

Abstract
The oral rexinoid bexarotene (Targretin) is widely used for treatment of cutaneous T-cell lymphomas (CTCL). We recently reported the first case of adult T-cell leukemia/lymphoma (ATLL) that responded rapidly to combination therapy of bexarotene and interferon (IFN)-α2b with complete clinical response. We demonstrated that bexarotene induced apoptosis of the patient's malignant peripheral blood T-cells in vitro. However, our patient developed skin and nodal relapse 180 days after starting treatment. We now demonstrate that his peripheral blood malignant T cells became resistant to bexarotene-induced apoptosis. We investigated potential mechanisms that may cause aberrations in the retinoid X receptor (RXR) subunits, RXR-α and RXR-β, to account for these findings. Sequence analysis did not reveal acquisition of mutations in the genes encoding RXR-α and RXR-β by resistant cells. We assessed RXR-α and RXR-β expression by Western blot analysis and found that resistant cells had significantly decreased RXR-α expression compared with pretherapy bexarotene-sensitive cells. Our findings indicate that reduced expression of the RXR-α receptor subunit may represent a mechanism for resistance to bexarotene in T-cell malignancies.
Introduction
Bexarotene is a third-generation novel retinoid X receptor (RXR)-selective retinoid (“rexinoid”) that is FDA-approved for the treatment of cutaneous T-cell lymphoma (CTCL).1 When used as a single agent in advanced refractory CTCL, oral bexarotene has a total response rate of 45% to 71%1,2 with a duration of response from 3 to 17 months. Similar to other vitamin A derivatives such as all-trans retinoic acid, bexarotene is known to induce apoptosis and affect cellular proliferation, differentiation, and cytokine production, although the precise molecular mechanisms remain unknown.3–6 This multitude of effects is likely because the RXRs, a family of nuclear hormone receptors, not only participate in retinoid signaling pathways as coregulators of retinoic acid receptors (RARs), but also form heterodimers with at least 20 other nuclear receptors.7
Recently we confirmed the apoptotic effects of bexarotene and showed a dose-dependent effect of bexarotene in vitro on malignant peripheral blood T cells from patients with Sézary syndrome, a leukemic variant of mycosis fungoides/CTCL. However, the malignant T cells of one-third of patients tested remained consistently resistant to bexarotene-induced apoptosis.6 The mechanism behind clinical and in vitro bexarotene resistance has not previously been reported. We sought to investigate bexarotene's apoptotic effects and RXR expression levels in a case of human T-lymphotropic virus (HTLV)-1–associated adult T-cell leukemia/lymphoma (ATLL) that initially responded dramatically to bexarotene and interferon- (IFN)-α2b, but became clinically resistant after 180 days.
Case report
We previously reported a case of a 48-year-old man who presented with several months of diffuse papular erythroderma (total body erythema), fatigue, and chills.8 Skin biopsies revealed CD4+ CTCL. He had bulky axillary and pelvic lymphadenopathy which was confirmed by computed tomographic scan. Furthermore, he had peripheral blood involvement with an elevated white blood cell count of 18 100/μL (47% polys, 42.4% lymphocytes, 10.1% mononuclear, 0.5% eosinophils, 0.1% basophils, absolute lymphocytes at 7700/μL), an elevated CD4:CD8 ratio of 7.4 (normal < 4:1), aberrant peripheral blood CD4+CD7− and CD4+CD26− T cell populations at 60% and 62%, respectively (normal populations typically < 10% of total T cells). Additional laboratory abnormalities included an elevated serum lactate dehydrogenase (LDH) level of 836 U/L (normal, 313-618 U/L) and positive serology for HTLV-1 antibody by both enzyme-linked immunosorbent assay (ELISA) and Western blot analysis.
Given the clinical and laboratory information, a diagnosis of ATLL was made. The patient started taking oral bexarotene at a dose of 150 mg daily and subcutaneous injections of interferon (IFN)–α2b at a dose of 1.5 million units 3 times a week. After 3 weeks of therapy, he had a near complete clinical response, with resolution of his papular erythroderma and his lymphadenopathy as well as normalization of his LDH and CD4:CD8 ratio. Extracorporeal photopheresis (ECP) and zidovudine were added to his regimen.
A recurrence of his diffuse erythematous rash with extensive lymphadenopathy occurred 180 days later. Bone marrow biopsy at time of relapse showed a hypercellular marrow but no involvement of lymphoma. A fine-needle aspirate of a submental lymph node showed an atypical lymphoid proliferation consistent with relapse of his ATLL with probable large cell transformation. Six cycles of cyclophosphamide, doxorubicin, prednisone, and vincristine (CHOP) chemotherapy was initiated. There was a partial response in skin and blood and a complete response in the lymph nodes. Unfortunately, the patient experienced recurrent disease 5 months later. The patient was restarted on ECP and IFN-γ was added. Denileukin diftitox and total skin electron beam were initiated 2 months later. Carboplatin, etoposide, and ifosfamide (ICE) and bortezomib were added as additional therapy. The patient died 18 months after initial diagnosis secondary to sepsis, dehydration, and hypercalcemia.
Methods
Patient
The patient was initially seen and followed in the CTCL clinic in the Department of Dermatology and the Division of Hematology and Oncology at the Hospital of the University of Pennsylvania. Blood samples were obtained in conformity with an Institutional Review Board protocol approved by the University of Pennsylvania. Informed consent was obtained in accordance with the Declaration of Helsinki.
Peripheral blood mononuclear cell isolation and culture
Peripheral blood mononuclear cells (PBMCs) were isolated twice, before initiating therapy and again at the time of disease recurrence at 180 days. Venous blood was collected into heparinized syringes following uniform standards. The heparinized blood was diluted 1:2 with phosphate-buffered saline (PBS), pH 7.2, and layered on a Ficoll-Hypaque gradient (Lymphoprep; Accurate Chemical, Westbury, NY). The gradient was centrifuged at 800g for 30 minutes at room temperature and the PBMC band was collected and washed twice in PBS.
Cells were cultured in medium (RPMI 1640; Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen) and penicillin/streptomycin at 37°C in 24-well plates.
Apoptosis assays
PBMCs were plated at a density of 2 × 106/mL per well with medium alone, bexarotene (10 μM; provided as a generous gift from Ligand Pharmaceuticals, San Diego, CA), IFN-α2b (1000 U/mL; Roferon A; Roche, Nutley, NJ), or a combination of bexarotene plus IFN-α2b for 96 hours. Bexarotene at a final concentration of 10 mM was used for our studies, as this concentration reflects levels achieved in the blood of patients receiving oral therapy. Camptothecin (Sigma-Aldrich, St Louis, MO) was used as a positive control for induction of apoptosis. To evaluate the effect of the specified treatments on apoptosis in vitro, a modification of the dUTP nick end labeling (TUNEL) assay was performed as previously described.9 Apoptotic cells were identified using a FACScan flow cytometer (Becton Dickinson, San Jose, CA). Apoptotic cells were distinguished from necrotic cells on the basis of their forward scatter/side scatter characteristics and ability to incorporate Cy5-dUTP label.
Sequencing/mutation analysis
Genomic DNA was extracted from the patient's peripheral blood using the QIAamp DNA Mini Kit (Qiagen, Valencia, CA). RXR-α and RXR-β coding exons were PCR-amplified using flanking intron-based primers (see “PCR/sequencing primers” for primer sequences). PCR reactions were performed in a volume of 50 μL using AmpliTaq Gold Polymerase (Applied Biosystems, Foster City, CA) under conditions of 94°C for 1 minute, 58°C for 1 minute, 72°C for 1 minute for 35 cycles. GC-rich exons 1, 2, and 4 for RXR-α and exons 1 and 2 for RXR-β were PCR-amplified using FastStart Taq DNA Polymerase (Roche Applied Science, Indianapolis, IN). Exon-derived PCR amplification products were resolved on 1.5% agarose gels and purified using the QIAquick Gel Extraction Kit (Qiagen). Samples were sequenced by the DNA Sequencing Facility at the University of Pennsylvania School of Medicine. Sequencing reactions were performed on an ABI GeneAmp 9700 thermal cycler (Applied Biosystems), resolved in an ABI 3730 DNA sequencer and analyzed by ABI Sequencing Analysis software version 5.1 (both from Applied Biosystems). Complete coding sequence data were obtained for both sense and antisense strands for all exons except for exon 1 of the RXR-α gene. Sequence data were generated only in the reverse direction of exon 1, because the high GC content immediately upstream of this exon required use of a forward primer situated in close proximity to the RXR-α initiation codon for PCR amplification and priming sequencing reactions. Therefore, reliable data were not obtained for sequences at the 5′ end of the exon 1 coding region primed in the forward direction. Sequence data were compared with GenBank database sequences for mutation analysis.
PCR/sequencing primers
RXR-α.
Exon 1 forward: GGCCGGGCATGAGTTAGT; reverse: CCGAACTCACGCGAAGGT. Exon 2 forward: AGGTGCGTGCATCTGTAGCT; reverse: CCTCAAGATGAGGCGGTCGT. Exon 3 forward: GACATAGGGACAAACCTGGT; reverse: AGACGTGAGAACCCTGGAAT. Exon 4 forward: AGACCAGCAGGTCCCTTTCT; reverse: ACTCCCTGTTGTCCATCTCG. Exon 5 forward: ATGCTGGTGTTGTGGGTGAG; reverse: TCACCTCAGGTAAGAGGCCG. Exon 6 forward: CGTATTCAGCGTCCTGCATG; reverse: TCCTGGTACGTGTCCCATCT. Exon 7 forward: TCAGGATGGGTCGGTGACAT; reverse: ATACTAGGCAGGATGTGCAC. Exon 8 forward: CTTGGGTATCTGGGGTGTG; reverse: CACATGTAGCCAGAGGCAAG. Exon 9 forward: AGGGTTCTGACCTGTGGCTT; reverse: GTGCCACCTTCTCATTCACA. Exon 10 forward: AGATTCAGGGCTACAGACCA; reverse: CACATCTCTTAGGCAGAGCA.
RXR-β.
Exon 1 forward: TGCTCAGCTAATCCTCCGAT; reverse: AGATAAAGCGGTCACTGGCT. Exon 2 forward: TGTACATACCCCTCCCTCAG; reverse: GGCCAGGCAGTAAGTTGGT. Exon 3 forward: TCTGTGACATTCTGCTTCCC; reverse: AATCCCACAGGTGATGATAC. Exon 4 forward: ATGGTGAAGGTGTCTCCATG; reverse: GAGAGGTACACAGTCTGAGT. Exon 5 forward: TTCATGGCTGGCTGCTGACT; reverse: GTAGAACAGACCTAGACTGC. Exon 6 forward: GACTCACTTACAGGGATTGG; reverse: TCAGCAAGTTTGGCTCCCTG. Exons 7 and 8 forward: CACTGATGTGCTTTGAATCC; reverse: GGGTAACTTAGGAGTCTCGG. Exons 9 and10 forward: GCATGGCCATCCTGATTTGG; reverse: CATCAAGGTTCTGGGAACATG.
Quantitative PCR
Messenger RNA was extracted from PBMCs before therapy and at time of relapse (180 days) using RNA STAT-60/mRNA isolation reagent (Tel-test, Friendswood, TX). cDNA was made using SUPERSCRIPT First-strand synthesis for RT-PCR (Invitrogen) using random hexamers.
Primers were designed using the Light Cycler Probe Design Software, Version 1.0 (Idaho Technology, Salt Lake City, UT) and synthesized at the Cell Center Facility at the University of Pennsylvania.
RT-PCR primers.
RXR-α forward: AAGATGCGGGACATGCAGAT; reverse: CGAGAGCCCCTTGGAGTCA. RXR-β forward: CCGATCCATTGATGTTCGAGAT; reverse: TCTGTCAGCACCCGATCAAAG.
Final reaction volume was 20 μL containing 10 μM forward/reverse primers, 50 mM Mg, 10 mM dNTP, 10 mM BSA, 1 unit of Platinum Taq polymerase (Invitrogen) and SYBR Green (1:1000; Biowhittaker Molecular, Rockland, ME). Reactions were performed in 96-well plates in replicates. Quantitative RT-PCR amplifications were performed in an Opticon Quantitative PCR Detection System (Bio-Rad, Hercules, CA). The thermal profile used for the SYBR Green real-time RT-PCR was 95°C for 10 minutes, followed by 40 cycles of 94°C for 50 seconds, 60°C for 50 seconds, and 72°C for 1 minute. After the run, the melting curve of each amplicon was examined to determine the specificity of the amplification. The data were analyzed using the Opticon 2 Monitor Software (Bio-Rad). Final data were normalized to GAPDH and expressed as the 2−ΔΔCT method of relative quantification.
Western blot analysis
Cell lysates from patient's PBMCs before therapy and at time of relapse (180 days) were prepared using a RIPA buffer with protease and phosphatase inhibitors. Lysates were incubated on ice for 15 minutes then cleared by centrifugation at 14 000g for 10 minutes at 4°C. Supernatants were assayed for protein content using the MicroBCA protein Assay kit (Pierce Chemical, Rockford, IL). Aliquots of lysate were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to PolyScreen (NEN Life Sciences Products, Waltham, MA). Western blotting was conducted in a standard manner using the indicated antibodies (rabbit polyclonal anti-RXR-α and anti-RXR-β) at 1:500 dilution (Santa Cruz Biotechnology, Santa Cruz, CA) and developed using enhanced chemiluminescence kit as described by the manufacturer (Lumilight Plus; Boehringer Mannheim, Indianapolis, IL). Values are standardized to β-actin levels.
Results
Reduction of bexarotene-induced apoptosis of HTLV-1–infected cells correlates with relapse of disease
Bexarotene induction of apoptosis of HTLV infected cells obtained from this patient at day 0 and day 180 after initiating therapy was analyzed by flow cytometry and TUNEL assay. Figure 1A shows that for cells obtained at baseline before therapy 17.9% of untreated PBMCs were apoptotic. However, after 96 hours of treatment with bexarotene at 10 μM in vitro, 37.8% of cells had undergone apoptosis. Thus, bexarotene treatment gave rise to an overall 52.6% increase in the rate of apoptosis.
Figure 1.

Bexarotene-induced apoptosis of HTLV-1–infected cells before therapy and after relapse of disease. Patient PBMCs were cultured in vitro with medium only, bexarotene (10 μM), IFN-α2b (1000 U/mL), or a combination of bexarotene (10 μM) and IFN-α2b (1000 U/mL) and subjected to flow cytometry. (A) Cells isolated at day 0, before therapy. The percentage of apoptotic cells more than doubled with bexarotene, increasing from 17.9% to 37.8%. Combination treatment with both bexarotene and IFN-α2b yielded a higher level of apoptosis than either individual treatment, but no synergistic effect was seen. (B) PBMCs isolated at day 180 after initiating therapy. The percentage of apoptotic cells increased from 8.0% to just 10.3% with bexarotene. Of note, combination treatment with both bexarotene and IFN-α2b did not increase the percentage of apoptotic cells compared with treatment with bexarotene alone.
For cells obtained at the time of disease recurrence 180 days after initiating therapy, 8.0% of untreated cells were apoptotic, whereas 10.3% of bexarotene-treated cells underwent apoptosis (Figure 1B). This change was minimal and represents a more than 50% reduction in the rate of apoptosis induced by bexarotene compared with cells obtained at day 0 (pretherapy time point). These findings indicate that clinical relapse of disease correlated with development of resistance of the patient's tumor cells to bexarotene-induced apoptosis.
Sequence analysis reveals no acquired mutations in RXR genes in tumor cells after bexarotene treatment
To determine whether resistance to bexarotene-induced apoptosis could be attributed to acquisition of a mutation in either the RXR-α gene or RXR-β gene, we performed sequence analysis of all coding exons for both genes using genomic DNA derived from high tumor burden peripheral blood obtained 180 days after initiating treatment. Coding or splice site mutations were not detected for any of the 10 exons that comprise either gene.
Expression analysis shows loss of RXR-α protein, but not mRNA, in tumor cells after bexarotene treatment
To determine whether resistance to bexarotene treatment could be attributed to variations in RXR expression, we studied expression of both RXR-α and RXR-β mRNA and protein in the patient's malignant T cells before and after bexarotene treatment. Total RNA was isolated from the patient's PBMCs at day 0 and day 180 and quantitative PCR was performed to detect expression of RXR-α and RXR-β mRNA. No mean fold change in the level of expression of mRNA for either gene was detected over time relative to pretherapy levels. RXR-β expression was scarcely detectable, as also evidenced by Western blot analysis (Figure 2).
Figure 2.

Transcription of RXR-α mRNA before therapy and after relapse of disease. Real-time PCR of RXR-α was performed from patient PBMCs collected at day 0 before therapy and day 180 after initiating therapy. No change in the relative mean fold change in expression over time was found when comparing to day 0 of therapy.
Protein extracts from the patient's PBMCs obtained at day 0 (before) and day 180 (after) initiating therapy were subjected to Western blot analysis. Expression of both proteins was normalized to β-actin expression. Strong expression of RXR-α was observed at day 0 (Figure 3). However, RXR-α expression was markedly diminished by day 180. In contrast, expression levels of RXR-β protein did not change significantly over time and were detectable only at very low levels at baseline.
Figure 3.
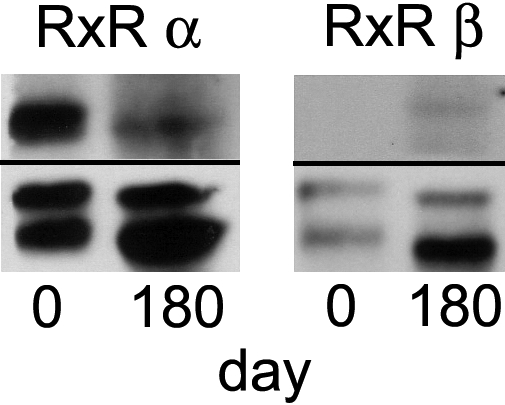
Expression of RXR-α and RXR-β subunits before therapy and after relapse of disease. Western blot analysis of protein extracts from patient PBMCs collected at day 0 before therapy and day 180 after initiating therapy revealed marked down-regulation of RXR-α protein after bexarotene therapy. Expression of both proteins was normalized to β-actin.
Discussion
Our patient with ATLL manifested a complete clinical response to low-dose bexarotene and IFN-α characterized by the regression of detectable skin, nodal, and blood disease. However, the patient suffered a relapse of his disease 180 days later despite continuous treatment. In vitro studies demonstrated significant levels of bexarotene-induced apoptosis of phenotypically abnormal (CD4+CD26−) T cells obtained before initiation of therapy. In contrast to pre-therapy findings, the patient's abnormal T cells were resistant to the apoptotic effects of bexarotene at the time of clinical relapse.
We investigated potential mechanisms involving the RXR-α and RXR-β receptors to account for the development of resistance to bexarotene by tumor cells. To determine whether either the RXR-α or RXR-β gene acquired a mutation altering receptor function, we sequenced all coding exons for both genes. Given that RXR subunits may form either homodimers or heterodimers with a variety of partners to generate functional receptors, dominant negative mutations could significantly impair overall levels of RXR function. Precedence for this type of phenomenon derives from findings that a dominant negative PML-RAR mutation was associated with retinoid resistance in an acute promyelocytic leukemia subclone.10 However, no coding mutations were identified for either gene, indicating that structural alterations in RXR-α or RXR-β are not responsible for bexarotene resistance in our patient's malignant T cells.
To assess whether altered levels of RXR-α or RXR-β may account for bexarotene resistance, we examined expression of both subunits using blood samples obtained before treatment and after relapse. Both RXR-α and RXR-β proteins were detectable by Western blot analysis of protein extracts from malignant T cells before initiating therapy. Baseline expression of RXR-β mRNA and protein was minimal relative to RXR-α and no change was discernible over time. However, expression of RXR-α protein was markedly diminished after relapse in comparison to pretherapy levels. Thus, the putative underlying mechanism for clinical progression of the patient's ATLL appears to be loss of responsiveness to bexarotene due to loss of the RXR-α subunit.
Loss of RXR-α expression appeared to have occurred through a posttranscriptional mechanism, given that levels of RXR-α mRNA did not appear to differ in the patient's malignant T cells before treatment and after relapse. Therefore, mechanisms such as mutation or alteration of transcriptional regulatory regions, allelic loss, or epigenetic phenomena (including aberrations in promoter methylation or histone acetylation) do not appear to account for loss of RXR-α expression in this case.
Nonetheless, the basis for loss of RXR-α receptor expression could be accounted for by several potential means. Numerous natural and synthetic ligands are known to down-modulate a variety of receptor systems during exposure to ligand.11,12 Thus, it is possible that the chronic exposure of our patient's transformed T cells to bexarotene could have led to the down-modulation of RXR-α expression. Under these circumstances, discontinuation of bexarotene would have been expected to result in up-regulation of RXR-α receptors. This likelihood was not explored using our patient's cells due to his deteriorating clinical status. Another mechanism for observed loss of RXR-α receptor expression could be related to selection of a subset of transformed T cells that may have lacked RXR receptor expression from the outset of treatment. This is supported by our flow cytometry results, which suggested that a single clonal population was present within the peripheral blood at the time of each study. Early during treatment with bexarotene, it is possible that the majority of bexarotene-sensitive cells were eliminated, leaving only transformed T cells lacking RXR-α receptors to proliferate in an uncontrolled manner.
The therapeutic implications of RXR receptor loss in T cells during therapy with bexarotene are numerous. The general frequency of receptor loss during therapy is unknown. Should receptor loss prove to be a common phenomenon, this mechanism could account for the ultimate loss of clinical responsiveness to bexarotene among those patients who experience relapse of disease. Moreover, should it be determined that the mechanism is the transient down-modulation of the receptor in response to the ligand, then treatment approaches could be developed that incorporate the cyclical use of bexarotene. In addition, studies of other agents used to treat cutaneous T-cell lymphoma, such as IFN-α, IFN-γ, and zidovudine, might shed light on whether they antagonize receptor down-modulation. If so, such findings might explain the possible synergistic effects of interferons when used together with bexarotene.13
Furthermore, our findings suggest a potential mechanism for why the cells of some Sézary syndrome patients demonstrate resistance to bexarotene-induced apoptosis altogether.14 It is possible that the malignant T cells of these patients may lack RXR receptors at baseline before initiating treatment with bexarotene. Should this be the case, assessment of RXR receptor expression could be used as a test to predict potential responsiveness to bexarotene before commencing a lengthy course of therapy and to facilitate patient selection for this mode of treatment.
Acknowledgments
This work was partially supported by a grant from the Leukemia & Lymphoma Society (A.H.R.) and by National Institutes of Health grant CA100499.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: J.H.L., E.J.K., A.H.R., and S.S.F. wrote the paper. J.S. and X.Y.C. performed the Western blot and quantitative PCR. S.S.F., S.Z., and A.B. performed the mutational analysis. S.K.R., M.W., and B.B. performed the apoptosis data. E.J.K., S.R., A.H.R., and S.N. cared for the patient.
Conflict-of-interest disclosure: A.H.R. is a member of the speakers' bureau for Eisai Pharmaceuticals. S.R. has been a consultant for Abbott Immunology. The remaining authors declare no competing financial interests.
Correspondence: Julie H. Lin, University of Pennsylvania, Department of Dermatology, 2 Maloney Building, 3600 Spruce Street, Philadelphia, PA 19104; e-mail: julie.lin@mail.mcgill.ca.
References
- 1.Duvic M, Hymes K, Heald P, et al. Bexarotene is effective and safe for treatment of refractory advanced-stage cutaneous T-cell lymphoma: multinational phase II-III trial results. J Clin Oncol. 2001;19:2456–2471. doi: 10.1200/JCO.2001.19.9.2456. [DOI] [PubMed] [Google Scholar]
- 2.Duvic M, Martin AG, Kim Y, et al. Phase 2 and 3 clinical trial of oral bexarotene (Targretin capsules) for the treatment of refractory or persistent early-stage cutaneous T-cell lymphoma. Arch Dermatol. 2001;137:581–593. [PubMed] [Google Scholar]
- 3.Agarwal VR, Bischoff ED, Hermann T, Lamph WW. Induction of adipocyte-specific gene expression is correlated with mammary tumor regression by the retinoid X receptor-ligand LGD1069 (targretin). Cancer Res. 2000;60:6033–6038. [PubMed] [Google Scholar]
- 4.Zhang C, Hazarika P, Ni X, Weidner DA, Duvic M. Induction of apoptosis by bexarotene in cutaneous T-cell lymphoma cells: relevance to mechanism of therapeutic action. Clin Cancer Res. 2002;8:1234–1240. [PubMed] [Google Scholar]
- 5.Wu K, Zhang Y, Xu XC, et al. The retinoid X receptor-selective retinoid, LGD1069, prevents the development of estrogen receptor-negative mammary tumors in transgenic mice. Cancer Res. 2002;62:6376–6380. [PubMed] [Google Scholar]
- 6.Budgin JB, Richardson SK, Newton SB, et al. Biological effects of bexarotene in cutaneous T-cell lymphoma. Arch Dermatol. 2005;141:315–321. doi: 10.1001/archderm.141.3.315. [DOI] [PubMed] [Google Scholar]
- 7.Szanto A, Narkar V, Shen Q, et al. Retinoid X receptors: X-ploring their (patho)physiological functions. Cell Death Differ. 2004;11(Suppl 2):S126–S143. doi: 10.1038/sj.cdd.4401533. [DOI] [PubMed] [Google Scholar]
- 8.Richardson S, Budgin JB, Junkins-Hopkins JM, et al. Low-dose bexarotene and low-dose interferon alfa-2b for adult T-cell leukemia/lymphoma associated with human T-lymphotropic virus 1. Arch Dermatol. 2005;141:301–304. doi: 10.1001/archderm.141.3.301. [DOI] [PubMed] [Google Scholar]
- 9.Sgonc R, Boeck G, Dietrich H, et al. Simultaneous determination of cell surface antigens and apoptosis. Trends Genet. 1994;10:41–42. doi: 10.1016/0168-9525(94)90140-6. [DOI] [PubMed] [Google Scholar]
- 10.Shao W, Benedetti L, Lamph WW, Nervi C, Miller WH., Jr A retinoid-resistant acute promyelocytic leukemia subclone expresses a dominant negative PML-RAR alpha mutation. Blood. 1997;89:4282–4289. [PubMed] [Google Scholar]
- 11.Broad A, Jones DE, Kirby JA. Toll-like receptor (TLR) response tolerance: a key physiological “damage limitation” effect and an important potential opportunity for therapy. Curr Med Chem. 2006;13:2487–2502. doi: 10.2174/092986706778201675. [DOI] [PubMed] [Google Scholar]
- 12.Dunlop EA, Maxwell AP, Lappin TR. Impaired downregulation following erythropoietin receptor activation in non-small cell lung carcinoma. Stem Cells. 2007;25:380–384. doi: 10.1634/stemcells.2006-0452. [DOI] [PubMed] [Google Scholar]
- 13.McGinnis KS, Junkins-Hopkins JM, Crawford G, et al. Low-dose oral bexarotene in combination with low-dose interferon alfa in the treatment of cutaneous T-cell lymphoma: clinical synergism and possible immunologic mechanisms. J Am Acad Dermatol. 2004;50:375–379. doi: 10.1016/j.jaad.2003.10.669. [DOI] [PubMed] [Google Scholar]
- 14.Brennand S, Sutton VR, Biagi J, et al. Lack of apoptosis of Sezary cells in the circulation following oral bexarotene therapy. Br J Dermatol. 2005;152:1199–1205. doi: 10.1111/j.1365-2133.2005.06539.x. [DOI] [PubMed] [Google Scholar]