropyl sidechain stabilizes the duplex by 6 °C (115). Based upon the limited number of sequences studied to date, we tentatively suggest that when the tethered cation can reach a G, the duplex is destabilized, and when it cannot, it is stabilized.
13. Significance
The SDEL provides a predictive and unifying model that reconciles and explains structural, reactivity and thermodynamic data. We believe that we have made a compelling case for the importance of the SDEL in the binding and bonding of cationic molecules (and reactive intermediates) with DNA. In turn, the intrinsic sequence-dependent reactivity toward electrophiles, in part, determines mutation patterns with molecules that react via charged intermediates. Moreover, the structure of the nucleobases and the base pairing schemes in DNA have remained invariant over billions of years of evolution, but the primary sequence and the resulting secondary and tertiary structures have co-evolved with the proteins that are involved in DNA metabolism (116). Whether the creation of sequence-dependent major (and minor) groove cation binding sites is part of the co-evolution process remains to be seen. Clearly basic amino acid sidechains that are introduced into DNA as a result of DNA-protein equilibrium binding would be expected to localize in these high cation occupancy sites due to the favorable enthalpic electrostatic interactions and the entropic release of localized cations. In addition, the formation or localization of cationic charge in the grooves of the double helix, due to covalent bonding or equilibrium binding by small molecules and proteins, can affect DNA topology. The latter is mechanistically linked to how some DNA binding proteins coordinate the transcriptional regulation of genes.
Acknowledgments
The work described was supported by grants from the National Institutes of Health, Department of Health and Human Services (RO1 CA76049 and PO1 CA36727).
Footnotes
Abbreviations: DDD, Drew-Dickerson dodecamer 5′-CGCGAATTCGCG′; DDD-1, 5′-CGCGAATXCGCG (X = 5-[3-aminopropyl]-2′-deoxyuridine]; DDD-2, 5′-CGCGAAXTCGCG; DDD-3, 5′-CGCGAAXXCGCG; DMS, dimethyl sulfate; ds-DNA, double-strand DNA; 7-mG, N7-methylguanine; 6-mG, O6-methylguanine; MNU, N-methyl-N-nitrosourea.
References
- 1.Lawley PD. Carcinogenesis by alkylating agents. In: Searle CE, editor. Chemical Carcinogens. Vol. 1. American Chemical Society; Washington, D.C: 1984. pp. 325–484. [Google Scholar]
- 2.Osborne MR. Carcinogenesis by alkylating agents. In: Searle CE, editor. Chemical Carcinogens. Vol. 1. American Chemical Society; Washington, D.C: 1984. pp. 485–524. [Google Scholar]
- 3.Beranek DT, Weis CC, Swenson DH. A comprehensive quantitative analysis of methylated and ethylated DNA using high pressure liquid chromatography. Carcinogenesis. 1980;1:595–605. doi: 10.1093/carcin/1.7.595. [DOI] [PubMed] [Google Scholar]
- 4.Den Engelse L, Menkveld GJ, De Brij RJ, Tates AD. Formation and stability of alkylated pyrimidines and purines (including imidazole ring-opened 7- alkylguanine) and alkylphosphotriesters in liver DNA of adult rats treated with ethylnitrosourea or dimethylnitrosamine. Carcinogenesis. 1986;7:393–403. doi: 10.1093/carcin/7.3.393. [DOI] [PubMed] [Google Scholar]
- 5.Rogozina IB, Pavlov YI. Theoretical analysis of mutation hotspots and their DNA sequence context specificity. Rev Mutat Res. 2003;544:65–85. doi: 10.1016/s1383-5742(03)00032-2. [DOI] [PubMed] [Google Scholar]
- 6.Lawley PD. Some chemical aspects of dose-response relationships in alkylation mutagenesis. Mutat Res. 1974;23:283–295. doi: 10.1016/0027-5107(74)90102-x. [DOI] [PubMed] [Google Scholar]
- 7.Lovell DP. Dose-response and threshold-mediated mechanisms in mutagenesis: statistical models and study design. Mutat Res. 2000;464:87–95. doi: 10.1016/s1383-5718(99)00169-2. [DOI] [PubMed] [Google Scholar]
- 8.Gao S, Drouin R, Holmquist GP. DNA repair rates mapped along the human PGK1 gene at nucleotide resolution. Science. 1994;263:1438–1440. doi: 10.1126/science.8128226. [DOI] [PubMed] [Google Scholar]
- 9.Tornaletti S, Pfeifer GP. Slow repair of pyrimidine dimers at p53 mutation hotspots in skin cancer. Science. 1994;263:1436–1438. doi: 10.1126/science.8128225. [DOI] [PubMed] [Google Scholar]
- 10.Pfeifer GP. Formation and processing of UV photoproducts: effects of DNA sequence and chromatin environment. Photochem Photobiol. 1997;65:270–283. doi: 10.1111/j.1751-1097.1997.tb08560.x. [DOI] [PubMed] [Google Scholar]
- 11.Warpehoski MA, Hurley LH. Sequence selectivity of DNA covalent modification. Chem Res Toxicol. 1988;1:315–333. doi: 10.1021/tx00006a001. [DOI] [PubMed] [Google Scholar]
- 12.Denissenko MF, Pao A, Tang M, Pfeifer GP. Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in p53. Science. 1996;274:430– 432. doi: 10.1126/science.274.5286.430. [DOI] [PubMed] [Google Scholar]
- 13.Miller EC, Miller JA. Mechanisms of chemical carcinogenesis: nature of proximate carcinogens and interactions with macromolecules. Pharmacol Rev. 1966;18:805– 838. [PubMed] [Google Scholar]
- 14.Lawley PD. Historical origins of current concepts of carcinogens. Adv Cancer Res. 1994;65:17–111. doi: 10.1016/s0065-230x(08)60065-2. [DOI] [PubMed] [Google Scholar]
- 15.Brosch D, Kirmse W. Stereochemistry of nucleophilic displacement on 1- alkanediazonium ions. J Org Chem. 1991;56:907–908. [Google Scholar]
- 16.Agnew TE, Kim HJ, Fishbein JC. Diazonium ion chemistry: replacement of H by alkyl at the central carbon accelerates an SN2 substitution reaction. J Phys Org Chem. 2005;17:483–488. [Google Scholar]
- 17.Loechler EL, Green CL, Essigmann JM. In vivo mutagenesis by O6- methylguanine built into a unique site in a viral genome. Proc Natl Acad Sci USA. 1984;81:6271–6275. doi: 10.1073/pnas.81.20.6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lukash LL, Boldt J, Pegg AE, Dolan ME, Maher VM, McCormick JJ. Effect of O6-alkylguanine-DNA alkyltransferase on the frequency and spectrum of mutations induced by N-methyl-N′-nitro-N-nitrosoguanidine in the HPRT gene of diploid human fibroblasts. Mutat Res. 1991;250:397–409. doi: 10.1016/0027-5107(91)90196-u. [DOI] [PubMed] [Google Scholar]
- 19.Pullman A, Pullman B. Molecular electrostatic potential of the nucleic acids. Quart Rev Biophys. 1981;14:289–380. doi: 10.1017/s0033583500002341. [DOI] [PubMed] [Google Scholar]
- 20.Preussmann R, Stewart BW. Carcinogenesis by alkylating agents. In: Searle CE, editor. Chemical Carcinogens. Vol. 2. American Chemical Society; Washington, D.C: 1984. pp. 643–828. [Google Scholar]
- 21.Preussmann R, Eisenbrand G. Carcinogenesis by alkylating agents. In: Searle CE, editor. Chemical Carcinogens. Vol. 2. American Chemical Society; Washington, D.C: 1984. pp. 829–868. [Google Scholar]
- 22.White EH. The chemistry of the N-alkyl-N-nitrosamides. II. A new method for the deamination of aliphatic amines. J Am Chem Soc. 1955;77:6011–6014. [Google Scholar]
- 23.Park KK, Wisnok JS, Archer MC. Mechanism of alkylation by N-nitroso compounds: detection of rearranged alcohol in the microsomal metabolism of N-nitroso- di-n-propylamine and base-catalyzed decomposition of N-n-propyl-N-nitrosourea. Chem- Biol Interact. 1977;18:349–354. doi: 10.1016/0009-2797(77)90020-5. [DOI] [PubMed] [Google Scholar]
- 24.Morimoto K, Tanaka A, Yamaha T. Reaction of 1-n-propyl-1-nitrosourea with DNA in vitro. Carcinogenesis. 1983;4:1455–1458. doi: 10.1093/carcin/4.11.1455. [DOI] [PubMed] [Google Scholar]
- 25.Hecht SS. DNA adduct formation from tobacco-specific N-nitrosamines. Mutat Res. 1999;424:127–142. doi: 10.1016/s0027-5107(99)00014-7. [DOI] [PubMed] [Google Scholar]
- 26.Maxam AM, Gilbert W. A new method for sequencing DNA. Proc Natl Acad Sci USA. 1977;74:560–564. doi: 10.1073/pnas.74.2.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mattes WB, Hartley JA, Kohn KW. Mechanism of DNA strand breakage by piperidine at sites of N7-alkylguanines. Biochim Biophys Acta. 1986;868:71–76. doi: 10.1016/0167-4781(86)90088-6. [DOI] [PubMed] [Google Scholar]
- 28.Maxam AM, Gilbert W. Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol. 1980;65:499–560. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- 29.Briscoe WT, Cotter LE. DNA sequence has an effect on the extent and kinds of alkylation of DNA by a potent carcinogen. Chem-Biol Interact. 1985;56:321–331. doi: 10.1016/0009-2797(85)90014-6. [DOI] [PubMed] [Google Scholar]
- 30.Spratt TE, Zydowsky TM, Floss HG. Stereochemistry of the in vitro and in vivo methylation of DNA by (R)- and (S)-N-[2H1,3H]methyl-N-nitrosourea and (R)- and (S)-N-nitroso-N-[2H1,3H]methyl-N-methylamine. Chem Res Toxicol. 1997;10:1412–1419. doi: 10.1021/tx970097b. [DOI] [PubMed] [Google Scholar]
- 31.Wurdeman RL, Gold B. The effect of DNA sequence, ionic strength and cationic DNA affinity binders on the methylation of DNA by N-methyl-N-nitrosourea. Chem Res Toxicol. 1988;1:146–147. doi: 10.1021/tx00003a003. [DOI] [PubMed] [Google Scholar]
- 32.Wurdeman RL, Church KM, Gold B. DNA methylation by N-methyl-N- nitrosourea, N-methyl-N′-nitro-N-nitrosoguanidine, N-nitroso(1-acetoxyethyl)methylamine and diazomethane. The mechanism for the formation of N7-methylguanine in sequence- characterized 5′-[32P]-end-labeled DNA. J Am Chem Soc. 1989;111:6408–6412. [Google Scholar]
- 33.Mattes WB, Hartley JA, Kohn KW. DNA sequence selectivity of guanine-N7 alkylation by nitrogen mustards. Nucl Acids Res. 1986;14:2971–2987. doi: 10.1093/nar/14.7.2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gibson NW, Mattes WB, Hartley JA. Identification of specific DNA lesions induced by three classes of chloroethylating agents: chloroethylnitrosoureas, chloroethylmethanesulfonates and chloroethylimidazotetrazines. Pharmac Ther. 1985;31:153– 163. doi: 10.1016/0163-7258(85)90042-7. [DOI] [PubMed] [Google Scholar]
- 35.Hartley JA, Gibson NW, Kohn KW, Mattes WB. DNA sequence selectivity of guanine-N7 alkylation by three antitumor chloroethylating agents. Cancer Res. 1986;46:1943–1947. [PubMed] [Google Scholar]
- 36.Mattes WB, Hartley JA, Kohn KW, Matheson DW. GC-rich regions in genomes as targets for DNA alkylation. Carcinogenesis. 1988;9:2065–2072. doi: 10.1093/carcin/9.11.2065. [DOI] [PubMed] [Google Scholar]
- 37.Kohn KW, Hartley JA, Mattes WB. Mechanisms of DNA sequence selective alkylation of guanine-N7 positions by nitrogen mustards. Biochem Pharmacol. 1988;37:1799–1800. doi: 10.1016/0006-2952(88)90452-2. [DOI] [PubMed] [Google Scholar]
- 38.Konakahara T, Wurdeman RL, Gold B. Synthesis of an N-methyl-N- nitrosourea linked to a methidium analogue and its reactions with [32P]-end-labeled DNA. Biochemistry. 1988;27:8606–8613. doi: 10.1021/bi00423a015. [DOI] [PubMed] [Google Scholar]
- 39.Church KM, Wurdeman RL, Zhang Y, Chen FX, Gold B. N-2- Chloroethyl-N-nitrosoureas covalently bound to non-ionic and monocationic lexitropsin dipeptides. Synthesis, DNA affinity binding characteristics and reactions with [32P]-end- labeled DNA. Biochemistry. 1990;29:6827–6838. doi: 10.1021/bi00481a011. [DOI] [PubMed] [Google Scholar]
- 40.Gold B, Church KM, Wurdeman RL, Zhang Y, Chen F-X. Control over the sequence specificity of DNA alkylation: syntheses and reactions with 32P-end- labeled DNA of N-alkyl-N-nitrosoureas linked to minor groove binding lexitropsins. In: O’Neill I, Chen J, Bartsch H, editors. Relevance to Human Cancer of N-Nitroso Compounds, Tobacco Smoke and Mycotoxins. IARC Scientific Publications, No. 105; Lyon, France: 1991. pp. 439–442. [PubMed] [Google Scholar]
- 41.Lavery R, Pullman A, Pullman B. The electrostatic field of B-DNA. Theoret Chim Acta (Berl) 1982;62:93–106. [Google Scholar]
- 42.Klopman G, Fierson MR, Rosenkrantz HS, McCoy EC. Reaction potentials of the constituent bases of DNA and the effects of base stacking. Cancer Biochem Biophys. 1985;7:275–290. [PubMed] [Google Scholar]
- 43.Kim HS, LeBreton PR. UV photoelectron and ab initio quantum mechanical characterization of valence electrons in Na+-water-2′-deoxyguanosine 5′-phosphate clusters: electronic influence on DNA alkylation by methylating and ethylating carcinogens. Proc Natl Acad Sci USA. 1994;91:3725–3729. doi: 10.1073/pnas.91.9.3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim NS, Zhu Q, LeBreton PR. Aqueous ionization and electron-donating properties of dinucleotides: sequence-specific electronic effects on DNA alkylation. J Am Chem Soc. 1999;121:11516–11530. [Google Scholar]
- 45.Zhu Q, LeBreton PR. DNA photoionization and alkylation patterns in the interior of guanine runs. J Am Chem Soc. 2000;122:12824–12834. [Google Scholar]
- 46.Cloutier JF, Drouin R, Castonguay A. Treatment of human cells with N- nitroso(acetoxymethyl)methylamine: distribution patterns of piperidine-sensitive DNA damage at the nucleotide level of resolution are related to the sequence context. Chem Res Toxicol. 1999;12:840–849. doi: 10.1021/tx990025f. [DOI] [PubMed] [Google Scholar]
- 47.Wurdeman RW, Douskey M, Gold B. DNA methylation by N-methyl-N- nitrosourea: Methylation pattern changes in single- and double-stranded DNA, and in DNA with mismatched or bulged guanines. Nucl Acids Res. 1993;21:4975–4980. doi: 10.1093/nar/21.21.4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen X, Burrows CJ, Rokita SE. Conformation-specific detection of guanine in DNA: ends, mismatches, bulges and loops. J Am Chem Soc. 1992;114:322–325. [Google Scholar]
- 49.Milligan JR, Hirani-Hojatti S, Catz-Biro L, Archer MC. Chem Biol Interact. 1989;72:175–189. doi: 10.1016/0009-2797(89)90026-4. [DOI] [PubMed] [Google Scholar]
- 50.Milligan JR, Skotnicki SM, Archer MC. The reaction of N-methyl-N- nitrosourea with DNA in B and non-B conformations. Carcinogenesis. 1990;11:947–951. doi: 10.1093/carcin/11.6.947. [DOI] [PubMed] [Google Scholar]
- 51.Shimizu M, Hanvey JC, Wells RD. Intramolecular DNA triplexes in supercoiled plasmids. I. Effect of loop size on formation and stability. J Biol Chem. 1989;264:5944–5949. [PubMed] [Google Scholar]
- 52.Rajalakshmi S, Rao PM, Sarma DSR. Studies on carcinogen chromatin DNA interaction: inhibition of N-methyl-N-nitrosourea-induced methylation of chromatin-DNA by spermidine and distamycin A. Biochemistry. 1978;17:4515–4518. doi: 10.1021/bi00614a024. [DOI] [PubMed] [Google Scholar]
- 53.Kelly J, Shah D, Chen FX, Wurdeman R, Gold B. Quantitative and qualitative analysis of DNA methylation at N3-adenine by N-methyl-N-nitrosourea and dimethyl sulfate. Chem Res Toxicol. 1998;11:1481–1486. doi: 10.1021/tx9801763. [DOI] [PubMed] [Google Scholar]
- 54.Howerton SB, Nagpal A, Williams LD. Surprising roles of electrostatic interactions in DNA–ligand complexes. Biopolymers. 2003;69:87–99. doi: 10.1002/bip.10319. [DOI] [PubMed] [Google Scholar]
- 55.Loveless A. Possible relevance of O-6 alkylation of deoxyguanosine to the mutagenicity and carcinogenicity of nitrosamines and nitrosamides. Nature. 1969;223:206–207. doi: 10.1038/223206a0. [DOI] [PubMed] [Google Scholar]
- 56.Glassner BJ, Weeda G, Allan JM, Broekhof JL, Carls NH, Donker I, Engelward BP, Hampson RJ, Hersmus R, Hickman MJ, Roth RB, Warren HB, Wu MM, Hoeijmakers JH, Samson LD. DNA repair methyltransferase (Mgmt) knockout mice are sensitive to the lethal effects of chemotherapeutic alkylating agents. Mutagenesis. 1999;14:339–347. doi: 10.1093/mutage/14.3.339. [DOI] [PubMed] [Google Scholar]
- 57.Shiraishi A, Sakumi K, Sekiguchi M. Increased susceptibility to chemotherapeutic alkylating agents of mice deficient in DNA repair methyltransferase. Carcinogenesis. 2000;21:1879–1883. doi: 10.1093/carcin/21.10.1879. [DOI] [PubMed] [Google Scholar]
- 58.Dolan ME, Oplinger M, Pegg AE. Sequence specificity of guanine alkylation and repair. Carcinogenesis. 1988;9:2139–2143. doi: 10.1093/carcin/9.11.2139. [DOI] [PubMed] [Google Scholar]
- 59.Ziegel R, Shallop A, Jones R, Tretyakova N. K-ras gene sequence effects on the formation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-DNA adducts. Chem Res Toxicol. 2003;16:541–550. doi: 10.1021/tx025619o. [DOI] [PubMed] [Google Scholar]
- 60.Lu X, Heilman JM, Blans P, Fishbein JC. The structure of DNA dictates purine atom site selectivity in alkylation by primary diazonium ions. Chem Res Toxicol. 2005;18:1462–1470. doi: 10.1021/tx0501334. [DOI] [PubMed] [Google Scholar]
- 61.Manning GS. The molecular theory of polyelectrolyte solutions with applications to the electrostatic properties of polynucleotides. Quart Rev Biophys. 1978;2:179–246. doi: 10.1017/s0033583500002031. [DOI] [PubMed] [Google Scholar]
- 62.Record MT, Jr, Anderson CF, Lohman TM. Thermodynamic analysis of ion effects on the binding and conformational equilibria of proteins and nucleic acids: the roles of ion association or release, screening, and ion effects on water activity. Quart Rev Biophys. 1978;2:103–179. doi: 10.1017/s003358350000202x. [DOI] [PubMed] [Google Scholar]
- 63.Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 64.see Protein database (http://www.rcsb.org/pdb).
- 65.Howerton SB, Sines CC, VanDerveer D, Williams LD. Locating monovalent cations in the grooves of B-DNA. Biochemistry. 2001;40:10023–10031. doi: 10.1021/bi010391+. [DOI] [PubMed] [Google Scholar]
- 66.Shui X, McFail-Isom L, Hu GG, Williams LD. The B-DNA dodecamer at high resolution reveals a spine of water on sodium. Biochemistry. 1998;37:8341– 8355. doi: 10.1021/bi973073c. [DOI] [PubMed] [Google Scholar]
- 67.McFail-Isom L, Sines C, Williams LD. DNA structure: cations in charge? Current Op Struct Biol. 1999;9:298–304. doi: 10.1016/S0959-440X(99)80040-2. [DOI] [PubMed] [Google Scholar]
- 68.Brown ID. What factors determine cation coordination numbers. Acta Crystallogr. 1988;B44:545–553. [Google Scholar]
- 69.Wulfsberg G. Principles of Descriptive Inorganic Chemistry. University Science Books; Sausalito, CA: 1991. [Google Scholar]
- 70.Pedersen PA, Nielsen JM, Rasmussen JH, Jorgensen PL. Contribution to Tl+, K+, and Na+ binding of Asn776, Ser775, Thr774, Thr772, and Tyr771 in cytoplasmic part of fifth transmembrane segment in alpha-subunit of renal Na, K- ATPase. Biochemistry. 1998;37:17818–17827. doi: 10.1021/bi981898w. [DOI] [PubMed] [Google Scholar]
- 71.Villeret V, Huang S, Fromm HJ, Lipscomb WN. Crystallographic evidence for the action of potassium, thallium, and lithium ions on fructose-1,6- bisphosphatase. Proc Natl Acad Sci USA. 1995;92:8916–8920. doi: 10.1073/pnas.92.19.8916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Loria JP, Nowak T. Conformational changes in yeast pyruvate kinase studied by 205Tl+ NMR. Biochemistry. 1998;37:6967–6974. doi: 10.1021/bi972454n. [DOI] [PubMed] [Google Scholar]
- 73.Basu S, Szewczak AA, Cocco M, Strobel SA. Direct detection of monovalent metal ion binding to a DNA G-quartet by 205Tl NMR. J Am Chem Soc. 2000;122:3240–3241. [Google Scholar]
- 74.Basu S, Rambo RP, Strauss-Soukup J, Cate JH, Ferre-D’Amare AR, Strobel SA, Doudna JA. A specific monovalent metal ion integral to the AA platform of the RNA tetraloop receptor. Nat Struct Biol. 1998;5:986–992. doi: 10.1038/2960. [DOI] [PubMed] [Google Scholar]
- 75.Badger J, Li Y, Caspar DL. Thallium counterion distribution in cubic insulin crystals determined from anomalous x-ray diffraction data. Proc Natl Acad Sci USA. 1994;91:1224–1228. doi: 10.1073/pnas.91.4.1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Badger J, Kapulsky A, Gursky O, Bhyravbhatla B, Caspar DL. Structure and selectivity of a monovalent cation binding site in cubic insulin crystals. Biophys J. 1994;66:286–292. doi: 10.1016/s0006-3495(94)80795-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gill HS, Eisenberg D. The crystal structure of phosphinothricin in the active site of glutamine synthetase illuminates the mechanism of enzymatic inhibition. Biochemistry. 2001;40:1903–1912. doi: 10.1021/bi002438h. [DOI] [PubMed] [Google Scholar]
- 78.Braunlin WH, Nordenskiöld L, Drakenberg T. The interaction of calcium (II) with DNA probed by 43Ca-NMR is not influenced by terminal phosphate groups at ends and nicks. Biopolymers. 1989;28:1339–1342. doi: 10.1002/bip.360280713. [DOI] [PubMed] [Google Scholar]
- 79.Braunlin WH, Nordenskiöld L, Drakenberg T. A reexamination of 25Mg2+ NMR in DNA solution: site heterogeneity and cation competition effects. Biopolymers. 1991;31:1343–1346. doi: 10.1002/bip.360311111. [DOI] [PubMed] [Google Scholar]
- 80.Braunlin WH, Drakenberg T, Nordenskiöld L. Ca2+ binding environment on natural and synthetic polymeric DNA’s. J Biomolec Struct Dyn. 1992;10:333–343. doi: 10.1080/07391102.1992.10508651. [DOI] [PubMed] [Google Scholar]
- 81.Marcus RA. Calculation of thermodynamic properties of polyelectrolytes. J Chem Phys. 1955;23:1057–1068. [Google Scholar]
- 82.Chiu TK, Dickerson RE. 1 Å Crystal structures of B-DNA reveal sequence-specific binding and groove-specific bending of DNA by magnesium and calcium. J Mol Biol. 2000;301:915–945. doi: 10.1006/jmbi.2000.4012. [DOI] [PubMed] [Google Scholar]
- 83.Sines CC, McFail-Isom L, Howerton SB, VanDerveer D, Williams LD. Cations mediate B-DNA conformational heterogeneity. J Am Chem Soc. 2000;122:11048–11056. [Google Scholar]
- 84.Minasov G, Tereshko V, Egli M. Atomic-resolution crystal structures of B-DNA reveal specific influences of divalent metal ions on conformation and packing. J Mol Biol. 1999;291:83–99. doi: 10.1006/jmbi.1999.2934. [DOI] [PubMed] [Google Scholar]
- 85.Braunlin WH, Xu Q. Hexaamminecobalt(III) binding environments on double-helical DNA. Biopolymers. 1992;32:1703–1711. doi: 10.1002/bip.360321212. [DOI] [PubMed] [Google Scholar]
- 86.Agrawal S. Functionalization of oligonucleotides with amino groups and attachment of amino specific reporter groups. Meth Molec Biol. 1994;26:93–120. doi: 10.1007/978-1-59259-513-6_3. [DOI] [PubMed] [Google Scholar]
- 87.Hashimoto H, Nelson MG, Switzer C. Formation of chimeric duplexes between zwitterionic and natural DNA. J Org Chem. 1993;58:4194–4195. [Google Scholar]
- 88.Strauss JK, Roberts C, Nelson MG, Switzer C, Maher LJ., III DNA bending by hexamethylene-tethered ammonium ions. Proc Natl Acad Sci USA. 1996;93:9515–9520. doi: 10.1073/pnas.93.18.9515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Strauss JK, Prakash TP, Roberts C, Switzer C, Maher LJ., III DNA bending by a phantom protein. Chem Biol. 1996;3:671–678. doi: 10.1016/s1074-5521(96)90135-0. [DOI] [PubMed] [Google Scholar]
- 90.Dande P, Liang G, Chen FX, Roberts C, Switzer C, Gold B. The regioselective effect of zwitterionic DNA substitutions on DNA alkylation: Evidence for a strong sidechain orientational preference. Biochemistry. 1997;36:6024–6032. doi: 10.1021/bi962602u. [DOI] [PubMed] [Google Scholar]
- 91.Liang G, Encell L, Switzer C, Gold B. The role of electrostatics in the sequence selective reaction of charged alkylating agents with DNA. J Am Chem Soc. 1995;117:10135–10136. [Google Scholar]
- 92.Zinkel SS, Crothers DM. DNA bend direction by phase sensitive detection. Nature. 1987;328:178–181. doi: 10.1038/328178a0. [DOI] [PubMed] [Google Scholar]
- 93.Williams SL, Parkhurst LK, Parkhurst LJ. Changes in DNA bending and flexing due to tethered cations detected by fluorescence resonance energy transfer. Nucl Acids Res. 2006;34:1028–1035. doi: 10.1093/nar/gkj498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Brookes P, Lawley PD. Reaction of mono- and difunctional alkylating agents with nucleic acids. Biochem J. 1961;80:496–503. doi: 10.1042/bj0800496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ojwang JO, Grueneberg DA, Loechler EL. Synthesis of a duplex oligonucleotide containing a nitrogen mustard interstrand DNA-DNA interstrand cross- link. Cancer Res. 1989;49:6529–6537. [PubMed] [Google Scholar]
- 96.Grueneberg DA, Ojwang JO, Benasutti M, Hartman S, Loechler EL. Construction of a human shuttle vector containing a single nitrogen mustard interstrand, DNA-DNA cross-link at a unique plasmid location. Cancer Res. 1991;51:2268–2272. [PubMed] [Google Scholar]
- 97.Millard JT, Paucher S, Hopkins PB. Mechlorethamine cross-links deoxyguanosine residues at 5′-GNC sequences in duplex DNA fragments. J Am Chem Soc. 1990;112:2459–2460. [Google Scholar]
- 98.Rink SM, Hopkins PB. A mechlorethamine-induced DNA interstrand cross-link bends duplex DNA. Biochemistry. 1995;34:1439–1445. doi: 10.1021/bi00004a039. [DOI] [PubMed] [Google Scholar]
- 99.Klostermeier D, Millar DP. Time-resolved fluorescence resonance energy transfer: a verstalile tool for the analysis of nucleic acids. Biopolymers. 2002;61:159–179. doi: 10.1002/bip.10146. [DOI] [PubMed] [Google Scholar]
- 100.Okonogi TM, Reese AW, Alley SC, Hopkins PB, Robinson BH. Flexibility of duplex DNA on the submicrosecond timescale. Biophys J. 1999;77:3256–3276. doi: 10.1016/S0006-3495(99)77157-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fan YH, Gold B. The sequence-specificity for DNA interstrand crosslinking by a, -alkanediol dimethylsulfonate esters: Evidence for DNA distortion by the initial monofunctional lesion. J Am Chem Soc. 1999;121:11942–11946. [Google Scholar]
- 102.Galton DAG. Myleran in chronic myeloid leukaemia; results of treatment. Lancet. 1953;264:208–213. doi: 10.1016/s0140-6736(53)90885-x. [DOI] [PubMed] [Google Scholar]
- 103.Ponti M, Souhami RL, Fox BW, Hartley JA. DNA interstrand crosslinking and sequence selectivity of dimethanesulphonates. Br J Cancer. 1991;63:743– 747. doi: 10.1038/bjc.1991.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Millard JT, White MM. Diepoxybutane cross-links DNA at 5′-GNC sequences. Biochemistry. 1993;32:2120–2124. doi: 10.1021/bi00059a034. [DOI] [PubMed] [Google Scholar]
- 105.Li Z, Huang L, Dande P, Gold B, Stone MP. Structure of a DNA duplex containing a nucleotide with a major groove cationic sidechain. J Amer Chem Soc. 2002;124:8553–8560. doi: 10.1021/ja0201707. [DOI] [PubMed] [Google Scholar]
- 106.Auffinger P, Westhof E. Water and ion binding around RNA and DNA (C, G) oligomers. J Mol Biol. 2000;300:1113–1131. doi: 10.1006/jmbi.2000.3894. [DOI] [PubMed] [Google Scholar]
- 107.Hamelberg C, McFail-Isom L, Williams LD, Wilson WD. Flexible structure of DNA: Ion dependence of minor-groove structure and dynamics. J Am Chem Soc. 2000;122:10513–10520. [Google Scholar]
- 108.Hamelberg D, Williams LD, Wilson WD. Influence of the dynamic positions of cations on the structure of the DNA minor groove: Sequence-dependent effects. J Am Chem Soc. 2001;123:7745–7755. doi: 10.1021/ja010341s. [DOI] [PubMed] [Google Scholar]
- 109.McConnell KJ, Beveridge DL. DNA structure: what’s in charge? J Mol Biol. 2000;304:803–820. doi: 10.1006/jmbi.2000.4167. [DOI] [PubMed] [Google Scholar]
- 110.Auffinger P, Westhof E. Water and ion binding around r(UpA)12 and d(TpA)12 oligomers--comparison with RNA and DNA (CpG)12 duplexes. J Mol Biol. 2001;305:1057–1072. doi: 10.1006/jmbi.2000.4360. [DOI] [PubMed] [Google Scholar]
- 111.Shikiya R, Li JS, Gold B, Marky LA. Incorporation of a cationic 3- aminopropyl chain into the Dickerson-Drew dodecamer: Correlation of energetic, structure and hydration. Biochemistry. 2005;44:12582–12588. doi: 10.1021/bi050897i. [DOI] [PubMed] [Google Scholar]
- 112.Marky LA, Blumenfeld KS, Kozlowski S, Breslauer KJ. Salt- dependent conformational transitions in the self-complementary deoxydodecanucleotide d(CGCGAATTCGCG): evidence for hairpin formation. Biopolymers. 1983;22:1247–1257. doi: 10.1002/bip.360220416. [DOI] [PubMed] [Google Scholar]
- 113.Rouzina I, Bloomfield VA. DNA bending by small, mobile multivalent cations. Biophys J. 1998;74:3152–3164. doi: 10.1016/S0006-3495(98)78021-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Moulaei T, Maehigashi T, Lountos G, Komeda S, Watkins D, Stone M, Marky L, Li JS, Gold B, Williams LD. Structure of B-DNA with cations tethered in the major groove. Biochemistry. 2005;44:7458–7468. doi: 10.1021/bi050128z. [DOI] [PubMed] [Google Scholar]
- 115.Soto AM, Kankia BI, Dande P, Gold B, Marky LA. Thermodynamic and hydration effects for the incorporation of a cationic 3-aminopropyl chain into DNA. Nucl Acids Res. 2002;30:3171–3180. doi: 10.1093/nar/gkf430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Trifonov EN. Consensus temporal order of amino acids and evolution of the triplet code. Gene. 2000;261:139–151. doi: 10.1016/s0378-1119(00)00476-5. [DOI] [PubMed] [Google Scholar]