

Abstract
Prion diseases are neurodegenerative infections with gliosis and vacuolation. The mechanisms of degeneration remain unclear, but chemokines may be important. In current experiments CCR1 knock-out (KO) mice succumbed more rapidly to scrapie infection than WT controls. Infected KO mice had upregulation of CCL3, a CCR1 ligand, and CCR5, a receptor with specificity for CCL3. Both infected KO and WT mice had upregulation of CCR5-mediated signaling involving activation of Erk1/2 in astrocytes; however, activation was earlier in KO mice suggesting a role in pathogenesis. In both mouse strains activation of the Erk1/2 pathway may lead to astrocyte dysfunction resulting in neurodegeneration.
Keywords: Prion, Knock-out mouse, Neurodegeneration, Chemokine, Scrapie, Transmissible Spongiform Encephalopathies
1. Introduction
Prion diseases, or transmissible spongiform encephalopathies (TSEs), are fatal infectious neurodegenerative disorders that include Creutzfeldt-Jakob disease (CJD) in humans, bovine spongiform encephalopathy (BSE) in cattle, chronic wasting disease (CWD) in North American deer and elk, and scrapie in sheep. TSEs are characterized by the conversion of normal protease-sensitive prion protein (PrP-sen) to the abnormal partially protease-resistant isoform of prion protein (PrP-res). This process is believed to be the result of an alteration in the normal folding of PrP-sen to create misfolded molecules which form self-aggregates of various sizes (for review, see (Caughey and Lansbury, 2003). Accumulation of aggregated PrP-res in the CNS is associated with pathological changes characteristic of prion diseases including gliosis, vacuolation of the neuropil and neuronal loss (Wells, 1993; Budka et al., 1995).
The role of PrP-res in the pathogenic process of prion diseases is poorly understood at this time. PrP-res may be able to damage neurons both directly through interactions with PrP-sen on the plasma membrane, and indirectly through interactions with glial cells, in particular, microglia and astroglia (Williams et al., 1997; Giese et al., 1998; Betmouni and Perry, 1999; Giese and Kretzschmar, 2001; Eikelenboom et al., 2002; Rock et al., 2004; Wojtera et al., 2005). Glial activation is a normal response of the brain to many types of damage, and this activity is part of the normal healing process. However, there is also evidence in a variety of diseases that activation of glia might also contribute to the pathological process, perhaps by the release of neurotoxic mediators. For example, in prion diseases, upregulation of several cytokines and chemokines, including tumor necrosis factor-alpha (TNF-α), interleukin 1 alpha (IL-1α), IL-1β, IL-10, IL-13, transforming growth factor beta (TGF-β), CCL5 (RANTES), CXCL9 (MIG), CXCL10 (IP-10) and CXCL13 (B cell-attracting chemokine) have been reported (Campbell et al., 1994; Kordek et al., 1996; Williams et al., 1997; Kim et al., 1999; Baker et al., 2002; Cunningham et al., 2002; Burwinkel et al., 2004; Thackray et al., 2004) . In addition, several chemokine receptors, such as CCR1, CCR3 and CCR5 have been found to be upregulated in scrapie disease (Lee et al., 2005a). We hypothesize that some of these molecules are involved in the pathogenic process of the disease, rather than merely being a host response to the damage induced by other mechanisms.
Most chemokines can interact with more than a single receptor. Thus, it is difficult to determine whether these molecules have a causative role in disease where upregulation has been detected. Our laboratory has previously used knockout (KO) mice deficient in cytokines or chemokine receptors to show an active role for these molecules in a retrovirus-induced CNS disease in vivo (Peterson et al., 2004b; Peterson et al., 2004a). In the present study, we used a similar strategy to compare scrapie infection in KO mice lacking CCR1 expression (CCR1 KO mice) versus wild-type (WT) control mice. In these experiments, the CCR1 KO mice died more rapidly from scrapie disease than did WT mice, in spite of the fact the PrP-res levels were lower in the KO mice. Our results suggested that in the absence of CCR1, scrapie disease was accelerated by an increased expression of CCR5 and its ligand CCL3. This ultimately led to the increased activation of the extracellular-signal regulated kinases 1/2 (Erk1/2), potentially leading to astrocyte dysfunction resulting in neurodegeneration.
2. Materials and Methods
Mice
CCR1 KO C57BL/6 mice were a gift from Phil Murphy (National Institute of Allergy and Infectious Diseases, National Institutes of Health) (Gao et al., 1997). WT controls shared the C57BL/6 background. All mice were maintained at Rocky Mountain Laboratories and handled according to the policies of the Rocky Mountain Laboratories Animal Care and Use Committee and all applicable federal guidelines. Adult (6 to 8 week old) animals were used for all experiments.
Scrapie inoculation
Mice were injected intracerebrally (i.c.) with 50 µl of a 1% (wt/vol) dilution of brain homogenate from a C57BL/6 mouse terminally ill from RML/Chandler scrapie (approximately 2.5 × 107 ID50). Brain homogenates were diluted in phosphate-buffered saline (PBS) pH 7.2, supplemented with 2% fetal bovine serum (Hyclone, Logan, UT). Mice were observed several times each week for clinical signs of scrapie, which include kyphosis, wasting, ataxia and an exaggerated high stepping gait most noticeable in the hind limbs (Race et al., 2000). Brain samples from mice in each group with clinical evidence of scrapie disease were analyzed by Western blot for PrP-res to confirm the clinical diagnosis.
Pathology and Immunohistochemistry
Brain tissue for histology analysis was procured by lightly anesthetizing mice, trans-cardial perfusion with 30 ml PBS, and removal of the intact brain. One-half of the sagitally cut brain was fixed in 3.7% phosphate-buffered formaldehyde for 3 to 5 days. Following fixation, tissue was dehydrated and paraffin-embedded using standard techniques. Serial 4-µm tissue sections were cut using a standard Leica microtome. Tissue sections were placed on positively-charged glass slides and dried overnight at 56°C. Immunohistochemical staining was performed using the Ventana automated Nexus stainer (Ventana, Tucson, AZ). All histology procedures were performed on both mock-infected and scrapie-infected animals, including appropriate immunohistochemical controls. Staining for GFAP was performed by re-hydrating sections to Tris buffer, pH 7.5; using anti-GFAP (1:1000-Dako), biotinylated goat anti–rabbit IgG (1:250-Vector Laboratories, Burlingame, CA) followed by Streptavidin-Alk-Phos (Ventana) and Alk-Phos Red (Ventana). Prior to staining for Iba-1(Ito et al., 1998), tissue sections were re-hydrated with 0.1 mM citrate buffer pH 6.0 and subjected to 122°C and 22psi for 30 seconds (Biocare de-cloaking chamber, Walnut Creek, CA). A standard avidin-biotin alkaline phosphatase (alk-phos) protocol was used with anti Iba-1 (1:1000-WAKO Chemicals USA, Richmond, VA), biotinylated goat anti-rabbit IgG (1:250- Vector Laboratories), Streptavidin-Alk-Phos (Ventana) and Alk-Phos Red (Ventana). For the detection of PrP-res, sections were re-hydrated with 0.1 mM citrate buffer, pH 6.0, for 20 minutes at 122°C and 22psi. Staining was performed with D13 (1:1500-In Pro Biotechnology, South San Francisco, CA). Detection was accomplished using biotinylated goat anti-human IgG F(ab) fragment (1:500-Jackson Immuno Research, West Grove, PA) followed by Supersensitive Streptavidin-HRP (1:3-Biogenex, San Ramon, CA) and amino-ethyl carbazole (AEC) (Ventana). To stain for p-Erk1/2, sections were re-hydrated with 0.1 mM citrate buffer, pH 6.0, for 5 minutes at 122°C and 22psi. A standard avidin-biotin peroxidase technique was employed using anti-p-Erk1/2 (1:1000-Cell Signaling, Danvers, MA), biotinylated goat anti-rabbit IgG (1:250-Vector), Supersensitive Streptavidin-HRP (1:3-Biogenex) and diaminobenzidine (DAB) (Ventana).
Western blot analysis
Brain tissue from CCR1 KO and WT mice was analyzed for the presence of using Western blotting techniques as previously described (Race et al., 1992) with some modifications. To detect PrP-res, 10% (w/v) brain tissue homogenates were made in modified RIPA buffer (5mM Tris-HCl, pH 7.4, 1% Triton X-100, 1% sodium deoxycholate, 150 mM NaCl, and 5 mM EDTA). To remove nucleic acids, the homogenates were treated with 20 U Benzoase nuclease (Novagen, Madison, WI) for 30 minutes at 37°C, and debris was removed by centrifugation at 1000 × g for 5 minutes at 4°C. Homogenates were treated with a final concentration of 50 µg/ml proteinase K for 1 hour at 37°C. Five-mg tissue equivalents were diluted in sample buffer, boiled for 5 minutes, then run on a 16% polyacrylamide gel. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes then probed with anti-PrP rabbit polyclonal antibody, R30 (1:3000), followed by donkey anti-rabbit IgG-HRP (1:3000) (Amersham Biosciences, Buckinghamshire, UK). The protein bands were then visualized using standard enhanced chemiluminescence (ECL) procedures (GE Healthcare, Piscataway, NJ). Detection for enhanced chemifluorescence (ECF) for quantitation was performed as described above, except using ECF Western blotting kit (GE Healthcare), as per instructions. The blots were dried, then scanned with a Storm phosphoimager and analyzed with ImageQuant V5.2 software (Molecular Dynamics, Sunnyvale, CA).
To detect Erk1, Erk2, p-Erk1/2, and p-Elk-1, homogenates were prepared as above, with the exceptions of omission of the proteinase K treatment, and the addition of Halt Protease Inhibitor Cocktail (Pierce, Rockford, IL). Homogenates were run on 12% polyacrylamide gels, and proteins were transferred to PVDF membranes. The blots were probed with either anti-p44 MAP Kinase (Erk1) antibody, anti-p42 MAP Kinase (Erk2) antibody, anti-phospho-p44/p42 Map Kinase (Thr202/204) antibody (p-Erk1/2), or anti-phospho-Elk-1 (p-Elk-1) at a dilution of 1:1000 (Cell Signaling Technology). Detection by ECL or ECF was as described above.
RT-PCR analysis
Quantitative real-time RT-PCR was performed as previously described, with some modifications (Dimcheff et al., 2003). Brain tissue was harvested and immediately placed in RNALater (Qiagen, Valencia, CA). The thalamus was then dissected from the rest of the brain, and total RNA was isolated using an RNeasy mini kit (Qiagen) followed by DNAse treatment. Each RNA sample was reverse transcribed into cDNA using reverse transcription reagents with random hexamers (Applied Biosystems, Foster City, CA). PCR was performed on the cDNAs in triplicate using TaqMan PCR Master Mix (Applied Biosystems). All primer and probes used were pre-developed gene expression assays (Applied Biosystems). The relative quantity of each gene tested was normalized against mouse β-actin for each sample. ΔCT values were calculated by subtracting the β-actin CT value from the CT value of the gene of interest. ΔΔCT values were calculated from subtracting the ΔCT value from the gene of interest in scrapie-infected mice from the ΔCT value of the mock-infected control.
3. Results
CCR1 mRNA expression upregulation in WT scrapie infected mice
To search for a possible role of CCR1 during scrapie disease, we evaluated whether CCR1 mRNA expression was altered after scrapie infection in C57BL/6 wild-type (WT) mice, as was seen by Lee et al. in the ME-7 scrapie system (Lee et al., 2005a). By quantitative RT-PCR, we found that CCR1 was upregulated (>2-fold) in the RML/Chandler scrapie-infected WT mice, as compared to the mock-infected WT controls, at the time of clinical disease (data not shown). The greater level of CCR1 in the WT scrapie-infected mice suggested that CCR1 might play a role during scrapie-induced pathogenesis.
Faster scrapie disease in CCR1 KO mice than in WT mice
To evaluate the role of the chemokine receptor CCR1 in scrapie pathogenesis, CCR1 KO and control C57BL/6 mice (WT) were challenged intracranially (i.c.) with the scrapie strain RML/Chandler. The CCR1 KO mice succumbed to scrapie disease faster than the WT mice, with a median incubation period of 154 days, versus 169 days in the WT controls (p < 0.0001) (Fig. 1). The shortened incubation period in the CCR1 KO mice suggested that CCR1 might play a neuroprotective role in scrapie-induced pathogenesis in the wild-type control mice.
Figure 1.
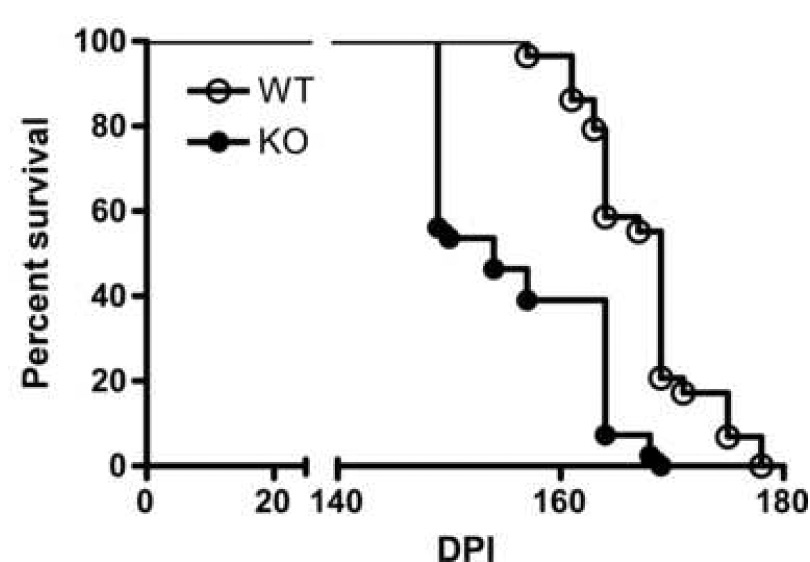
Survival curve of WT and CCR1 KO mice following i.c. scrapie inoculation. The data described is a percentage of mice alive vs. days post inoculation. These data were pooled from two independent experiments (WT mice: n = 29 mice, CCR1 KO mice: n = 41 mice). The median survival time for WT mice was 169 days post inoculation, vs. 154 days for the CCR1 KO mice. As determined by the logrank test, this difference was statistically significant (p<0.0001).
Similar brain pathology and PrP-res deposition in CCR1 KO and WT mice
Generation of PrP-res in the brain is often correlated with the tempo of clinical prion disease. Therefore, scrapie-infected CCR1 KO and WT control mice were evaluated by immunohistochemistry for PrP-res accumulation, gliosis, and spongiform pathology at preclinical and clinical timepoints. The distribution of PrP-res deposition throughout the brain was similar in both strains of mice (thalamus shown in Fig 2). The level of astrocyte and microglial activation was also analyzed immunohistologically by staining the brain sections with anti-glial fibrillary acidic protein (GFAP) and anti-ionizing calcium-binding adaptor molecule 1 (Iba-1) antibodies, respectively. The distribution and extent of both astroglial and microglial activation was similar in the CCR1 KO and WT mice at the preclinical and clinical time points (Fig 2). There were also no differences observed in the spongiform pathology between the two strains of mice (Fig. 2). The similarities observed immunohistochemically in the CCR1 KO and the WT mice did not provide a clear explanation for the differences in time of onset of clinical disease.
Figure 2.
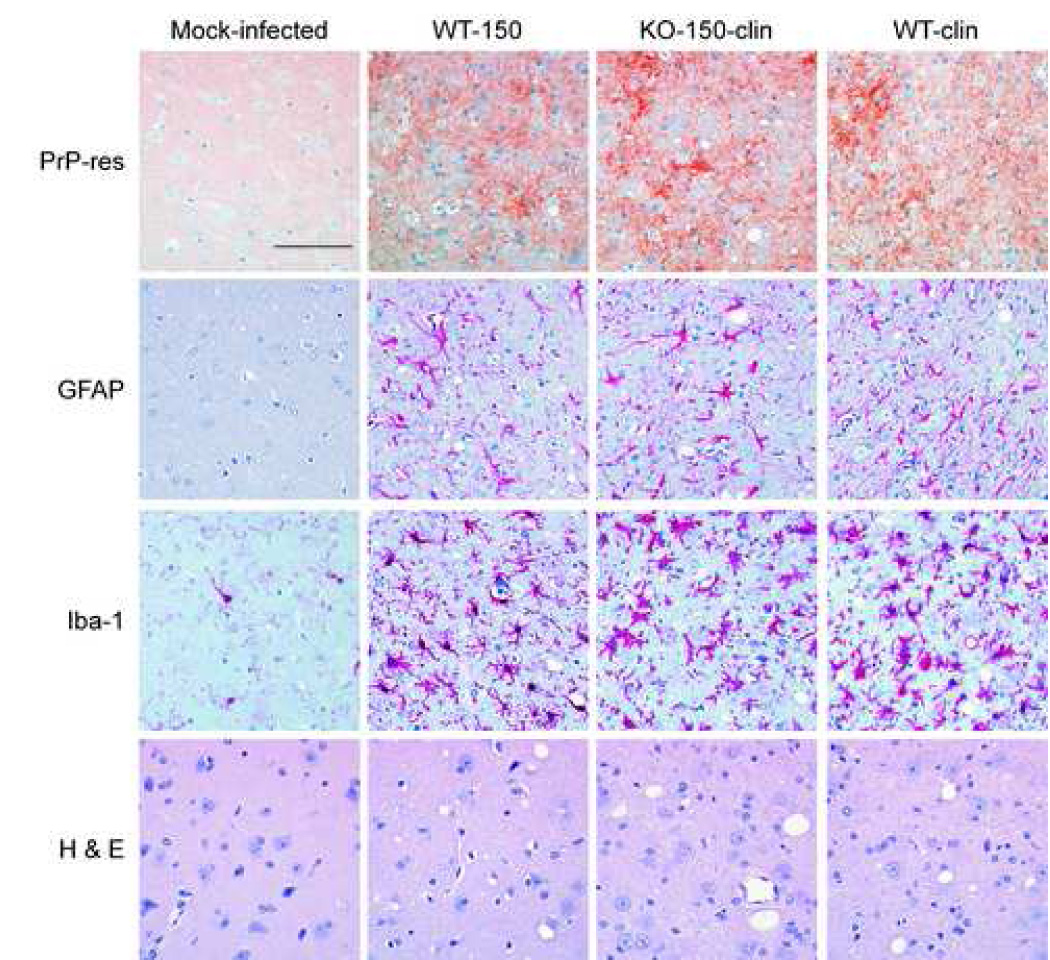
Histopathological analysis of WT and CCR1 KO scrapie-infected mice. Shown are representative sections of the thalamus of a mock-infected control, a WT scrapie-infected mouse at 150 dpi, a clinical CCR1 KO mouse at 150 dpi, as well as a clinical WT mouse at 169 dpi. Although the CCR1 KO and WT mice have similar PrP-res deposition (D13 anti-PrP), gliosis (anti-GFAP and anti-Iba-1), and vacuolization (H&E), the CCR1 KO mice succumb to scrapie disease faster than the WT controls. Magnification bar represents 100 µm.
Reduced PrP-res in CCR1 KO mice compared to WT mice
To further evaluate the amount of PrP-res accumulation in the brains of scrapie-infected CCR1 KO and WT mice, brain extracts were analyzed by immunoblotting. By Western blot analysis, CCR1 KO mice seemed to have less PrP-res at the time of clinical disease than the WT mice (Fig. 3A). The amount of PrP-res was quantitated by enhanced chemifluorescence, and it was determined that clinical CCR1 KO mice (150 dpi) had slightly lower PrP-res levels than WT preclinical mice examined at the same time, but the difference was not statistically significant (Fig. 3B). However, clinical CCR1 KO mice (150 dpi) did have significantly lower levels of PrP-res than the clinical WT mice (169 dpi) (Fig. 3B). Thus, these lower PrP-res levels in CCR1 KO mice did not correlate with the accelerated disease course. Furthermore, the low PrP-res levels in CCR1 KO mice did not appear to be due to lower PrP expression in these mice, as measured by quantitative RT-PCR (Fig. 3C).
Figure 3.
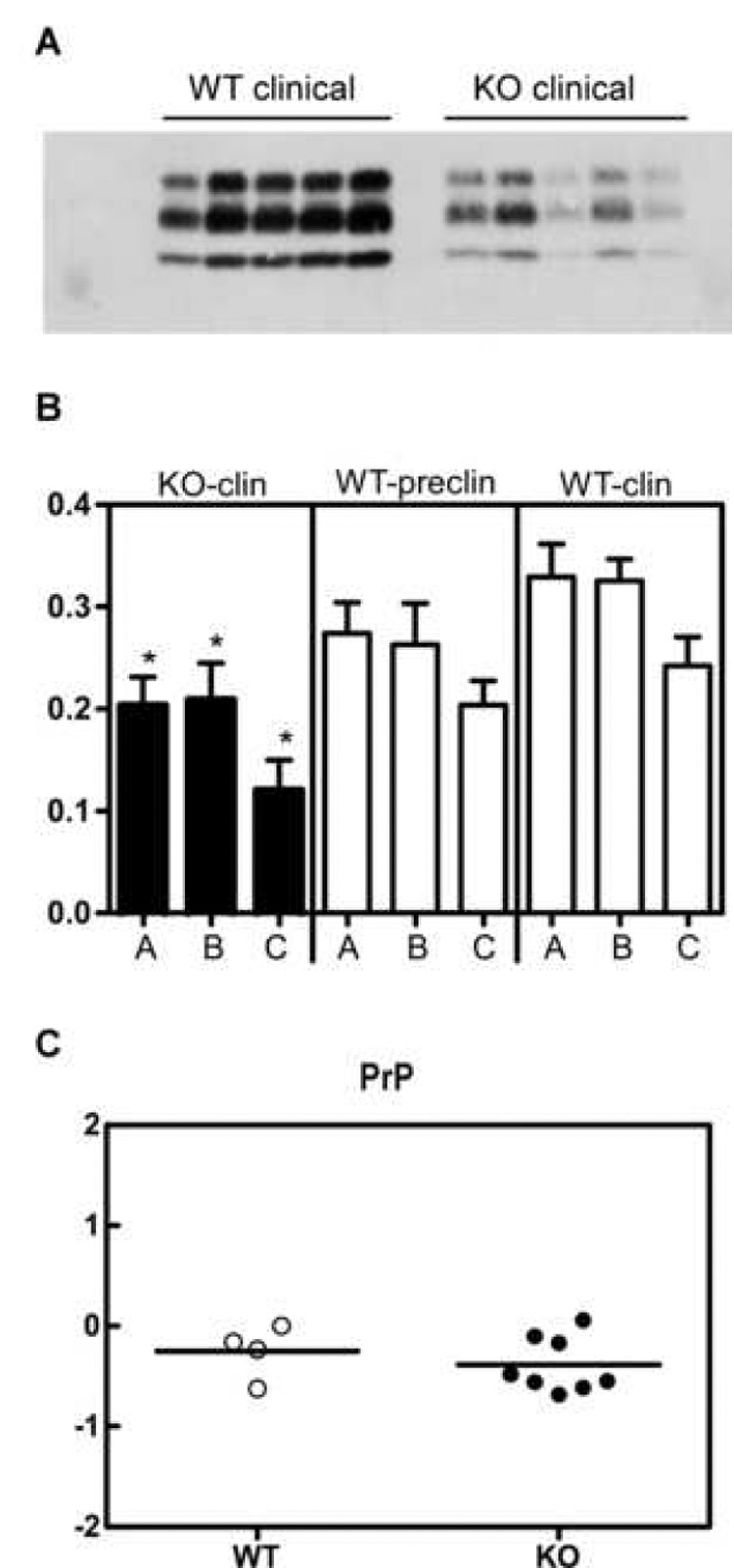
Quantification of PrP in the brains of WT and CCR1 KO mice. (A) Brains from scrapie-infected mice were analyzed for amounts of PrP-res by Western blot using R30 rabbit anti-PrP peptide serum. (B) The amount of PrP-res was quantitated by enhanced chemifluorescence. The different glycosylated bands of PrP-res are indicated as a = diglycosylated, b = monoglycosylated, and c = unglycosylated. The data represents the average of 3–6 mice per group. The data indicate that at the time of clinical disease, CCR1 KO mice had similar to slightly-reduced levels of PrP-res when compared to the non-clinical WT mice at 150 dpi, but had significantly reduced levels of PrP-res as compared to the clinical WT mice (* = p<0.05) . (C) Real time RT-PCR of WT and CCR1 mice for PrP-sen expression. Shown are the relative amounts of PrP-sen RNA expression in scrapie-infected WT vs. CCR1KO mice. All RNA samples were normalized to β-actin, and then subtracted from the respective mock-infected controls to give the ΔΔCT values. The data indicates that the relative expression of PrP-sen is similar in both types of mice.
Comparable astrocyte and microglial activation in CCR1 KO and WT mice
By immunohistochemical evaluation, the amount of astrocyte and microglial activation looked very similar in both the CCR1 KO and WT scrapie-infected mice. However, a more quantitative RT-PCR assay was performed to compare the levels of different glial activation marker mRNAs present in the thalamus of the scrapie-infected brains at the clinical time points for the CCR1 KO mice (median of 154 dpi) and the WT controls (median of 169 dpi). In agreement with the extensive astrogliosis seen after scrapie infection, there was a large increase in GFAP expression in scrapie-infected mice as compared to mock-infected controls (represented by dotted line); however, there was no significant difference after scrapie infection between the CCR1 KO and the WT mice (Fig.4). Similarly, the levels of several microglial activation markers, CD14, ITGAM (CD11b) and emr1 (F4/80), were upregulated in the scrapie-infected mice as compared to the mock-infected controls, but there was no significant difference between the CCR1 KO and WT mice (Fig. 4). Thus, differential glial activation did not appear to be responsible for the shortened incubation time observed in CCR1 KO mice.
Figure 4.
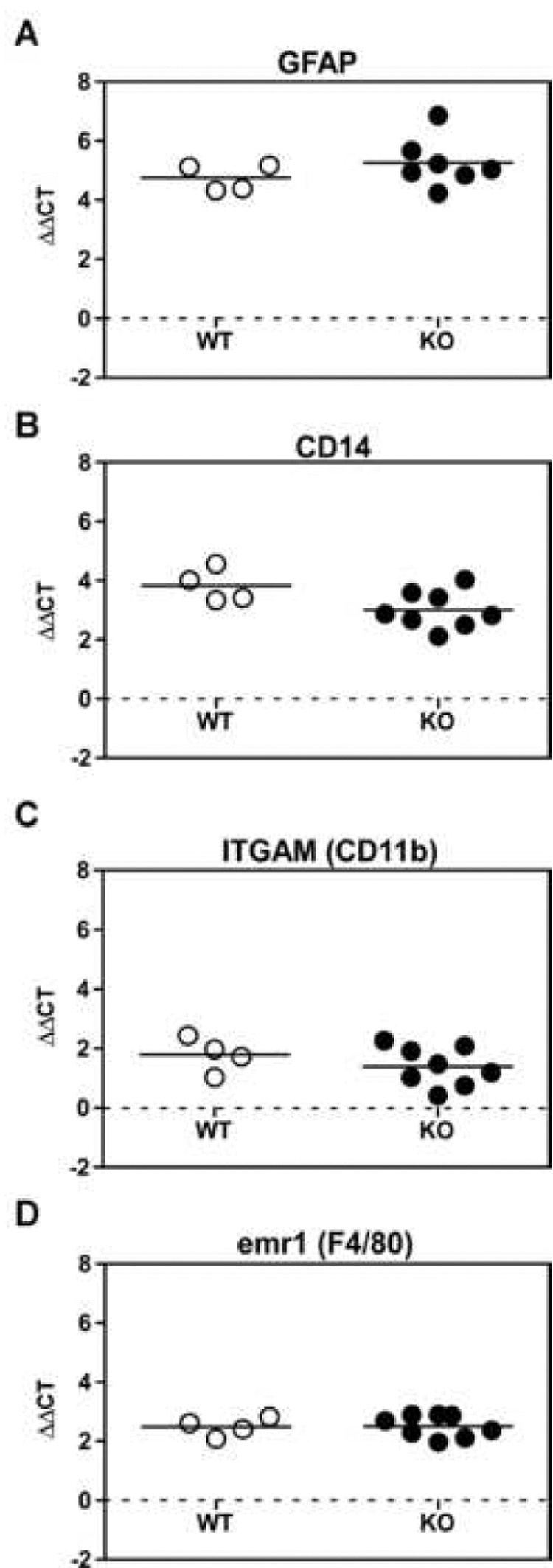
Real time RT-PCR of WT and CCR1 KO thalamus RNA for glia marker expression. All RNA samples from the thalamus of WT and CCR1 KO mice at the time of clinical disease were normalized to β-actin, and then subtracted from the respective mock-infected controls to give the ΔΔCT values. Each RNA sample was examined for the activated astrocyte marker GFAP, as well as three microglial markers, CD14, ITGAM and emr1. All scrapie-infected mice exhibited an increase in both astrocyte and microglial activation markers as compared to the mock-infected controls (as shown by dotted line). However, no significant difference was observed between scrapie-infected CCR1 KO and WT controls.
Upregulation of CCR5 and CCL3 in scrapie-infected CCR1 KO mice
There is often significant overlap in the binding specificities of chemokine receptors and their ligands. For example, ligands of CCR1, such as CCL5 (RANTES), CCL7 (MCP-3) and CCL3 (MIP-1α), can also bind to CCR3 and/or CCR5 receptors. In CCR1 KO mice, these receptors might be upregulated to compensate for the lack of CCR1. Therefore, mRNA levels for CCR3 and CCR5 were evaluated by quantitative RT-PCR. CCR3 expression could not be detected in the brains of WT or CCR1 KO mice. However, CCR5 expression was detectable in both strains of mice, and at the time of clinical disease, the scrapie-infected CCR1 KO mice were found to express significantly higher levels of CCR5 than the scrapie-infected WT controls (Fig. 5A). In contrast, CCR5 levels were similar in the mock-infected KO and WT mice (data not shown). Thus, scrapie infection appeared to be the cause of the increase in CCR5 expression in the CCR1 KO mice.
Figure 5.
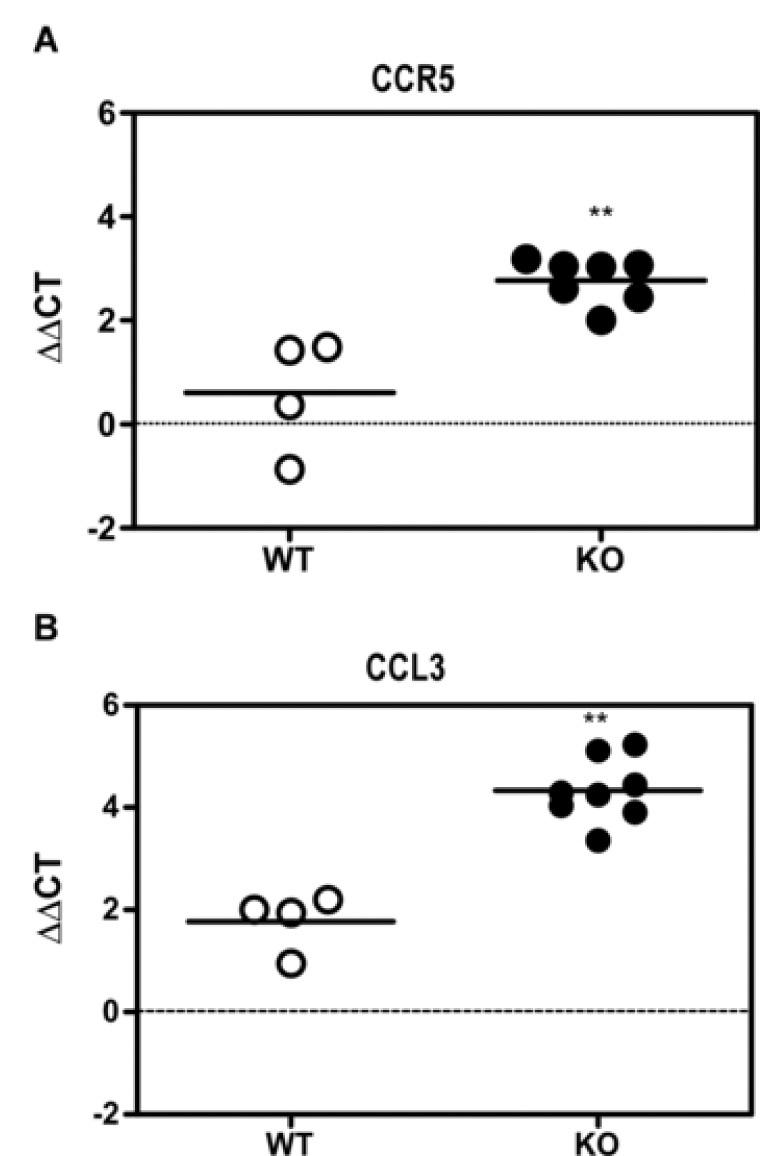
Real time RT-PCR for chemokines and cytokines. RNA from the thalamus of the WT and CCR1 KO mice were evaluated for changes in mRNA expression of chemokines and cytokines following scrapie infection. Shown are the ΔΔCTs for the thalamus RNA of clinical scrapie-infected CCR1KO vs. WT mice. The data shown indicate that there is a significant increase in the expression of CCR5 and its ligand, CCL3 (Mann-Whitney test; p = 0.0061 and 0.004, respectively) in the CCR1 KO mice as compared to the WT controls. Additional chemokines and cytokines tested, but not significantly different between scrapie-infected CCR1KO and WT mice include CCL2 (MCP-1), CCL7 (MCP-3), CCL12 (MCP-5), CCL4 (MIP-1β), CCL5 (RANTES), CXCL10 (IP-10), IL-1α and TNFα.
To determine whether CCR5 upregulation correlated with upregulation of specific chemokines, we evaluated the expression of several chemokines that are potential ligands for either CCR1 or CCR5, as well as others implicated in the pathogenesis of various neurodegenerative diseases. No difference was found in expression of a number of cytokines and chemokines, including CCL2 (MCP-1), CCL7 (MCP-3), CCL12 (MCP-5), CCL4 (MIP-1β), CCL5 (RANTES), CXCL10 (IP-10), IL-1α and TNFα between the scrapie-infected CCR1 KO mice and the WT controls (data not shown). However, in the CCR1 KO mice, a significant increase in the level of the chemokine macrophage inflammatory protein 1 alpha (CCL3) was observed (Fig. 5B). These data suggested that during scrapie infection, in the absence of CCR1, expression of both the CCR5 receptor and the common ligand, CCL3, were upregulated.
Increase in phospho-Erk1/2 in scrapie-infected mice
To investigate further the possible role of CCR5 and CCL3 in this model, we studied the extracellular signal-regulated protein kinase (Erk) 1/2 signaling pathway. The Erk signaling pathway can be activated through several different cellular receptors, including the G protein-coupled receptors (GPCR), such as CCR5. Erk1/2 may play a role in several neurodegenerative diseases, including Alzheimer’s (Perry et al., 1999; Rapoport and Ferreira, 2000; Dineley et al., 2001; Russo et al., 2002), Parkinson’s disease (Kulich and Chu, 2001; Gomez-Santos et al., 2002; Zhu et al., 2002), as well as scrapie (Lee et al., 2005b; Marella et al., 2005; Nordstrom et al., 2005). Activation of this pathway involves phosphorylation of the Erk1/2 proteins (p44/p42) which can be detected by immunoblotting. To determine whether CCR1 KO mice have increased activation of the Erk1/2 pathway, brain homogenates of CCR1 KO and WT mice were analyzed by Western blot. The level of constitutively expressed Erk1/2 proteins, as well as the amount of phosphorylated Erk1/2 (p-Erk1/2) proteins, was determined. Constitutive expression of Erk1 and Erk2 was similar in the CCR1 KO and WT mice (Fig. 6). In contrast, p-Erk1/2 was significantly increased in the clinical CCR1 KO mice as compared to the WT control mice at 150 dpi, and 169 dpi (clinical) (Fig. 6). However, in WT mice at 150 dpi, p-Erk1/2 was also significantly increased above levels seen at 120 dpi. Since chronic activation of the Erk 1/2 signaling pathway has been shown to play a role in neurodegeneration (Colucci-D'Amato et al., 2003), the activation of the Erk1/2 pathway in the scrapie-infected CCR1 KO and WT mice may be involved in the scrapie disease process. Therefore, the acceleration of this process in CCR1 KO mice might account for the more rapid disease course seen in the current experiments.
Figure 6.
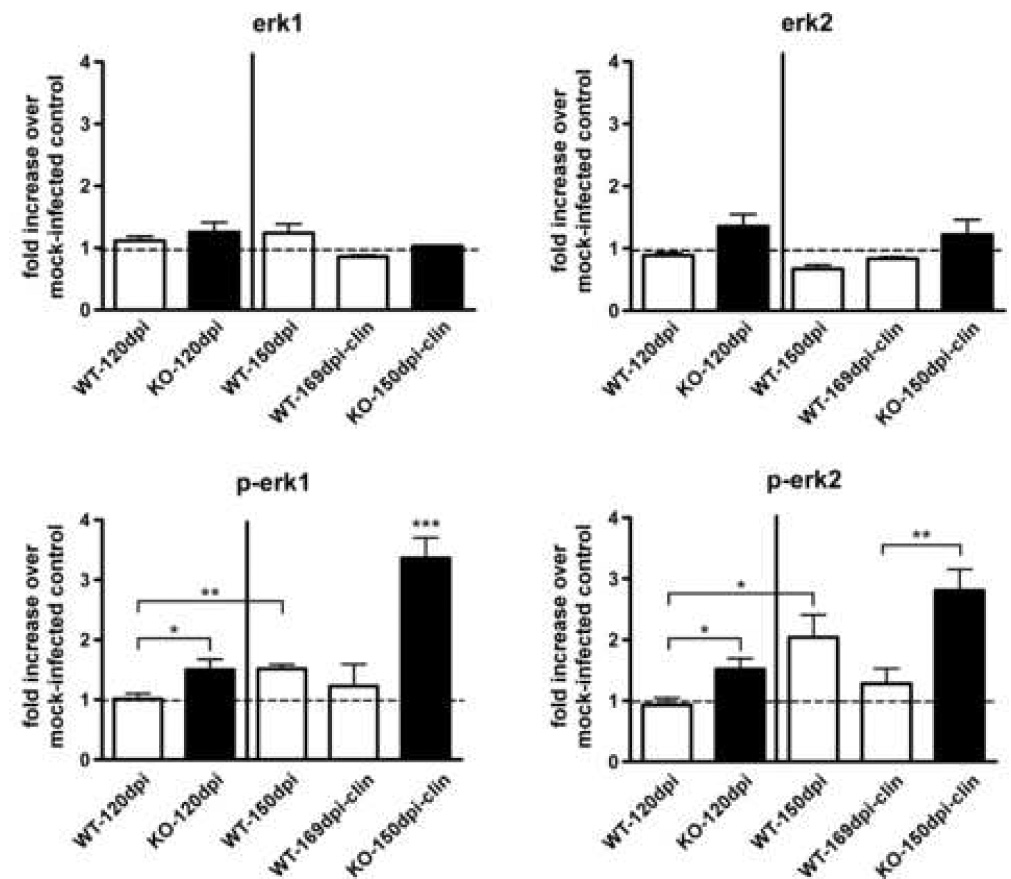
Quantitation of Erk1, Erk2 and p-Erk1/2 in scrapie-infected CCR1 KO and WT mice. Brains from scrapie-infected mice were analyzed for amounts of constitutively expressed Erk1/2 and activated Erk1/2 (p-Erk1/2) by Western blot and quantitated by enhanced chemifluorescence. Data is expressed as fold increase over mock-infected controls. p-Erk1/2 is significantly increased in the CCR1KO mice as compared to WT controls at the preclinical time point (120 dpi), as well as at the clinical time points. In addition, p-Erk1/2 is significantly increased in the 150 dpi WT mice as compared to the preclinical (120dpi) WT mice (Mann-Whitney test; *= p<0.05, **= p<0.005 and ***= p<0.001).
p-Erk1/2 positive cells are primarily astrocytes
To determine what cell types within the brain contain the activated p-Erk1/2 proteins, CCR1 KO mice were evaluated by immunohistochemistry at the onset of clinical disease for the presence of p-Erk1/2. Positive staining for p-Erk1/2 was observed in numerous areas of the brain, as shown in Fig. 7A. The sections were then co-stained with either anti-GFAP or anti-Iba-1 to determine if the p-Erk1/2 staining colocalized with either astrocytes or microglia, respectively. As shown in Fig. 7B, the p-Erk1/2 stain colocalized with the anti-GFAP stain, but not with the anti-Iba-1 stain, indicating that the cells primarily expressing p-Erk1/2 are activated astrocytes. This increase in signaling through the Erk1/2 pathway in astrocytes could potentially lead to astrocyte dysfunction, ultimately resulting in neurodegeneration.
Figure 7.
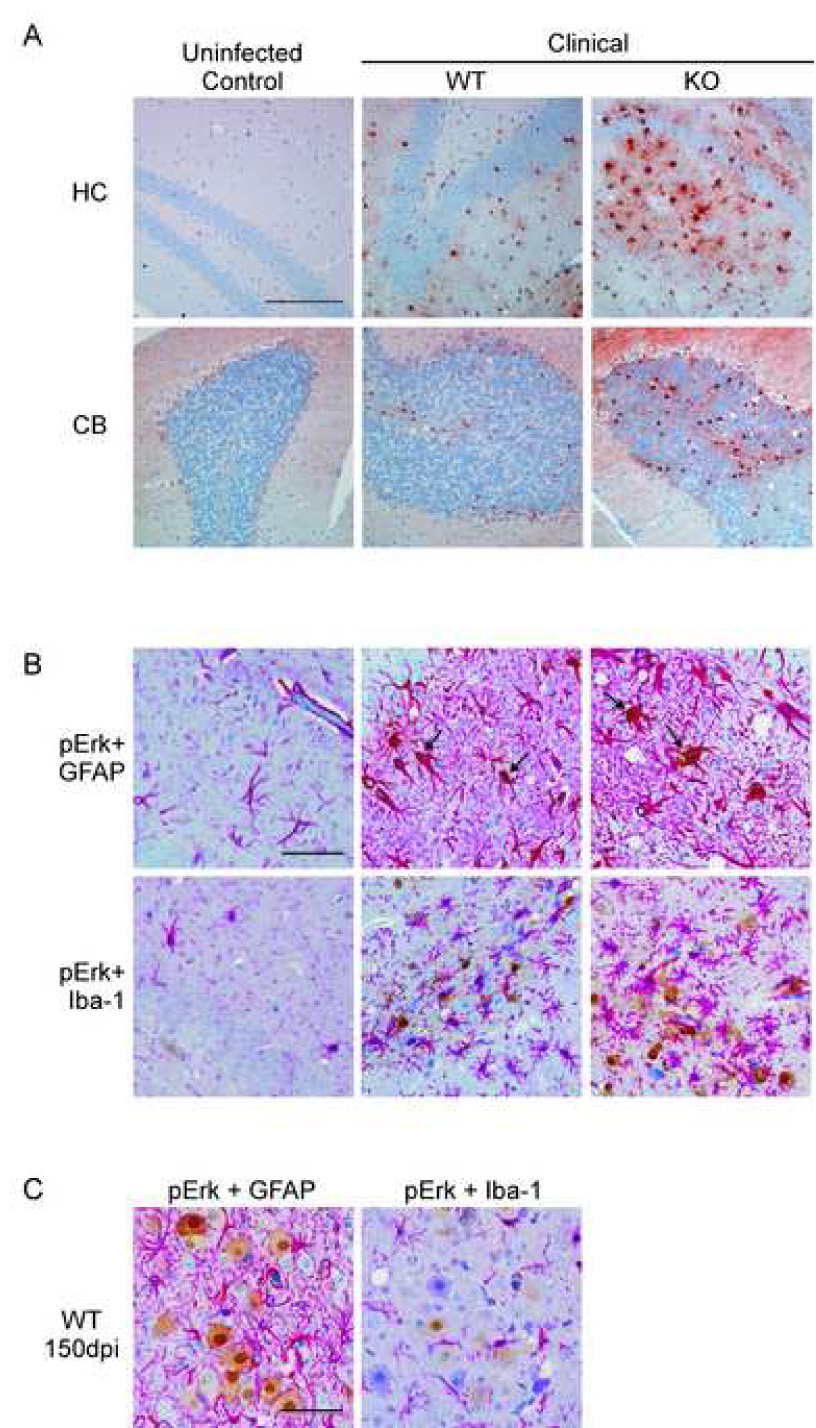
Determination of cells expressing p-Erk1/2 in the brains of scrapie-infected mice. (A) Shown are representative sections of the hippocampus (HC) and cerebellum (CB) from mock-infected controls and scrapie-infected clinical CCR1 KO mice stained for p-Erk1/2 expression. Magnification bar represents 200µm. (B) Sections were then co-stained with either GFAP or Iba-1, which stains the cytoplasms of activated astrocytes or microglia, respectively (bright pink around the nuclei). Cells expressing p-Erk1/2 are stained with DAB, (shown in brown). All sections were counterstained with hematoxylin. Arrows indicate colocalization of p-Erk1/2 expression with the anti-GFAP stain. This co-localization was not seen with anti-Iba-1. Magnification bar represents 100µm. (C) Dual staining of scrapie-infected WT cortex at 150dpi showing p-Erk1/2 staining in large neuron-like cells which were not co-stained by anti-GFAP or anti-Iba-1. Magnification bar represent 100µm.
Interestingly, in pre-clinical WT mice there were areas where p-Erk1/2 staining did not colocalize with either GFAP or Iba-1 staining, suggesting that these cells were most likely neurons (Fig. 7C). The expression of p-Erk1/2 in neurons might suggest a partial protective role for p-Erk1/2 in this situation (Roux and Blenis, 2004; Wada and Penninger, 2004). However, at the time of clinical disease in the WT mice, most p-Erk1/2 staining appeared to be in astrocytes, similar to CCR1 KO mice.
p-Erk1/2 is associated with the activation of transcription factor Elk-1
Upon activation, the p-Erk1/2 proteins phosphorylate either cytoplasmic targets, or migrate to the nucleus to phosphorylate and activate transcription factors. A member of the ets-family of transcription factors, Elk-1, can be phosphorylated by p-Erk1/2 and has been shown to induce apoptosis in cancer cells (Davis, 1993; McCarthy et al., 1997; Shao et al., 1998), has been implicated in pathways leading to neuronal death (Vanhoutte et al., 1999; Lee et al., 2003; Choe et al., 2004), as well as been shown to be activated in neuronal cultures exposed to PrP-res (Marella et al., 2005). To determine whether Elk-1 may be activated during scrapie infection, we evaluated brain homogenates for the presence of phosphorylated Elk-1 (p-Elk-1) protein. There was a marked increase in p-Elk-1 in the clinical CCR1 KO mice, as compared to the WT controls, suggesting that the p-Erk1/2 proteins are activating the Elk-1 transcription factor (Fig. 8). Similar to p-Erk1/2, the activation of Elk-1 could possibly lead to astrocyte dysfunction and neuronal damage and/or death.
Figure 8.
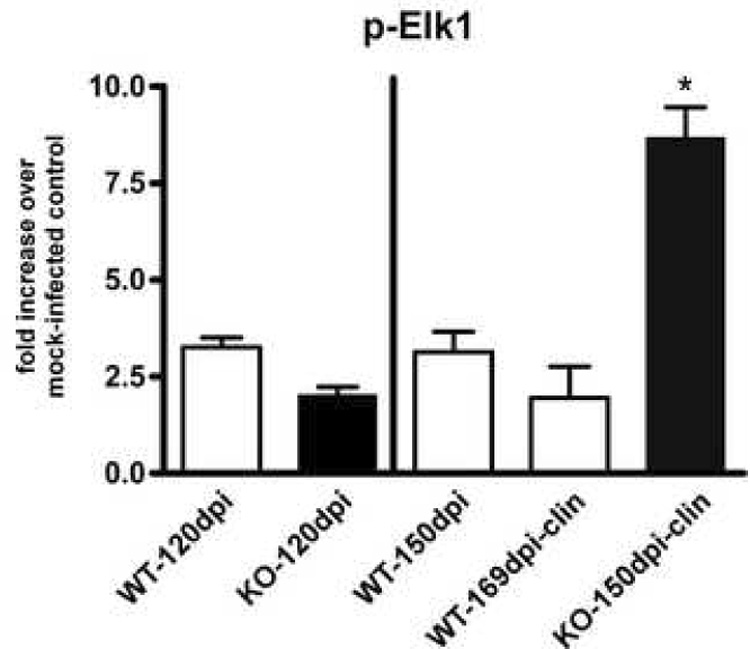
Quantitation of p-Elk-1 in scrapie-infected CCR1 KO and WT mice. Brains from scrapie-infected mice were analyzed for amounts of phosphorylated Elk-1 protein by Western blot and quantitated by enhanced chemifluorescence. Data is expressed as fold increase over mock-infected controls. p-Elk-1 is significantly increased in the clinical CCR1KO mice as compared to WT controls at 150 dpi as well as at the clinical time point (Mann-Whitney test; p<0.05).
4. Discussion
An increase in chemokine receptor expression in the CNS has been associated with neuropathogenesis in a number of neurodegenerative diseases. During Alzheimer’s disease (AD), expression of chemokine receptor CCR1 has been shown to be increased in neurons and associated with amyloid plaques (Halks-Miller et al., 2003). In addition to an increase in CCR1 expression in the neurons of AD brains, there is also an increase in the expression of CCR3 and CCR5 in reactive microglia (Xia et al., 1998). Similarly, during scrapie infection in mice, an increase in CCR1, CCR3 and CCR5 expression in reactive astrocytes and increased CCR5 expression in reactive microglia has been observed (Lee et al., 2005a). However, in these experiments it was unclear whether CCR1, CCR3 and CCR5 upregulation was primary or secondary to CNS damage.
To further evaluate the role of chemokine receptors in scrapie disease, we utilized a CCR1 chemokine receptor KO mouse in the present study. We found that in the absence of CCR1, the kinetics of scrapie disease was accelerated. Although the CCR1 KO mice succumbed to scrapie disease faster than the WT controls, disease progression did not correlate with an increase in the amount of PrP-res, or with a difference in PrP-res distribution. In addition, the acceleration in disease progression in the CCR1 KO mice did not correlate with an increase in microglial or astroglial activation, since CCR1 KO and WT mice were indistinguishable by histopathology, as well as by real-time RT-PCR for astrocyte and microglial activation markers. These data suggest that neither an increase in PrP-res, nor an increase in glial activation is responsible for the accelerated disease progression in the CCR1 KO mice. It is also possible that the size of PrP-res aggregates and sub-cellular location might influence disease progression, but these were not measured in the present study.
There are many possible effects of the lack of CCR1 expression in this system. To evaluate whether other chemokine receptors might be compensating for the lack of CCR1, we determined the relative expression of two receptors, CCR3 and CCR5, which also bind common ligands of CCR1. We found that CCR5 was significantly increased in scrapie-infected CCR1 KO mice as compared to the WT controls, and this increase of CCR5 expression was specific to scrapie infection, as the mock-infected CCR1 KO and WT controls had similar levels of the chemokine receptor. In addition to the increase of CCR5 expression, we found a significant increase in the expression of the chemokine CCL3. Interestingly, CCL3 is a common ligand to both CCR1 and CCR5. Whether the increased expression of CCL3 is linked to the increased expression of CCR5 (or vice versa), or merely a result of the lack of CCR1 expression, has yet to be determined. However, we speculate that the increased expression of both the CCR5 receptor, as well as its ligand, could potentially increase signaling through the CCR5 receptor, and may alter disease progression (Fig. 9A).
Figure 9.
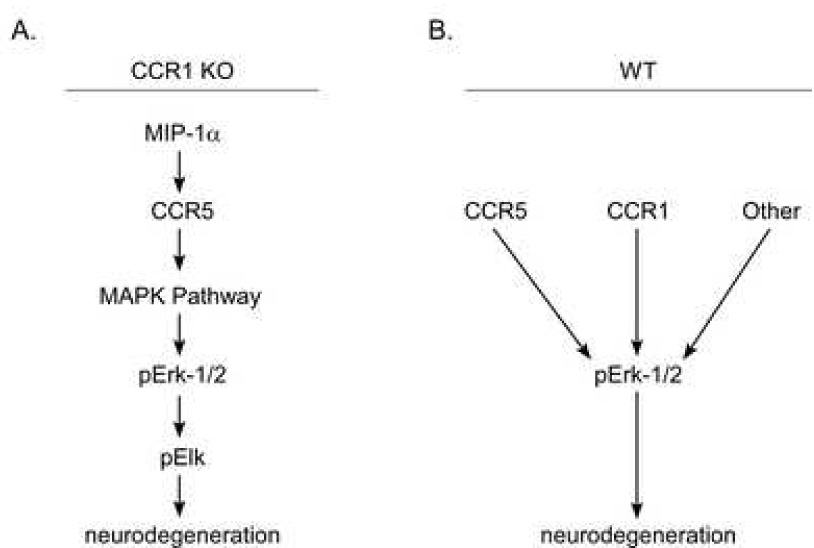
Possible pathways of scrapie-induced neurodegeneration in CCR1 KO and WT mice.
G-coupled protein receptors, such as CCR5, can ultimately signal through several different pathways, depending on the cell that is receiving the signal, as well as the activation state of the cell. One such pathway is the mitogen activated protein kinase (MAPK) pathway, which can induce a variety of cellular functions in different cell types (Fig. 9A). Erk1/2, p38 kinases, and the c-Jun NH2-terminal kinases (JNK) are the three major MAPK subfamilies (Davis, 1993). Traditionally, activation of the Erk1/2 pathway in response to stress or growth factors is believed to be involved in proliferation and survival of the cells (reviewed in (Roux and Blenis, 2004; Wada and Penninger, 2004). However, chronic activation of the Erk1/2 pathway has also been found to play a role in neurodegeneration (Colucci-D'Amato et al., 2003), and has been implicated in several neurodegenerative diseases, including Alzheimer’s (Perry et al., 1999; Rapoport and Ferreira, 2000; Dineley et al., 2001; Russo et al., 2002) and Parkinson’s disease (Kulich and Chu, 2001; Gomez-Santos et al., 2002; Zhu et al., 2002). In addition, upregulation of the Erk1/2 pathway was also seen in scrapie-infected hamsters (Lee et al., 2005b).
The present experiments using CCR1 KO mice demonstrated that upregulation of p-Erk1/2 can play a primary role in accelerating prion disease pathogenesis. Since this upregulation was also seen in WT mice at the clinical time-point, it is likely that p-Erk1/2 plays a role in WT mice as well (Fig. 9B). Astrocytes appeared to be the main sites of p-Erk1/2 upregulation, and it is likely that this might lead to astrocyte dysfunction resulting in neuronal damage. However, some other cell population, such as microglia, might also be involved in this process. Microglia might express increased levels of the CCR5 receptor seen in CCR1 KO mice. Signaling through this receptor by the upregulated CCL3 might lead to microglial activation and production of various cytokines and chemokines, which in turn could activate the Erk1/2 pathway in the astrocytes. Since proper astrocyte function is essential for neuronal health, chronic activation of the Erk1/2 pathway could potentially lead to neuronal excitotoxicity and ultimately neurodegeneration.
Acknowledgements
We thank Richard Race, Sonja Best, Gerry Baron and Kristin McNally for critical reading of the manuscript and helpful suggestion. We acknowledge Anita Mora and Gary Hettrick for excellent technical assistance with graphics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baker CA, Martin D, Manuelidis L. Microglia from Creutzfeldt-Jakob disease-infected brains are infectious and show specific mRNA activation profiles. J Virol. 2002;76:10925–10913. doi: 10.1128/JVI.76.21.10905-10913.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betmouni S, Perry VH. The acute inflammatory response in CNS following injection of prion brain homogenate or normal brain homogenate. Neuropathol Appl Neurobiol. 1999;25:20–28. doi: 10.1046/j.1365-2990.1999.00153.x. [DOI] [PubMed] [Google Scholar]
- Budka H, Aguzzi A, Brown P, Brucher JM, Bugiani O, Gullotta F, Haltia M, Hauw JJ, Ironside JW, Jellinger K, et al. Neuropathological diagnostic criteria for Creutzfeldt-Jakob disease (CJD) and other human spongiform encephalopathies (prion diseases) Brain Pathol. 1995;5:459–466. doi: 10.1111/j.1750-3639.1995.tb00625.x. [DOI] [PubMed] [Google Scholar]
- Burwinkel M, Riemer C, Schwarz A, Schultz J, Neidhold S, Bamme T, Baier M. Role of cytokines and chemokines in prion infections of the central nervous system. Int J Dev Neurosci. 2004;22:497–505. doi: 10.1016/j.ijdevneu.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Campbell IL, Eddleston M, Kemper P, Oldstone MB, Hobbs MV. Activation of cerebral cytokine gene expression and its correlation with onset of reactive astrocyte and acute-phase response gene expression in scrapie. J Virol. 1994;68:2583–2387. doi: 10.1128/jvi.68.4.2383-2387.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- Choe ES, Parelkar NK, Kim JY, Cho HW, Kang HS, Mao L, Wang JQ. The protein phosphatase 1/2A inhibitor okadaic acid increases CREB and Elk-1 phosphorylation and c-fos expression in the rat striatum in vivo. J Neurochem. 2004;89:383–390. doi: 10.1111/j.1471-4159.2003.02334.x. [DOI] [PubMed] [Google Scholar]
- Colucci-D'Amato L, Perrone-Capano C, di Porzio U. Chronic activation of ERK and neurodegenerative diseases. Bioessays. 2003;25:1085–1095. doi: 10.1002/bies.10355. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Boche D, Perry VH. Transforming growth factor beta1, the dominant cytokine in murine prion disease: influence on inflammatory cytokine synthesis and alteration of vascular extracellular matrix. Neuropathol Appl Neurobiol. 2002;28:107–119. doi: 10.1046/j.1365-2990.2002.00383.x. [DOI] [PubMed] [Google Scholar]
- Davis RJ. The mitogen-activated protein kinase signal transduction pathway. J Biol Chem. 1993;268:14553–14556. [PubMed] [Google Scholar]
- Dimcheff DE, Askovic S, Baker AH, Johnson-Fowler C, Portis JL. Endoplasmic reticulum stress is a determinant of retrovirus-induced spongiform neurodegeneration. J Virol. 2003;77:12617–12629. doi: 10.1128/JVI.77.23.12617-12629.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer's disease. J Neurosci. 2001;21:4125–4133. doi: 10.1523/JNEUROSCI.21-12-04125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eikelenboom P, Bate C, Van Gool WA, Hoozemans JJ, Rozemuller JM, Veerhuis R, Williams A. Neuroinflammation in Alzheimer's disease and prion disease. Glia. 2002;40:232–239. doi: 10.1002/glia.10146. [DOI] [PubMed] [Google Scholar]
- Gao JL, Wynn TA, Chang Y, Lee EJ, Broxmeyer HE, Cooper S, Tiffany HL, Westphal H, Kwon-Chung J, Murphy PM. Impaired host defense, hematopoiesis, granulomatous inflammation and type 1-type 2 cytokine balance in mice lacking CC chemokine receptor 1. J Exp Med. 1997;185:1959–1968. doi: 10.1084/jem.185.11.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese A, Kretzschmar HA. Prion-induced neuronal damage--the mechanisms of neuronal destruction in the subacute spongiform encephalopathies. Curr Top Microbiol Immunol. 2001;253:203–217. doi: 10.1007/978-3-662-10356-2_10. [DOI] [PubMed] [Google Scholar]
- Giese A, Brown DR, Groschup MH, Feldmann C, Haist I, Kretzschmar HA. Role of microglia in neuronal cell death in prion disease. Brain Pathol. 1998;8:449–457. doi: 10.1111/j.1750-3639.1998.tb00167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Santos C, Ferrer I, Reiriz J, Vinals F, Barrachina M, Ambrosio S. MPP+ increases alpha-synuclein expression and ERK/MAP-kinase phosphorylation in human neuroblastoma SH-SY5Y cells. Brain Res. 2002;935:32–39. doi: 10.1016/s0006-8993(02)02422-8. [DOI] [PubMed] [Google Scholar]
- Halks-Miller M, Schroeder ML, Haroutunian V, Moenning U, Rossi M, Achim C, Purohit D, Mahmoudi M, Horuk R. CCR1 is an early and specific marker of Alzheimer's disease. Ann Neurol. 2003;54:638–646. doi: 10.1002/ana.10733. [DOI] [PubMed] [Google Scholar]
- Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res. 1998;57:1–9. doi: 10.1016/s0169-328x(98)00040-0. [DOI] [PubMed] [Google Scholar]
- Kim JI, Ju WK, Choi JH, Choi E, Carp RI, Wisniewski HM, Kim YS. Expression of cytokine genes and increased nuclear factor-kappa B activity in the brains of scrapie-infected mice. Brain Res Mol Brain Res. 1999;73:17–27. doi: 10.1016/s0169-328x(99)00229-6. [DOI] [PubMed] [Google Scholar]
- Kordek R, Nerurkar VR, Liberski PP, Isaacson S, Yanagihara R, Gajdusek DC. Heightened expression of tumor necrosis factor alpha, interleukin 1 alpha, and glial fibrillary acidic protein in experimental Creutzfeldt-Jakob disease in mice. Proc Natl Acad Sci U S A. 1996;93:9754–9758. doi: 10.1073/pnas.93.18.9754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulich SM, Chu CT. Sustained extracellular signal-regulated kinase activation by 6-hydroxydopamine: implications for Parkinson's disease. J Neurochem. 2001;77:1058–1066. doi: 10.1046/j.1471-4159.2001.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Choi SS, Han KJ, Han EJ, Suh HW. Cycloheximide inhibits neurotoxic responses induced by kainic acid in mice. Brain Res Bull. 2003;61:99–107. doi: 10.1016/s0361-9230(03)00078-9. [DOI] [PubMed] [Google Scholar]
- Lee HP, Jun YC, Choi JK, Kim JI, Carp RI, Kim YS. The expression of RANTES and chemokine receptors in the brains of scrapie-infected mice. J Neuroimmunol. 2005a;158:26–33. doi: 10.1016/j.jneuroim.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Lee HP, Jun YC, Choi JK, Kim JI, Carp RI, Kim YS. Activation of mitogen-activated protein kinases in hamster brains infected with 263K scrapie agent. J Neurochem. 2005b;95:584–593. doi: 10.1111/j.1471-4159.2005.03429.x. [DOI] [PubMed] [Google Scholar]
- Marella M, Gaggioli C, Batoz M, Deckert M, Tartare-Deckert S, Chabry J. Pathological prion protein exposure switches on neuronal mitogen-activated protein kinase pathway resulting in microglia recruitment. J Biol Chem. 2005;280:1529–1534. doi: 10.1074/jbc.M410966200. [DOI] [PubMed] [Google Scholar]
- McCarthy SA, Chen D, Yang BS, Garcia Ramirez JJ, Cherwinski H, Chen XR, Klagsbrun M, Hauser CA, Ostrowski MC, McMahon M. Rapid phosphorylation of Ets-2 accompanies mitogen-activated protein kinase activation and the induction of heparin-binding epidermal growth factor gene expression by oncogenic Raf-1. Mol Cell Biol. 1997;17:2401–2412. doi: 10.1128/mcb.17.5.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordstrom EK, Luhr KM, Ibanez C, Kristensson K. Inhibitors of the mitogen-activated protein kinase kinase 1/2 signaling pathway clear prion-infected cells from PrPSc. Journal of Neuroscience. 2005;25:8451–8456. doi: 10.1523/JNEUROSCI.2349-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry G, Roder H, Nunomura A, Takeda A, Friedlich AL, Zhu X, Raina AK, Holbrook N, Siedlak SL, Harris PL, Smith MA. Activation of neuronal extracellular receptor kinase (ERK) in Alzheimer disease links oxidative stress to abnormal phosphorylation. Neuroreport. 1999;10:2411–2415. doi: 10.1097/00001756-199908020-00035. [DOI] [PubMed] [Google Scholar]
- Peterson KE, Hughes S, Dimcheff DE, Wehrly K, Chesebro B. Separate sequences in a murine retroviral envelope protein mediate neuropathogenesis by complementary mechanisms with differing requirements for tumor necrosis factor alpha. J Virol. 2004a;78:13104–13112. doi: 10.1128/JVI.78.23.13104-13112.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson KE, Errett JS, Wei T, Dimcheff DE, Ransohoff R, Kuziel WA, Evans L, Chesebro B. MCP-1 and CCR2 contribute to non-lymphocyte-mediated brain disease induced by Fr98 polytropic retrovirus infection in mice: role for astrocytes in retroviral neuropathogenesis. J Virol. 2004b;78:6449–6458. doi: 10.1128/JVI.78.12.6449-6458.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Race R, Oldstone M, Chesebro B. Entry versus blockade of brain infection following oral or intraperitoneal scrapie administration: role of prion protein expression in peripheral nerves and spleen. J Virol. 2000;74:828–833. doi: 10.1128/jvi.74.2.828-833.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Race R, Ernst D, Jenny A, Taylor W, Sutton D, Caughey B. Diagnostic implications of detection of proteinase K-resistant protein in spleen, lymph nodes, and brain of sheep. Am J Vet Res. 1992;53:883–889. [PubMed] [Google Scholar]
- Rapoport M, Ferreira A. PD98059 prevents neurite degeneration induced by fibrillar beta-amyloid in mature hippocampal neurons. J Neurochem. 2000;74:125–133. doi: 10.1046/j.1471-4159.2000.0740125.x. [DOI] [PubMed] [Google Scholar]
- Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, Peterson PK. Role of microglia in central nervous system infections. Clin Microbiol Rev. 2004;17:942–964. doi: 10.1128/CMR.17.4.942-964.2004. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68:320–344. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo C, Dolcini V, Salis S, Venezia V, Zambrano N, Russo T, Schettini G. Signal transduction through tyrosine-phosphorylated C-terminal fragments of amyloid precursor protein via an enhanced interaction with Shc/Grb2 adaptor proteins in reactive astrocytes of Alzheimer's disease brain. J Biol Chem. 2002;277:35282–35288. doi: 10.1074/jbc.M110785200. [DOI] [PubMed] [Google Scholar]
- Shao N, Chai Y, Cui JQ, Wang N, Aysola K, Reddy ES, Rao VN. Induction of apoptosis by Elk-1 and deltaElk-1 proteins. Oncogene. 1998;17:527–532. doi: 10.1038/sj.onc.1201931. [DOI] [PubMed] [Google Scholar]
- Thackray AM, McKenzie AN, Klein MA, Lauder A, Bujdoso R. Accelerated prion disease in the absence of interleukin-10. J Virol. 2004;78:13697–13707. doi: 10.1128/JVI.78.24.13697-13707.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhoutte P, Barnier JV, Guibert B, Pages C, Besson MJ, Hipskind RA, Caboche J. Glutamate induces phosphorylation of Elk-1 and CREB, along with c-fos activation, via an extracellular signal-regulated kinase-dependent pathway in brain slices. Mol Cell Biol. 1999;19:136–146. doi: 10.1128/mcb.19.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–2849. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- Wells GA. Pathology of nonhuman spongiform encephalopathies: variations and their implications for pathogenesis. Dev Biol Stand. 1993;80:61–69. [PubMed] [Google Scholar]
- Williams A, Van Dam AM, Ritchie D, Eikelenboom P, Fraser H. Immunocytochemical appearance of cytokines, prostaglandin E2 and lipocortin-1 in the CNS during the incubation period of murine scrapie correlates with progressive PrP accumulations. Brain Res. 1997;754:171–180. doi: 10.1016/s0006-8993(97)00067-x. [DOI] [PubMed] [Google Scholar]
- Wojtera M, Sikorska B, Sobow T, Liberski PP. Microglial cells in neurodegenerative disorders. Folia Neuropathol. 2005;43:311–321. [PubMed] [Google Scholar]
- Xia MQ, Qin SX, Wu LJ, Mackay CR, Hyman BT. Immunohistochemical study of the beta-chemokine receptors CCR3 and CCR5 and their ligands in normal and Alzheimer's disease brains. Am J Pathol. 1998;153:31–37. doi: 10.1016/s0002-9440(10)65542-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JH, Kulich SM, Oury TD, Chu CT. Cytoplasmic aggregates of phosphorylated extracellular signal-regulated protein kinases in Lewy body diseases. Am J Pathol. 2002;161:2087–2098. doi: 10.1016/S0002-9440(10)64487-2. [DOI] [PMC free article] [PubMed] [Google Scholar]