

Abstract
Polyketide synthases (PKSs) catalyze the production of numerous biologically important natural products via repeated decarboxylative condensation reactions. Modular PKSs, such as the 6-deoxyerythronolide B synthase (DEBS), consist of multiple catalytic modules, each containing a unique set of covalently linked catalytic domains. To better understand the engineering opportunities of these assembly lines, the extender unit and acyl carrier protein (ACP) specificity of ketosynthase (KS) domains from modules 3 and 6 of DEBS were analyzed. These studies were undertaken with a newly developed didomain [KS][AT] construct, which lacks its own ACP domain and can therefore be interrogated with homologous or heterologous ACP or acyl-ACP substrates. By substituting the natural methylmalonyl extender unit with a malonyl group, a modest role was demonstrated for the KS in recognition of the nucleophilic substrate. The KS domain from module 3 of DEBS was found to exhibit a distinct ACP-recognition profile from the KS domain of module 6. Based on the above kinetic insights, a hybrid module was constructed ([KS3][AT3][KR5][ACP5][TE]) which displayed substrate recognition and elongation capabilities consistent with the natural module 3 protein. Unlike module 3 however, which lacks a ketoreductase (KR) domain, the hybrid module was able to catalyze reduction of the β-ketothioester product of chain elongation. The high expression level and functionality of this hybrid protein demonstrates the usefulness of kinetic analysis for hybrid module design.
INTRODUCTION
Polyketide natural products exhibit antibiotic, antifungal, anticancer, and other important biological activities.1–5 Polyketide synthases (PKSs) catalyze repeated decarboxylative condensation of coenzyme A (CoA)-derived thioesters to synthesize polyketide compounds. Polyketide synthase modules consist of large polypeptides composed of a unique set of covalently fused enzymatic domains. Together, these domains catalyze one cycle of chain elongation before passing the intermediate to the next module in the assembly line. Chain elongation occurs minimally through the combined action of the β-keto synthase (KS), acyl transferase (AT), and acyl carrier protein (ACP) domains. The growing polyketide chain (electrophile) is first tethered onto the KS via a thioester linkage while the AT captures a nucleophilic β-carboxyacyl-CoA extender unit and transfers it to the phosphopantetheine arm of the ACP domain. Once both the growing chain and the extender units are covalently loaded onto the module, KS-mediated decarboxylative condensation occurs to form a β-ketothioester-ACP intermediate. In addition to these essential domains responsible for chain elongation, optional tailoring domains including ketoreductase (KR), dehydratase (DH), and enoylreductase (ER) domains control the final oxidation state of the β-carbon. Once processed, the polyketide chain is either passed to the KS domain of the downstream module or cyclized and released by the thioesterase (TE) domain at the C-terminus of the PKS. A thorough quantitative understanding of substrate specificity and the modular organization of PKSs will greatly enhance the progress in protein engineering for the biosynthesis of novel natural product analogs.6,7
The 6-deoxyerythronolide B synthase (DEBS) from Saccharopolyspora erythraea is a well-studied, modular PKS responsible for production of 6-deoxyerythronolide B (6dEB), the polyketide portion of the antibiotic erythromycin.8,9 The assembly of 6dEB is mediated by three large polypeptides (DEBS1–3), which together append six methylmalonyl extender units onto the propionate starter unit (Figure 1). The TE domain at the C-terminus of DEBS3 then cyclizes the heptaketide into the final macrolide aglycone product, 6dEB. Although each DEBS module has traditionally been regarded as a single structural and functional unit (Figure 2), recent limited proteolysis studies revealed two highly conserved interdomain boundaries, at which individual modules could be further disconnected to yield catalytically functional PKS subunits. Taking advantage of one of these boundaries (amino acid sequence YRVXW), it is possible to express standalone [KS][AT] didomain proteins.10 By separating the [KS][AT] didomain from the ACP domain, the substrate specificity of both KS-catalyzed acylation and condensation can now be interrogated independently in this deconstructed PKS module system.11 Alternative polyketide chain extenders can be readily introduced onto ACP domains by direct modification of apo-ACP domains using the Sfp-catalyzed phosphopantetheinyl transferase reaction. Furthermore, different combinations of [KS][AT] didomains and individual ACP domains from different modules can be examined to explore interdomain recognition (Figure 3).
Figure 1.

Modular organization of 6-deoxyerythronolide B synthase (DEBS). Each catalytic domain is represented by a block diagram. KS: ketosynthase; AT: acyl transferase; ACP: acyl carrier protein; DH: dehydratase; ER: enoyl reductase; KR: ketoreductase; and TE: thioesterase. KR0 denotes an inactive ketoreductase domain. The phosphopantetheine prosthetic group of ACP is drawn as a curly line. Inter-protein linkers are shown as matching tabs.
Figure 2.

Chain elongation cycle catalyzed by DEBS module 3 + TE (M3TE). AT is acylated with a methylmalonyl extender unit from its CoA-derivative, which gets transferred to the downstream ACP. KS is primed with a 2-methyl-3-hydroxy pentanoyl unit from diketide compound 1. Condensation takes place in the active site of KS with the release of carbon dioxide. TE catalyzes lactone formation with release of the final product.
Figure 3.

The deconstructed module system: chain elongation performed by [KS][AT] didomain and standalone ACP. Non-cognate nucleophile can also be introduced via acyl phosphopantetheinylation of apo-ACPs catalyzed by Sfp enzyme.
This report describes our efforts toward determining (a) tolerance and specificity of KS domains for non-cognate nucleophilic substrates, (b) tolerance and specificity of KS domains for heterologous ACP domains, and (c) the effect of C-terminal peptide linkers on polyketide chain elongation. Based on these insights we describe a new approach for engineering hybrid PKS modules based upon kinetic analysis of the substrate and ACP preferences of individual KS domains (Figure 4), as well as the influence of interdomain linkers within a PKS module.
Figure 4.
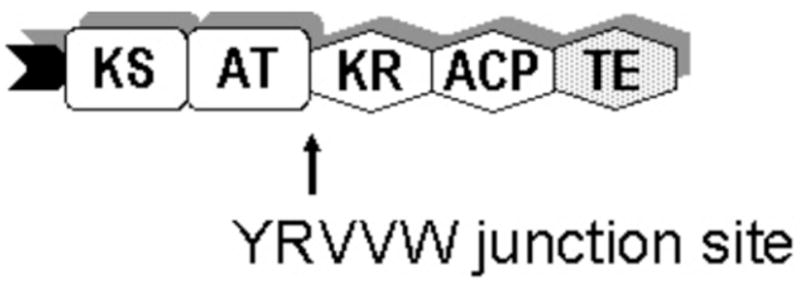
The hybrid module containing [KS3][AT3] and [KR5][ACP5][TE].
RESULTS
The deconstructed module system described by Kim et al enabled us to directly examine nucleophilic substrate preference and KS-ACP pairing specificity in chain elongation.10 The previously characterized [KS][AT] didomain protein from DEBS module 3 was chosen for the study.10, 11 The [KS6][AT6] didomain was used for comparison because DEBS module 6 has similar domain organization to DEBS module 3, and both KS3 and KS6 are known to process diketide 1.12
Expression and purification of recombinant [KS][AT] didomains and ACPs
[KS3][AT3] and [KS6][AT6] were expressed as soluble proteins at a yield of 20 mg purified protein per liter culture. Similarly, the ACP domains from all six DEBS elongation modules were expressed as soluble standalone proteins at a yield of 10 mg purified protein per liter culture. Alternative forms of ACP2, ACP3, and ACP4 were also constructed to include a C-terminal peptide linker, and were designated ACP2(2), ACP3(2), and ACP4(4), respectively. They were expressed as soluble proteins at a yield of 10 mg purified protein per liter culture (Figure 5). The malonyl- and methylmalonyl-ACP3 and ACP6 were analyzed via ESI mass spectrometry to confirm loading of the malonyl and methylmalonyl units.
Figure 5.

Proteins used in the assays (left to right): [KS3][AT3], [KS6][AT6], ACP1(0), ACP2(0), ACP3(0), ACP4(0), ACP5(0), ACP6(0), ACP2(2), ACP3(2), ACP4(4), M3TE, and M3M5TE.
Extender unit preference of ketosynthase domain
Although it is well understood that, in a PKS module, the AT domain serves as the principal gatekeeper for the loading of individual extender units, it remains unknown whether the KS domain is also sensitive to the structure of the chain elongation co-substrate. A better understanding of the specificity and tolerance of the KS domain for variations in chain extender structure would aid in the rational engineering of AT domain substitutions. Previous qualitative studies have shown that the KS domain of module 3 can catalyze condensation between a bound acyl chain and either a methylmalonyl or malonyl extender unit bound to the cognate ACP domain.10 Here we quantified the extender unit preference of KS domains from both DEBS module 3 and module 6 for their natural as well as unnatural nucleophilic substrates. In both cases, the respective malonyl- or methylmalonyl substrate was loaded onto the natural ACP partner, and the steady-state kinetics of formation of the corresponding triketide ketolactone products were analyzed using a radio-TLC assay. The condensation velocities for each combination are tabulated in Table 1. As might be expected, the methylmalonyl extender unit exhibited a higher rate of condensation than the alternative malonyl nucleophilic substrate. This difference suggests that in addition to the strict substrate specificity of the AT domain for its respective methylmalonyl- or malonyl-CoA substrate, the DEBS KS domain has a modest preference for its natural methylmalonyl chain extender unit.
Table 1.
Velocity of [KS][AT]-catalyzed chain elongation against methylmalonyl- and malonyl-ACP.
| Condensation Velocity
(μM/min) |
Relative % | |
|---|---|---|
| [KS3][AT3] | ||
| Methylmalonyl-ACP3 | 0.37 | 100 % |
| Malonyl-ACP3 | 0.10 | 27 % |
| [KS6][AT6] | ||
| Methylmalonyl-ACP6 | 0.042 | 100 % |
| Malonyl-ACP6 | 0.029 | 70 % |
Specificity of [KS][AT] for alternative ACPs in the condensation reaction
Chain elongation involves recognition of both the electrophilic and nucleophilic substrates as well as effective protein-protein interaction between the KS and ACP domains of a given module. By varying the ACP partner while holding the substrates constant, we are able to probe the KS-ACP interaction in condensation. To quantify the specificity of [KS3][AT3] and [KS6][AT6] didomains from DEBS for alternative ACPs, the steady-state formation of triketide ketolactone product was analyzed via radio-TLC assays. Because the KS domain was pre-acylated with the diketide and the AT-catalyzed CoA → AT → ACP acyl transfer reactions are fast compared to KS-catalyzed chain elongation, the observed rate of triketide formation should reflect the rate of condensation13 (supporting information). Figure 6A presents the rate of [KS3][AT3]-catalyzed chain elongation as a function of ACP concentration for each of the six DEBS ACPs. The steady-state kinetic parameters are tabulated in Table 2A. As might be expected, the specificity of [KS3][AT3], indicated by the kcat/Km parameter, is the highest for ACP3 (0.011 μM−1min−1). The kcat/Km values for ACP4 and ACP5 are within ten-fold of that for ACP3, with most of the penalty due to increases in Km. In contrast, although the kcat for ACP2 is reduced by a factor of 5 compared to ACP3, the observed kcat/Km for ACP2 is 30% that of ACP3, reflecting an offsetting decrease in Km. The ability of [KS3][AT3] to use either ACP1 or ACP6 was most severely compromised, with a 500~1000-fold reduction in kcat/Km. Similar experiments were conducted for [KS6][AT6] in combination with each of the six DEBS ACPs (Figure 6B and Table 2B). As expected, ACP6 is the best partner for [KS6][AT6] (kcat/Km = 0.0019 μM−1min−1). Of the heterologous ACPs, ACP5 is a moderate substrate, whereas the others are more severely compromised, both with respect to kcat/Km and maximum rate. Notably, while modules 3 and 6 exhibit predominantly mutually exclusive ACP domain preferences, both KS3 and KS6 can catalyze condensation using methylmalonyl-ACP5. The observed range of catalytic efficiencies indicates a significant sensitivity of KS-catalyzed chain elongation to the structure of the ACP domain carrying the nucleophilic chain extension substrate.
Figure 6.

(A) [KS3][AT3]-and (B) [KS6][AT6]-catalyzed chain elongation velocity as a function of ACP concentration for each of the six ACP domains from DEBS.
Table 2.
Kinetic parameters of (A) [KS3][AT3]- and (B) [KS6][AT6]-catalyzed chain elongation for each of the six DEBS ACPs. In the cases where condensation was too slow to determine kcat, kcat/Km values were derived from the highest [ACP] (200 uM) time course data assuming Km,ACP ≫ 200 uM.
| A
| ||||||
|---|---|---|---|---|---|---|
| ACP1 | ACP2 | ACP3 | ACP4 | ACP5 | ACP6 | |
| Km (μM) | N/A | 9 | 16 | 179 | 56 | N/A |
| kcat (1/min) | N/A | 0.03 | 0.18 | 0.21 | 0.16 | N/A |
| Relative kcat # | N/A | 17% | 100% | 118% | 87% | N/A |
| kcat/Km (1/min-μM) × 1000 | 0.014 | 3.2 | 11.0 | 1.2 | 2.8 | 0.025 |
| Relative kcat/Km * | 0.1% | 30% | 100% | 11% | 25% | 0.2% |
|
B
| ||||||
| ACP1 | ACP2 | ACP3 | ACP4 | ACP5 | ACP6 | |
|
| ||||||
| Km (μM) | 16 | 31 | N/A | 119 | 37 | 27 |
| kcat (1/min) | 0.005 | 0.012 | N/A | 0.008 | 0.021 | 0.050 |
| Relative kcat # | 9% | 23% | N/A | 16% | 41% | 100% |
| kcat/Km (1/min-μM) × 1000 | 0.28 | 0.39 | 0.0055 | 0.07 | 0.56 | 1.85 |
| Relative kcat/Km * | 15% | 21% | 0.3% | 4% | 30% | 100% |
(kcat)x / (kcat)homologous ACP
(kcat /Km)x / (kcat /Km)homologous ACP
Effect of the DEBS C-terminal Peptide Linker on the [KS][AT]-Catalyzed Condensation
In a natural PKS system, the KS domain must interact specifically with both intermodular upstream and intramodular downstream ACP domains in order to effect polyketide chain transfer and elongation. Earlier studies have shown that the specific interactions of the C- and N-terminal peptide linkers of adjacent PKS polypeptides play a critical role in intermodular polyketide chain transfer.14,15 In this study we tested the specificity of [KS3][AT3] for ACP2 and ACP3, each fused to the C-terminal DEBS1 (module 2) linker, as well as ACP4 fused to the C-terminal DEBS2 (module 4) linker. The corresponding ACP constructs were denoted ACP2(2), ACP3(2), and ACP4(4), respectively. The kcat/Km values of ACP2(2) and ACP3(2) decreased relative to their linker-less counterparts, displaying both lower kcat and increased Km (Figures 7A, 7B, Table 3). ACP4(4) displayed a kcat/Km value comparable to that of ACP4, although both kcat and Km were reduced (Figure 7C, Table 3).
Figure 7.

[KS3][AT3]-catalyzed chain elongation velocity as a function of ACP concentration for two forms of (A) ACP2, (B) ACP3, and (C) ACP4. Solid line: ACP without C-terminal linker; dashed line: ACP with C-terminal linker.
Table 3.
Kinetic parameters of [KS3][AT3]-catalyzed chain elongation for ACP2, 3, and 4 with C-terminal linkers. In the ACP2(2) case where condensation was too slow to determine kcat, kcat/Km values were derived from the highest [ACP] (200 uM) time course data assuming Km,ACP2(2) ≫ 200 uM.
| ACP2(2) | ACP3(0) | ACP3(2) | ACP4(4) | |
|---|---|---|---|---|
| Km (μM) | N/A | 16 | 32 | 60 |
| kcat (1/min) | N/A | 0.18 | 0.16 | 0.08 |
| Relative kcat # | N/A | 100% | 88% | 45% |
| kcat/Km (1/min-μM) × 1000 | 0.031 | 11.0 | 4.9 | 1.4 |
| Relative kcat/Km * | 0.2% | 100% | 45% | 12% |
(kcat)x / (kcat)ACP3
(kcat /Km)x / (kcat /Km)ACP3
Recombinant Hybrid Module M3M5TE
A hybrid module was constructed in which ACP5 was chosen to replace ACP3 based on its observed compatibility with the [KS3][AT3] didomain. The replacement was achieved by swapping [KR5][ACP5] and [KR30][ACP3] using the YRVVW junction sequence between AT and KR.10 A TE domain was included at the C-terminus of the protein. The hybrid module M3M5TE was expressed as soluble protein at an unexpectedly high yield of 20 mg purified protein per liter culture, comparable to the wild-type M3TE (Figure 5). To evaluate the catalytic performance of the hybrid protein, its domain acylation and chain elongation activities were first examined using M3TE and the [KS3][AT3] + ACP5 system as references.
KS Acylation Activity of M3M5TE
In polyketide chain elongation, the KS domain of a module receives the growing polyketide chain as an acylthioester attached to either an upstream ACP or a surrogate N-acetylcysteamine. We evaluated both the ability of the KS3 domain in the hybrid module M3M5TE to be acylated by [14C]-1, as well as the extent of back-transfer of KS-bound diketide to a stand-alone ACP domain. Radio-SDS PAGE and phosphorimaging showed that the KS3 in M3M5TE was indeed acylated by diketide 1 (Figure 8A lane 1), and could also transfer the diketide unit to ACP2(2) (Figure 8A lane 2). The KS-mediated ACP acylation, as the backward reaction of KS acylation, indicated the ability of KS in receiving a growing chain from the particular ACP.
Figure 8.

(A) Radio SDS-PAGE: [14C]Compound 1 labeling of hybrid module M3M5TE and ACP2(2). (B) Radio SDS-PAGE: [14C]Methylmalonyl-CoA labeling of M3M5TE and ACP5. (C) Radio TLC: Triketide lactone formation by M3M5TE. Lane 1, positive control reaction (module 3+TE, [14C]methylmalonyl-CoA, compound 1); lane 2, chain elongation by M3M5TE ([14C]methylmalonyl-CoA, compound 1); lane 3, chain elongation and reduction by M3M5TE ([14C]methylmalonyl-CoA, compound 1, NADPH).
AT Activity of M3M5TE
The AT domain of a DEBS module first captures the methylmalonyl-CoA extender unit and then transfers the methylmalonyl unit to the phosphopantetheinyl side chain of the ACP in the same module. To confirm the acylation and transacylation activities of AT3 in M3M5TE, the hybrid protein was incubated with either radiolabeled methylmalonyl-CoA alone or with methylmalonyl-CoA plus ACP5. Radio-SDS PAGE and phosphorimaging showed that the AT3 in M3M5TE was indeed acylated by methylmalonyl-CoA (Figure 8B lane 1) and could also transfer the methylmalonyl unit to the separate ACP5 domain (Figure 8B lane 2).
Condensation and Reduction Activities of M3M5TE
The hybrid module was incubated with unlabeled 1 and radiolabeled methylmalonyl-CoA to test its condensation capability, with or without cofactors NADPH. M3TE was used in parallel to produce triketide ketolactone standard. A radio-TLC assay was used to detect the formation of triketide condensation products. In the absence of cofactors, the hybrid module was indeed able to generate the expected triketide ketolactone (Figure 8C, lanes 1 and 2). Since DEBS KR5, unlike KR3, is an active ketoreductase, when the cofactors are present in the condensation reaction, the hybrid module was observed to produce both triketide ketolactone and its reduced β-hydroxy analogue (Figure 8C, lane 3). A time course was conducted to determine the condensation kinetics of the hybrid module relative to the intact module M3TE. The results are tabulated in Table 4, with the hybrid module achieving 70% of the original condensation velocity.
Table 4.
Velocity of hybrid module M3M5TE-catalyzed chain elongation.
| Condensation Velocity
(μM/min) |
Relative % | |
|---|---|---|
| holoM3M5TE | 0.044 | 70 % |
| holoM3TE | 0.063 | 100 % |
DISCUSSION
The use of separate [KS][AT] and ACP domains simplifies individual PKS domains relative to the properties of domains within an intact module. Domain acylation and ketide-unit elongation can now be studied as discrete catalytic events, and detailed substrate specificity can be examined for individual catalytic steps. Analytical techniques such as tandem proteolysis-mass spectrometry may also be simplified with smaller proteins relative to their larger counterparts. Finally, genetic manipulations can be used to generate new combinations of PKS domains originating from different modules, using either the YRVXW motif downstream of the AT (or DH) domain or the RLAGL motif upstream of the ACP domain. Earlier studies demonstrated the stability and functionality of DEBS [KS][AT] didomains constructed using the conserved YRVXW site. Preliminary evidence also indicated a preferred specificity for KS–ACP pairing in KS-catalyzed condensation. In this report we examined the extender unit preference of the KS domains from DEBS modules 3 and 6, determined ACP specificity of the two KS domains, and constructed a fully functional hybrid module accordingly.
Much prior effort has gone into bypassing the intrinsic substrate specificity of the AT domain, which plays a critical gatekeeper role in polyketide chain elongation.13,16–18 By using separate AT and ACP domains and directly loading the malonyl- or methylmalonyl chain extension unit on the ACP domain, it was possible to examine the extender unit specificity of the KS-catalyzed condensation reaction independent of the kinetics of acylation of both the AT and KS domains. At comparable acyl-ACP concentrations, the condensation rate in the presence of malonyl-ACP was 27–70% that of the natural methylmalonyl-ACP for both KS3 and KS6. Although both KS3 and KS6 are able to catalyze condensation with a non-cognate extender unit with only relatively modest kinetic penalties, it is possible that specific AT domain substitutions could result in comparable decreases in overall rates of polyketide chain elongation. Identification of critical active-site residues on the KS may provide a means to overcome such eventual kinetic bottlenecks.
Most studies to date of polyketide chain elongation have utilized KS and ACP domains from the same PKS module, leaving the role of KS–ACP protein-protein recognition unexplored. Using discrete [KS][AT] didomains, we can now probe a variety of protein–protein recognition elements to identify potential pitfalls in the engineering of catalytically efficient hybrid PKS modules. For both DEBS modules 3 and 6, the [KS][AT] didomain exhibits the highest specificity toward its cognate ACP domain, with varying degrees of kinetic attenuation toward heterologous ACPs, indicating that KS-catalyzed decarboxylative condensation is relatively insensitive to the structure of the nucleophilic malonyl- or methylmalonyl component while reflecting a clear specificity for the structure of the ACP to which it is attached. These observations suggest that inappropriate combinations of KS domains and nucleophile-carrying ACP domains can result in catalytically inefficient or even inactive hybrid PKS modules. By contrast, the intermodular transfer of polyketides from upstream ACP(n-1) to acceptor KS(n) domains has been shown to be relatively insensitive toward ACP structure, provided that the acceptor and donor domains carry a complementary pair of C-terminal and N-terminal linker peptides (docking domains), respectively.14,19,20
In addition to the range of observed kcat/Km values, the data also reveal that there is no simple correlation between relative kcat/Km and relative kcat values, due to offsetting variations in Km. For example, for the [KS3][AT3] domain, the kcat value for methylmalonyl-ACP4 (0.21 min−1) is 7 times higher than that of ACP2 (0.03 min−1), while the corresponding kcat/Km values show a 3:1 preference for ACP2. A difference in the relative order of kcat and kcat/Km values was also observed for the [KS6][AT6] didomain with ACP1 and ACP4. One might potentially overcome the higher Km of ACP4 while exploiting the higher kcat by incorporating ACP4 and KS3 into a single hybrid module.
The presence of a C-terminal peptide linker on a methylmalonyl-ACP domain in general decreases both the observed catalytic efficiency (kcat/Km) and the Km of the [KS][AT]-catalyzed condensation reaction. Possible explanations for the deleterious effect of the peptide linker are (a) the attached peptide linker might disrupt the normal interaction of the ACP with either the AT or KS domain, or both, affecting methylmalonyl transfer and/or condensation, or (b) the presence of the peptide linker may better position the ACP for electrophilic acyl transfer of an attached polyketide to the KS active site Cys than for condensation with the nucleophilic methylmalonyl residue. The small effect observed for ACP4 vs. ACP4(4) may reflect an inherently poor interaction with the didomain, independent of the structure of the appended linker peptide.
M3M5TE Hybrid
Previously described hybrid PKS modules have frequently exhibited low levels of protein expression levels as well as low PKS catalytic activity, presumably due to suboptimal choices of domain boundaries or the intrinsic incompatibility of the domain components.13 Taking advantage of the conserved YRVXW junction between the AT and KR domains, we constructed the first hybrid module in which the choice of heterologous domains was guided by experimental insight into both PKS domain compatibility and optimization of kinetic parameters. The [KS3][AT3] didomain was covalently linked to the [KR5][ACP5][TE] fragment of DEBS module 5+TE. The fully functional hybrid protein, M3M5TE, displayed expression levels as high as those observed for DEBS M3TE (~20 mg purified protein/L culture). The chimeric M3M5TE converted diketide to a mixture of both the triketide ketolactone and the expected reduced product, triketide hydroxylactone, presumably due to the competition between TE-mediated cyclization and KR reduction. The hybrid module retained 70% condensation activity relative to the intact module M3TE, consistent with the relative kcat value (89%) of methylmalonyl-ACP5 in combination with the [KS3][AT3] didomain. These results illustrate the utility of the YRVXW junction sequence as a handle for PKS engineering and present a strong kinetic correlation between intact and deconstructed PKS modules.
To improve engineering capabilities in modular PKS systems, extensive efforts have focused on determining domain specificity, relevant boundaries and precise mechanisms of interdomain communication. Recent studies on domain structure and interdomain junctions have greatly increased our understanding of how each domain should be defined. As more functional domains are isolated and tested among PKS modules, we anticipate significant strides toward practical combinatorial polyketide biosynthesis.
MATERIALS AND METHODS
Reagents and Chemicals
DL-[2-Methyl-14C]-methylmalonyl-CoA was from American Radiolabeled Chemicals. [14C](2S,3R)-2-Methyl-3-hydroxypentanoyl-N-acetylcysteamine thioester (1) was prepared by custom synthesis by Amersham Pharmacia. Unlabeled diketide 1 was prepared by established methods.21–23 All other chemicals were from Sigma. Thin-layer chromatography (TLC) plates were from J. T. Baker. SDS PAGE-gradient gels (4–15% acrylamide) were from BioRad. Ni-NTA affinity resin was from Qiagen. HiTrap-Q anion exchange column was from Amersham Pharmacia.
Methods
Standard recombinant DNA and protein manipulations were carried out according to published procedures as previously described.10 The sequence of all newly prepared expression plasmid constructs was verified directly.
Expression and Purification of [KS][AT] didomain from DEBS modules 3 and 6
For the expression of the KS-AT didomain from DEBS module 3, pAYC02 was constructed as follows. A fragment from the natural BsiWI site to the Tyr-Arg junction site was amplified by PCR using primers 5′ – GCCGTACGGCGTGGAGCCCGCGGCGGTCGTGG – 3′ and 5′ – GCGCGGTAGGCCAGCTCGTCGGAGGCGGCGGC – 3′. The PCR product was cloned into commercially available blunt-end vector pCR-BluntII-TOPO. The resulting intermediate plasmid was digested with BsiWI and EcoRI and cloned into the corresponding restriction sites in pRSG34 containing pET21 expression vector and DEBS module 3 + TE.15 The final plasmid, pAYC02, encodes a C-terminal His6-tagged [KS3][AT3] didomain ending at the Tyr-Arg junction site. The [KS][AT] didomain from DEBS module 6 was similarly constructed. A fragment from the natural AatII site to the Tyr-Arg junction site was amplified using primers 5′ – GCGGGTCGACGTCGTACAGCCGGTGTTGTT – 3′ and 5′ – ACGCGGTAGCGGCTGTCGGCGAGCTGGT – 3′ and cloned into pCR-BluntII-TOPO. The resulting intermediate plasmid was digested with AatII and EcoRI and cloned into the corresponding restriction sites in pRSG54 containing pET21 expression vector and DEBS module 6 + TE.15 The final plasmid, pAYC11, encodes a C-terminal His6-tagged [KS6][AT6] didomain ending at the Tyr-Arg junction site. Plasmids pAYC02 and pAYC11 were transformed into E. coli BL21(DE3) for protein expression. Cells were grown at 37 °C in LB medium with 100 μg/mL carbenicillin to an OD600 = 0.6, at which point they were cooled to 18 °C and induced with 0.2 mM IPTG for 20 h. The cells were harvested by centrifugation (4,500g, 15 min) and resuspended in lysis/wash buffer (75 mM phosphate, pH 7.6, 300 mM NaCl, 10 mM imidazole). Cells were lysed with sonication (5 × 1 min) and cellular debris was removed by centrifugation (17,000g, 50 min). Nickel-NTA agarose resin was added directly to the supernatant (2 mL resin/L culture) and mixed for 3 h at 4 °C. This resin was poured into a fritted column, washed with 10 resin volumes of lysis/wash buffer, and eluted with 3 resin volumes of elution buffer (150 mM phosphate, pH 7.6, 50 mM NaCl, 150 mM imidazole). The eluted protein was applied to a HiTrap-Q anion exchange column and eluted at approximately 370 mM NaCl.
Expression and Purification of ACPs
The DNA sequence encoding DEBS ACP1 was amplified by PCR as an NdeI-EcoRI fragment using primers 5′-AAAAAACATATGGCGCTGGCGTCGCTGCCCGCG - 3′ and 5′-TTTTGAATTCACGAGTTCGGCGGCCAGGTGCGC - 3′. This NdeI-EcoRI fragment was cloned into pET28a expression vector to yield plasmid pAYC43, encoding DEBS ACP1 with N- and C-terminal His6-tags. The DNA sequence encoding DEBS ACP5 was similarly amplified using primers 5′-AAAAAACATATGCGGCTCGCGGCGCTGTCGACG - 3′ and 5′-TTTTGAATTCACGAGCCGCTCCAGGTAGTGGTC - 3′. The NdeI-EcoRI fragment was cloned into pET28a to yield plasmid pAYC38, encoding DEBS ACP5 with N- and C-terminal His6-tags. The DNA sequence encoding DEBS ACP3 with DEBS1 linker subsequent to ACP2, denoted ACP3(2), was constructed as follows. The ACP domain was amplified by PCR as an NdeI-SpeI fragment using primers 5′-AAAAAACATATGCGGCTCGCGGGGCTTTCC - 3′ and 5′-TTTTTTACTAGTGGCGTCACCGACGAGCCGGGC - 3′. This NdeI-SpeI fragment was cloned into the pET21-derived pKW210 vector upstream of the region encoding the DEBS1 (module 2) C-terminal peptide linker to yield plasmid pNAS3, encoding DEBS ACP3(2) with N- and C-terminal His6-tags.
The construction of genes encoding ACP2 (pNW7), ACP2(2) (pNW6), ACP3 (pVYA05), ACP4 (pNW9), ACP4(4) (pNW8), and ACP6 (pPK223) has been previously described.10,19 All nine plasmids were transformed into E. coli BAP1 for holo-ACP protein expression. The E. coli BAP1 cell line contains a plasmid encoding the Sfp phosphopantetheinyl transferase and was used to effect phosphopantetheinylation of ACP.24 Apo-ACP3 and ACP6 were expressed in E. coli BL21 cells. Cells were grown at 37 °C in LB medium with 50 μg/mL kanamycin or 100 μg/mL carbenicillin to an OD600 = 0.6, at which point they were cooled to 18 °C and induced with 0.2 mM IPTG for 20 h. The cells were harvested by centrifugation and disrupted via sonication. ACPs were purified using nickel affinity chromatography then HiTrap-Q anion-exchange column. The apo-ACPs then underwent in vitro treatment with either methylmalonyl-CoA or malonyl-CoA substrates in the presence of recombinant Sfp (600 μM apo-ACP, 60 μM Sfp, 2 mM CoA substrate, 10 mM MgCl2, and 100 mM phosphate at pH 7.2), yielding methylmalonyl- or malonyl-ACP.
Expression and Purification of DEBS Module 3 + TE (M3TE) and hybrid module [KS3][AT3][KR5][ACP5] + TE (M3M5TE)
The construction of genes encoding M3TE (pRSG34) has been previously described.15 For the expression of the hybrid module containing KS3, AT3, KR5, ACP5, and TE from DEBS, denoted M3M5TE, pAYC46 was constructed using the gene SOEing method.25 First round PCR was used to produce two fragments, one encoding module 3 from the natural BsiWI site to the Tyr-Arg junction site (using primers 5′ – GCCATATGGTGGCGCTCGTACGGCGTGGAGCCC – 3′ and 5′ – GCTTCCCGCCATACGACGCGGTAGGCCAGCTCG – 3′), and another encoding module 5 from the Val-Val-Trp junction site to the natural BbvCI site with an EcoRI site added (using primers 5′ – CGAGCTGGCCTACCGCGTCGTATGGCGGGAAGC – 3′ and 5′ – GCGAATTCAGCTCCTCCCTCAGCTCGGACGCGC – 3′). The two PCR products, with 33-base-pair overlap, then primed each other in a second round of PCR, generating a BsiWI-EcoRI fragment. The fragment was digested with BsiWI and EcoRI and cloned into the corresponding restriction sites in pRSG34, sewing the module 3-derived porton of the hybrid to the PCR bridging fragment. The derived plasmid was digested with BbvCI and EcoRI and cloned into the corresponding restriction sites in pRSG46 containing pET21 and DEBS module 5 + TE15, thereby introducing the module 5 component of the chimeric module. The final plasmid pAYC46, along with pRSG34, was transformed into E. coli BAP1, and hybrid holoM3M5TE and holoM3TE proteins were expressed and purified similarly to the KS-AT didomain constructs.
Acylation of Ketosynthase domain in M3M5TE by [14C]-diketide 1
HoloM3M5TE (20 μM, in 100 mM phosphate at pH 7.2) was incubated with 50 μM [14C]-1 and 5 mM tris-2-carboxyethyl-phosphine (TCEP) for 2 h at room temperature. Samples were quenched with SDS-PAGE loading buffer lacking any reducing agents and loaded directly onto a SDS-PAGE gel. The gel was dried using a BioRad gel-drying system and analyzed using a phosphorimager.
Back Transfer of [14C]-diketide 1 from Ketosynthase of M3M5TE to standalone ACP
HoloM3M5TE (20 μM, in 100 mM phosphate at pH 7.2) was incubated with 50 μM [14C]-1 and 5 mM TCEP for 1 h at room temperature. HoloACP2(2) (105 μM, in 100 mM phosphate at pH 7.2) was then added to the reaction mixture and incubated for an additional hour. Samples were quenched and processed as described above.
Acylation of Acyl Transferase domain in M3M5TE by [14C]Methylmalonyl-CoA
HoloM3M5TE (20 μM, in 100 mM phosphate at pH 7.2) alone or holoM3M5TE together with holoACP5 (20 and 150 μM, respectively, in 100 mM phosphate at pH 7.2) were incubated on ice with 50 μM [14C]methylmalonyl-CoA and 5 mM TCEP for 10 min. Samples were quenched and processed as described above.
Radioactive Triketide Lactone Formation
The reaction volume was 10 μL for all single time point assays. For nucleophile preference of KS in condensation: [KS][AT] (20 μM, in 100 mM phosphate at pH 7.2) was incubated with 500 μM [14C]-1 for 1 h to acylate the KS domain. Malonyl- or methylmalonyl-ACP (200 μM) were then added and allowed to react at room temperature for 20, 40, or 60 min. At each time point the reaction was quenched by adding 20 μL of 0.5 M potassium hydroxide and heating the mixture for 20 min at 65°C. Hydrochloric acid (10 μL of 1.5 M) was then added, and the mixture was dried in a Speed-vac for 2 hours. The pellet was resuspended in 10 μL of ethyl acetate and spotted onto a TLC plate. A 60:40 mixture of ethyl acetate/hexane was used for radioactive TLC, with visualization and quantitation using a Packard phosphorimager. Turnover numbers were determined from the radioactivity of the triketide products. For ACP specificity of [KS][AT] in condensation: [KS][AT] (2 μM, in 100 mM phosphate at pH 7.2) was incubated with 5 mM unlabeled 1 and 5 mM TCEP for 1 h to acylate KS to completion. HoloACP (four ACP concentrations ranging from 2 to 200 μM) and 200 μM DL-[2-methyl-14C]methylmalonyl-CoA was then added and allowed to react at room temperature for 10, 20, 30, 40, 60, or 90 min. At each time point the reaction was quenched and processed as described above. For M3M5TE Triketide Formation: HoloM3M5TE or holoM3TE (2 μM, in 100 mM phosphate at pH 7.2) was incubated with 5 mM unlabeled 1, 5 mM TCEP, and 200 μM DL-[2-methyl-14C]methylmalonyl-CoA for 10, 20, 30, or 180 min. In the assay testing the KR5 domain of holoM3M5TE, NADPH (200 μM) was added to the mixture described above. Reactions were quenched and processed as described.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH (CA 66736 to C.K. and GM 22172 to D.E.C.). A.Y.C. is a recipient of a Stanford Graduate Fellowship, and N.A.S. is a recipient of an NIH postdoctoral fellowship (5F32GM 072293-02).
References
- 1.Cane DE, Walsh CT, Khosla C. Science. 1998;282:63–68. doi: 10.1126/science.282.5386.63. [DOI] [PubMed] [Google Scholar]
- 2.Khosla C, Gokhale RS, Jacobsen JR, Cane DE. Annu Rev Biochem. 1999;68:219–253. doi: 10.1146/annurev.biochem.68.1.219. [DOI] [PubMed] [Google Scholar]
- 3.Staunton J, Weissman KJ. Nat Prod Rep. 2001;18:380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 4.Walsh CT. ChemBioChem. 2002;3:124–134. [Google Scholar]
- 5.Walsh CT. Science. 2004;303:1805–1810. doi: 10.1126/science.1094318. [DOI] [PubMed] [Google Scholar]
- 6.Donadio S, Sosio M. Combinatorial Chem High Throughput Screening. 2003;6:489–500. doi: 10.2174/138620703106298671. [DOI] [PubMed] [Google Scholar]
- 7.Kumar P, Khosla C, Tang Y. Meth Enzymol. 2004;388:269–293. doi: 10.1016/S0076-6879(04)88023-6. [DOI] [PubMed] [Google Scholar]
- 8.Cortes J, Haydock SF, Roberts GA, Bevitt DJ, Leadlay PF. Nature. 1990;348:176–178. doi: 10.1038/348176a0. [DOI] [PubMed] [Google Scholar]
- 9.Donadio S, Staver MJ, McAlpine JB, Swanson SJ, Katz L. Science. 1991;252:675–679. doi: 10.1126/science.2024119. [DOI] [PubMed] [Google Scholar]
- 10.Kim CY, Alekseyev YV, Chen AY, Tang Y, Cane DE, Khosla C. Biochemistry. 2004;43:13892–13898. doi: 10.1021/bi048418n. [DOI] [PubMed] [Google Scholar]
- 11.Schnarr NA, Chen AY, Cane DE, Khosla C. Biochemistry. 2005;44:11836–11842. doi: 10.1021/bi0510781. [DOI] [PubMed] [Google Scholar]
- 12.Wu N, Kudo F, Cane DE, Khosla C. J Am Chem Soc. 2000;122:4847–4852. [Google Scholar]
- 13.Hans M, Hornung A, Dziarnowski A, Cane DE, Khosla C. J Am Chem Soc. 2003;125:5366–5374. doi: 10.1021/ja029539i. [DOI] [PubMed] [Google Scholar]
- 14.Tsuji SY, Wu N, Khosla C. Biochemistry. 2001;40:2317–2325. doi: 10.1021/bi002462v. [DOI] [PubMed] [Google Scholar]
- 15.Gokhale RS, Tsuji SY, Cane DE, Khosla C. Science. 1999;284:482–485. doi: 10.1126/science.284.5413.482. [DOI] [PubMed] [Google Scholar]
- 16.Stassi DL, Kakavas SJ, Reynolds KA, Gunawardana G, Swanson S, Zeidner D, Jackson M, Liu H, Buko A, Katz L. Proc Natl Acad Sci. 1998;95:7305–7309. doi: 10.1073/pnas.95.13.7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lau J, Fu H, Cane DE, Khosla C. Biochemistry. 1999;38:1643–1651. doi: 10.1021/bi9820311. [DOI] [PubMed] [Google Scholar]
- 18.Kato Y, Bai LQ, Xue Q, Revill WP, Yu TW, Floss HG. J Am Chem Soc. 2002;124:5268–5269. doi: 10.1021/ja0127483. [DOI] [PubMed] [Google Scholar]
- 19.Wu N, Cane DE, Khosla C. Biochemistry. 2002;41:5056–5066. doi: 10.1021/bi012086u. [DOI] [PubMed] [Google Scholar]
- 20.Wu N, Tsuji SY, Cane DE, Khosla C. J Am Chem Soc. 2001;123:6465–6474. doi: 10.1021/ja010219t. [DOI] [PubMed] [Google Scholar]
- 21.Cane DE, Kudo F, Kinoshito K, Khosla C. Chem Biol. 2002;9:131–142. doi: 10.1016/s1074-5521(02)00089-3. [DOI] [PubMed] [Google Scholar]
- 22.Jacobsen JR, Hutchinson CR, Cane DE, Khosla C. Science. 1997;277:367–369. doi: 10.1126/science.277.5324.367. [DOI] [PubMed] [Google Scholar]
- 23.Harris RC, Cutter AL, Weissman KJ, Hanefeld U, Timoney MC, Staunton J. J Chem Res. 1998;283:1230–1247. doi: 10.1016/s1074-5521(98)90666-4. [DOI] [PubMed] [Google Scholar]
- 24.Walsh CT, Gehring AM, Weinreb PH, Quadri LEN, Flugel RS. Curr Opin Chem Biol. 1997;1:309–315. doi: 10.1016/s1367-5931(97)80067-1. [DOI] [PubMed] [Google Scholar]
- 25.Horton RM. Mol Biotechnol. 1995;3:93–99. doi: 10.1007/BF02789105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.