

Abstract
The prevention of autoimmunity requires elimination of self-reactive T cells during their development and maturation. Expression of diverse self-antigens by stromal cells in the thymus is essential to this process, and depends, in part, on the activity of the Autoimmune Regulator (Aire) gene. Here we report the identification of extrathymic Aire-expressing cells (eTACs) resident within the secondary lymphoid organs. These stromally-derived eTACs express a diverse array of unique self-antigens and are capable of interacting with and deleting naïve autoreactive T cells. Using two-photon microscopy we observe stable, antigen-specific interactions between eTACs and autoreactive T cells. We propose that such a secondary network of self-antigen-expressing stromal cells may help reinforce immune tolerance by preventing the maturation of autoreactive T cells that escape thymic negative selection.
Immunological tolerance to self is essential in the prevention of autoimmune disease. Mechanisms of central tolerance are mediated in part through the expression of a wide array of otherwise tissue-specific self-antigens (TSAs) such as insulin and thyroglobulin in specialized medullary thymic epithelial cells (mTECs; 1, 2, 3). The thymic expression of many of these TSAs is dependent on the Autoimmune Regulator (Aire) gene (4, 5), and mutations in Aire lead to severe, multi-organ, tissue-specific autoimmunity in both mice (4, 6) and humans (7, 8). Although these results reveal a role for thymic Aire, self-tolerance must continue to be enforced after T cells leave the thymus. Consistent with this, Aire expression is also detectable outside the thymus, notably in the secondary lymphoid tissues (4,9), although the identity and function of such extrathymic Aire-expressing cells remains unclear (10, 11). Here we identify a population of extrathymic Aire-expressing cells and examine a potential role for Aire in maintaining peripheral tolerance.
To accurately label Aire-expressing cells in vivo, we employed a bacterial artificial chromosome (BAC)-transgenic approach (12) using the murine Aire locus modified to drive expression of green fluorescent protein (Gfp) fused to an autoimmune diabetes-related self-antigen gene, islet-specific glucose-6-phosphatase related protein (Igrp, Fig. 1A, 13). IGRP is a pancreatic β-cell specific protein against which autoreactive CD8 T cells are produced in both mouse and human autoimmune diabetes (14, 15, 16, 17). We elected to include Igrp in our transgenic construct because it is not detectable in the thymus (Fig. S1A) and because an IGRP-specific T-cell-receptor transgenic line (8.3; 14) can be used to monitor interactions of Igrp-Gfp-expressing cells. To verify the fidelity of the BAC transgene in recapitulating endogenous Aire expression in the resultant Adig (Aire-Driven Igrp-Gfp) transgenic mice, thymic sections were co-stained for Aire and GFP, revealing thymic GFP expression highly restricted to Aire-expressing cells in the medulla (Fig. 1B, Fig. S1B). By flow cytometry GFP+ cells were detectable specifically within the mTEC compartment (Fig. 1C), with approximately 30-40% of mTECs being GFP-positive. GFP+ mTECs expressed uniformly high levels of class II MHC and CD80 (Fig. 1D), similar to previous studies of Aire-expressing mTECs (3, 11, 18)
Fig. 1.
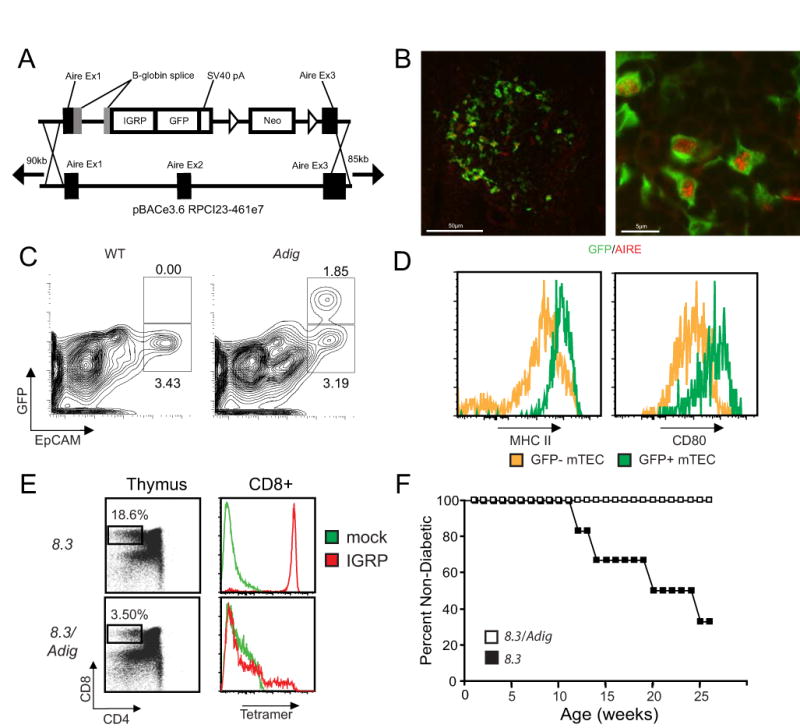
The Adig transgene recapitulates Aire expression in the thymus and mediates negative selection of autoreactive T cells. (A) Schematic of Igrp-gfp transgene targeting into the Aire BAC. Targeting replaces Aire exon 2, the coding portion of exon 1 and part of exon 3, such that the transgene does not make functional Aire (B) Immunofluorescent staining of GFP (green) and Aire (red) in thymic frozen sections of Adig mice. (C) Flow cytometry of CD45-, DAPI-, Ly51lo-gated thymic stromal cells from wildtype (left) and Adig (right) NOD mice. (D) Flow cytometry of CD45-, DAPI-, Ly51lo, EpCAM+ mTECs from Adig NOD mice gated as GFP- (yellow) or GFP+ (green) and stained for MHC II (left) and CD80 (right). (E) Thymocytes from 8.3 TCR-transgenic mice (top panels) or double-transgenic 8.3/Adig mice (bottom panels) stained for CD4/CD8 (left panels), or pre-gated as CD4- CD8+ and stained for IGRP-mimetope (red) or mock (green) peptide/Kd tetramer reactivity (right panels). (F) Diabetes incidence curves for 8.3 TCR-transgenic mice (N=6) and 8.3/Adig double-transgenic mice (N=10).
To test how the introduction of IGRP into the thymic medullary epithelium might affect T-cell selection, we compared 8.3 TCR-transgenic and 8.3/Adig double-transgenic mice. Tetramer staining confirmed that the 8.3/Adig double-transgenic mice showed a significant decrease in the percent and avidity of IGRP-specific CD8+ T cells in the thymus (Fig. 1E) and in the periphery (Fig. S1C). Further, while IGRP-reactive CD8+ T cells were readily detected in the polyclonal wildtype NOD background, they were completely absent in Adig NOD mice (Fig. S1D). To test the functional impact of this negative selection, 8.3 and 8.3/Adig mice were followed for diabetes incidence, and 8.3/Adig mice were completely protected from disease (Fig. 1F).
In a broad tissue survey using immunofluorescent anti-GFP staining, expression of the transgene was undetectable in most tissues, but distinct populations of extrathymic transgene-expressing cells were observed within the lymph nodes and spleen (Fig. S2A). These extra-Thymic Aire-expressing Cells (eTACs) were generally confined to the T-cell zones of the secondary lymphoid organs and preferentially localized to T cell-B cell boundary regions (Fig. 2A, B). Immunofluorescent costains showed these cells to be negative for B cell (B220), fibroblastic reticular cell (gp38, ERTR-7), and dendritic cell (CD11c) markers, but positive for class II MHC (Fig. 2A, B). Reciprocal bone-marrow chimeras demonstrated that many of these cells were stromal in origin (Fig. S3A). Given this, we isolated secondary lymphoid stroma for flow cytometry and found that, as in the thymus, a unique population of GFP+ cells was present that was CD45-, MHC II+ (Fig. 2C). These CD45- eTACs shared some characteristics with mTECs (e.g. MHC II+, PDL1+, EpCAM+), but were distinct in that eTACs did not express the costimulatory molecules CD80 and CD86 (Fig. 2D). While GFP+ eTACs represented a significant percentage of the EpCAM+ stromal cells in the periphery (8.5% ±2.4%), eTACs failed to bind the mTEC marker UEA-I or the FRC marker gp38, suggesting they are distinct from previously described self-antigen-expressing stromal populations (9, Fig. S4). eTACs also appeared to be ubiquitous in lymphoid organs, as they were detected in mesenteric lymph nodes, Peyer's patches (Fig. S2B) and in the tertiary lymphoid structures that form in the infiltrated pancreatic islets of NOD mice (Fig. S2C). By flow cytometry GFP+ cells were also detected in the CD45+ compartment that expressed CD11c, although the level of GFP expression in these cells was significantly lower than in the CD45- cells and less enriched for Aire message (Fig. 2E, S4).
Fig. 2.
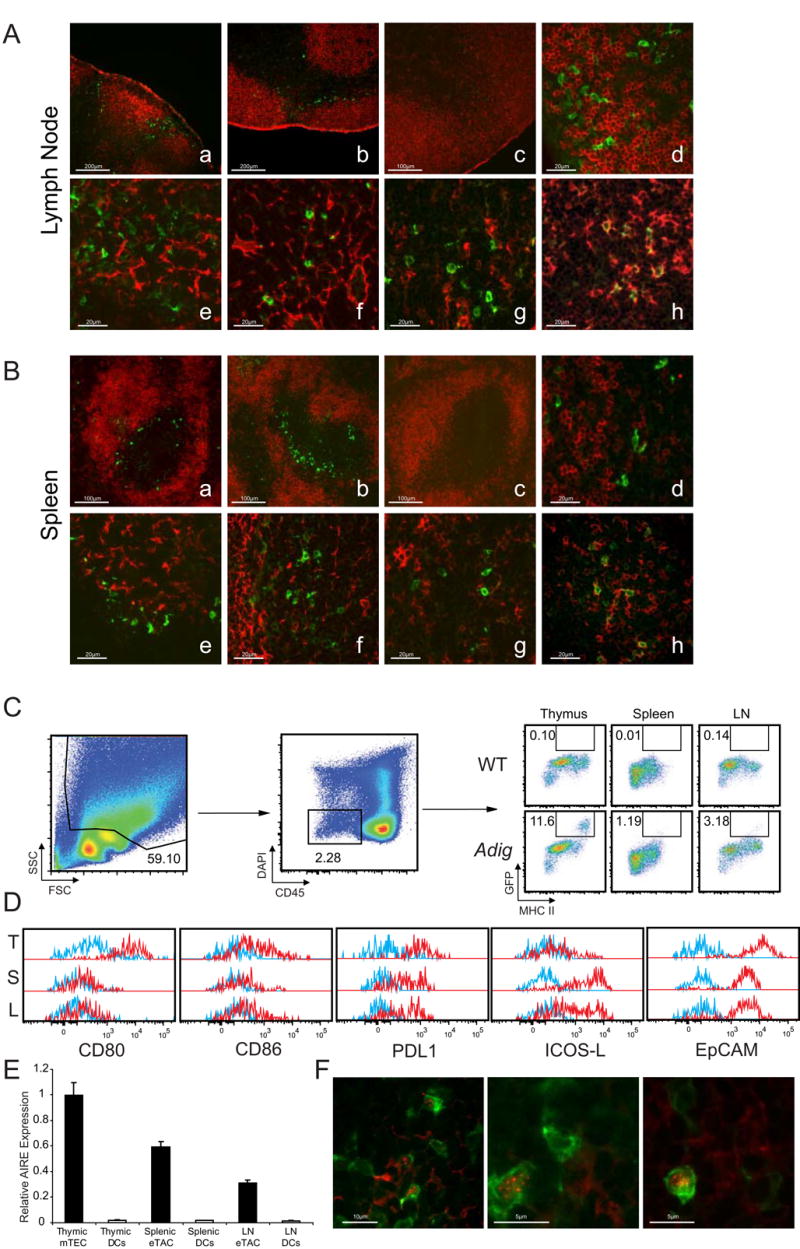
Aire-expressing stromal cells exist in the secondary lymphoid organs. (A and B) Representative immunofluorescent costains of lymph node (A) and spleen (B) sections co-stained for GFP (green, all sections) and B220 (a-d), gp38 (e), ERTR-7 (f), CD11c (g), or MHC II (h; all red). Images a, b, and d-h are from Adig NOD mice, images c are from wildtype NOD mice. (C) Flow cytometric analysis and gating from Adig and wildtype NOD thymus, spleen, and lymph node stroma analyzed for CD45, DAPI, MHC II, and GFP. (D) Flow cytometric analysis of Adig NOD thymus (T), spleen (S), and lymph nodes (L), showing expression of indicated markers (red) or isotype staining (blue) in mTECs and eTACs respectively, defined as CD45-, DAPI-, MHC II+, GFP+. (E) Real-time PCR analysis of Aire expression relative to endogenous control in cell-sorted mTECs/eTACs (CD45-, PI-, MHC II+, CD11c-, EpCAM+) and DCs (CD45+, PI-, MHC II+, CD11c+, EpCAM-) of nontransgenic NOD thymus, spleen, and lymph node. (F) Immunofluorescent GFP (green)/Aire (red) costains of lymph nodes from Adig NOD mice.
To validate that Igrp-Gfp transgene expression in eTACs reflected endogenous Aire expression, the eTAC surface markers identified in Adig mice (CD45-, MHC II+, EpCAM+) were used to sort eTACs from non-transgenic mice, and Aire transcript was indeed found to be abundant in these cells, confirming that this stromal population expressed high levels of Aire (Fig. 2E). Co-staining of secondary lymphoid organs for Aire and GFP also demonstrated Aire protein localized to perinuclear speckles within a subset of eTACs (Fig. 2F). The number of GFP+ cells in which Aire protein can be detected was smaller in the periphery (24.8% ±4.6%) than in the thymus (85.0% ±4.5%), and the Aire staining much weaker—near the limit of detection—which may explain why previous attempts to identify these cells in the absence of the transgenic reporter have been difficult (11).
Because Aire has been shown to play an important role in the transcriptional regulation of self-antigens in mTECs, we sought to define its function as a transcriptional regulator in eTACs. GFP+ eTACs were sorted for microarray analysis from the spleens and pooled lymph nodes of Adig mice crossed onto the Aire+/+ or Aire-/- background. As in mTECs, the number of genes upregulated by Aire in eTACs was greater than the number downregulated (Fig. 3A, Tables S1, S2). Both the total number of Aire-regulated genes in eTACs and the fold change of expression of those genes were smaller than has been observed in mTECs, perhaps reflecting the lower and potentially transient expression of Aire in the periphery. Strikingly, there was little overlap between Aire-regulated genes in eTACs and those in mTECs (Fig. 3B), suggesting that Aire in the periphery may regulate expression of a unique set of self-antigens. Despite these differences, however, we found a significant enhancement for TSAs among the positively Aire-regulated genes in eTACs (Fig. 3C), several of which were confirmed by quantitative RT-PCR (Fig. 3D). The list of genes regulated by Aire in eTACs also included a number of self-antigens whose human homologs have been described as autoantigens in human autoimmune diseases, including desmoglein 1a (pemphigus foliaceus; 19), ladinin 1 (linear IgA dermatosis, 20), and the NMDA receptor 2C (systemic lupus erythematosis, 21). Like other professional APCs, eTACs also expressed a large number of antigen-processing and presentation genes, suggesting a likely role for T-cell interaction (Fig. 3E). Comparison of global gene expression profiles between eTACs, mTECs, cTECs, thymic DCs and macrophages indicated that eTACs were most similar to DCs and mTECs (Fig. 3F).
Fig. 3.

Aire regulates the expression of a unique set of tissue-specific antigens in eTACs. (A) Heat map and unsupervised clustering of Aire-regulated genes in eTACs. Pooled eTACs were sorted from lymph nodes and spleens from cohorts of 3-6-week-old Adig Aire+/+ and Adig Aire-/- NOD mice. Each of the 8 arrays represents 3-5 pooled mice. (B) Schematic diagram of the unique and common genes regulated by Aire in eTACs and mTECs. (C) Classification of Aire-regulated genes in eTACs based on tissue-specificity, as compared to mTECs and to a random gene set. (D) Real-time PCR analysis of Aire-regulated TSAs in eTACs, normalized to endogenous control. eTACs were sorted from pooled nontransgenic Aire+/+ (black bars) and Aire-/- (white bars) NOD spleens based on the surface marker profile CD45-, PI-, CD11c-, MHC II+, EpCAM+, and characterized for expression of glutamate receptor NMDA2C (Grin2c), ras-related associated with diabetes (Rrad), ladinin (Lad1), gulonolactone (L-) oxidase (Gulo), and desmoglein 1 alpha (Dsg1a) (E) Expression of antigen processing and presentation genes in eTACs relative to other lymphoid cell populations after median-centered normalization to the expression of all genes in each array. (F) Global expression profile similarity of eTACs (left) and mTECs (right) to other relevant cell types based on Pearson correlation values calculated for population-specific centroids.
To directly test the ability of eTACs to promote tolerance by interacting with and deleting autoreactive T cells, adoptive co-transfer of CFSE-labeled congenic 8.3 and polyclonal CD8+ T cells was employed. Upon transfer into wildtype hosts, 8.3 CD8+ T cells proliferated only in the pancreatic lymph nodes, and persisted in all lymph nodes for up to two weeks (Fig. 4A). When transferred into Adig hosts, however, the entire population of 8.3 T cells proliferated rapidly in all secondary lymphoid organs by three days (Fig 4A), and had nearly disappeared by two weeks (Fig. 4A, C). To confirm that the absence of 8.3 T cells in transgenic recipients was due to cell death and not egress, these experiments were repeated in the presence of the S1P1 inhibitor FTY 720 (22, Fig. S5). Further, to determine whether a stromal eTAC population was sufficient to directly mediate this deletion, irradiated wildtype and Adig mice were reconstituted with bone marrow deficient for β2-microglobulin (β2M-/-) so that only stromal cells were capable of interacting with CD8+ lymphocytes. Chimerism was confirmed both by blood typing and functionally by observing that 8.3 T cells failed to proliferate in the pancreatic lymph nodes of nontransgenic β2M-/- reconstituted mice (Fig. 4B). In contrast, 8.3 T cells continued to proliferate in all secondary lymphoid organs of Adig β2M-/- reconstituted mice at 3 days, and all divided 8.3 T cells had been deleted by 14 days (Fig. 4B, C). While 8.3 cell division was less robust at day 3 in β2M-/- chimeric Adig mice when compared to unirradiated Adig mice, antigen-specific cell death at day 14 was even more dramatic in this setting (Fig. 4A vs. B).
Fig. 4.
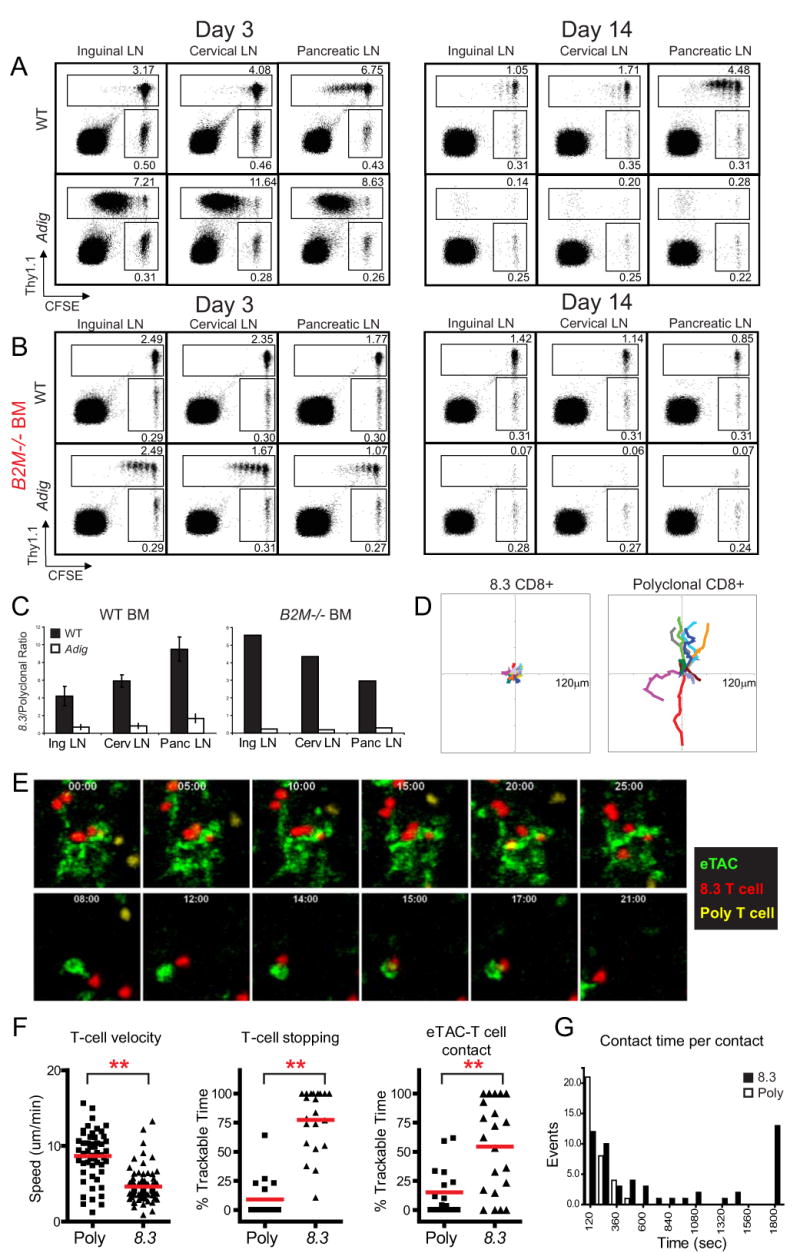
eTACs directly interact with autoreactive lymphocytes and mediate deletional tolerance. (A) Flow cytometry of CFSE-labeled and adoptively cotransferred 8.3 CD8+ T cells (Thy1.1) and polyclonal CD8+ T cells (Thy 1.2). Cells were harvested at day 3 (left) and day 14 (right) post-transfer (B) Adoptive transfer of the same donor populations as (A) into lethally irradiated wildtype (top) and Adig (bottom) recipients reconstituted with β2M-/- bone marrow. (C) Quantitation of antigen-specific deletion after adoptive transfer at day 14 in (A) and (B), showing the ratio of 8.3:polyclonal CD8+ T cells in wildtype (black) and Adig (white) NOD recipients. Representative of at least three mice each. (D-G) Two-photon imaging of 8.3 and polyclonal CD8+ T cells in axillary lymph nodes 4 hours after adoptive transfer into Adig NOD recipients. (D) 10-minute displacement analysis of all 8.3 CD8+ T cell tracks (left) and polyclonal CD8+ T cell tracks (right). (E) Representative images of interaction between 8.3 CD8+ T cells (red), polyclonal CD8+ T cells (yellow) and eTACs (green) (F) Average T cell track speed (left), percent of time in which a T cell is stopped (middle), and percent of time in which a T cell is making contact with an eTAC (right) among polyclonal (Poly) and 8.3 (8.3) CD8 T cells. ** P<0.001. (G) Histogram displaying the duration of individual T cell-eTAC interaction times per contact for polyclonal (black bars) and 8.3 (white bars) CD8+ T cells.
To clearly delineate whether eTACs can directly interact with T cells, two-photon microscopy of explanted lymph nodes was employed. GFP+ cells were observed uniquely in all examined lymph nodes of Adig mice (Movie S1), in a distribution that mirrored the localization of eTACs observed by immunofluorescent staining (Movie S2, Fig. 2A). Adoptive cotransfer of fluorophore-labeled 8.3 and polyclonal CD8+ T cells demonstrated sustained antigen-specific association between naïve 8.3 T cells and eTACs as early as 4 hours post-transfer (Fig. 4D, 4E, Movies S3-S4). 8.3 T cells exhibited distinct reductions in speed and total displacement relative to polyclonal CD8+ T cells (Fig. 4D, F), and 8.3 T cells spent significantly more time both stopped and in direct contact with eTACs (Fig. 4F). Indeed, the upper limit of this interaction time is unknown, as many 8.3 T cells spent the entire 30-minute duration of the acquisition periods in continuous contact (Fig. 4G, Movie S5). Some GFP+ cells in the lymph node appeared highly motile, but 8.3 T cells were able to maintain antigen-specific interactions despite this motility (Fig. 4E, Movie S6). Together, these results suggested that eTACs can form early, stable, long-term contacts with naïve autoreactive T cells entering the lymph node, and that such interaction leads to rapid proliferation and deletion of these T cells.
Here we have identified a novel population of extrathymic Aire-expressing cells (eTACs) that may play a significant and previously uncharacterized role in self-tolerance via the deletion of autoreactive T cells. Indeed eTACs share certain characteristics with mTECs, including being equipped to act as professional antigen-presenting cells and the Aire-regulated expression of unique tissue-specific antigens (TSAs). Interestingly, the set of Aire-regulated TSAs expressed in eTACs appears to have little overlap with thymic Aire-regulated antigens, which may explain why previous efforts examining known thymic TSAs for expression in secondary lymphoid organs has been inconsistent or conflicting. The lack of overlap suggests that there may be a higher order of Aire-dependent transcriptional regulation of TSA expression, whether direct or indirect, that differs between the thymus and the periphery. Many important questions remain, including the developmental origin of eTACs, and their relationship with other possible stromal APC populations (9). In this regard, the precise identification of eTACs will provide the framework for further exploring these issues. Further, it will be important to determine the physiologic relevance of this cell population in a non-transgenic setting, given the modest levels of TSA expression and and Aire-dependent gene upregulation in eTACs (approximately two-fold). However, it is notable that similar analyses of Aire-expressing thymic epithelial cells (mTECs) have demonstrated that TSAs are expressed at low levels in these cells (2), but even this low level of expression has proven critical for the maintenance of immune tolerance (4, 23). Indeed, our findings suggest that eTACs may represent a “safety net” within the entire immunologic periphery which functions to screen out naïve autoreactive T cell clones that escape thymic negative selection. Finally, we speculate that eTACs may play an increasingly significant role with advancing age, as the thymus involutes and the burden of maintaining self-tolerance shifts to the periphery.
Supplementary Material
Acknowledgments
We thank J. Bluestone, A. Abbas, N. Killeen, and J. Cyster for helpful discussion. J.G. is funded by the UCSF/NIH Medical Scientist Training Program and the American Diabetes Association; J.D. by the Giannini Foundation; R.F. by the Larry L. Hillblom Foundation; D.W. by a Dermatology Foundation Research Career Development Award. M.S.A. is supported by the Pew Scholars Program, the Sandler Foundation, and the Burroughs Wellcome Fund. This work was supported in part by the NIH (to M.S.A.). The authors have no competing financial interests.
Footnotes
This manuscript has been accepted for publication in Science. This version has not undergone final editing. Please refer to the complete version of record at http://www.sciencemag.org/. Their manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of the AAAS.
References and Notes
- 1.Smith KM, Olson DC, Hirose R, Hanahan D. Int Immunol. 1997;9:1355. doi: 10.1093/intimm/9.9.1355. [DOI] [PubMed] [Google Scholar]
- 2.Derbinski J, Schulte A, Kyewski B, Klein L. Nature Immunol. 2001;2:1032. doi: 10.1038/ni723. [DOI] [PubMed] [Google Scholar]
- 3.Derbinski J, et al. J Exp Med. 2005;202:33. doi: 10.1084/jem.20050471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson MS, et al. Science. 2002;298:1395. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- 5.Liston A, Lesage S, Wilson J, Peltonen L, Goodnow CC. Nature Immunol. 2003;4:350. doi: 10.1038/ni906. [DOI] [PubMed] [Google Scholar]
- 6.Ramsey C, et al. Hum Mol Genet. 2002;11:397. doi: 10.1093/hmg/11.4.397. [DOI] [PubMed] [Google Scholar]
- 7.Nagamine K, et al. Nature Genet. 1997;17:393. doi: 10.1038/ng1297-393. [DOI] [PubMed] [Google Scholar]
- 8.Finnish-German APECED Consortium. Nature Genet. 1997;17:399. doi: 10.1038/ng1297-399. [DOI] [PubMed] [Google Scholar]
- 9.Lee JW, et al. Nature Immunol. 2007;8:181. doi: 10.1038/ni1427. [DOI] [PubMed] [Google Scholar]
- 10.Halonen M, et al. J Histochem Cytochem. 2001;49:197. doi: 10.1177/002215540104900207. [DOI] [PubMed] [Google Scholar]
- 11.Hubert FX, et al. J Immunol. 2008;180:3824. doi: 10.4049/jimmunol.180.6.3824. [DOI] [PubMed] [Google Scholar]
- 12.Yu W, et al. Science. 1999;285:1080. doi: 10.1126/science.285.5430.1080. [DOI] [PubMed] [Google Scholar]
- 13.Materials and Methods are available as supporting material Science online.
- 14.Nagata M, Santamaria P, Kawamura T, Utsugi T, Yoon JW. J Immunol. 1994;152:2042. [PubMed] [Google Scholar]
- 15.Han B, et al. J Clin Invest. 2005;115:1879. doi: 10.1172/JCI24219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lieberman SM, et al. Proc Natl Acad Sci USA. 2003;100:8384. doi: 10.1073/pnas.0932778100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jarchum I, Nichol L, Trucco M, Santamaria P, Dilorenzo TP. Clin Immunol. 2008;127:359. doi: 10.1016/j.clim.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gray D, Abramson J, Benoist C, Mathis D. J Exp Med. 2007;204:2521. doi: 10.1084/jem.20070795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin MS, et al. J Clin Invest. 2000;105:207. doi: 10.1172/JCI8075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marinkovich MP, Taylor TB, Keene DR, Burgeson RE, Zone JJ. J Invest Dermatol. 1996;106:734. doi: 10.1111/1523-1747.ep12345782. [DOI] [PubMed] [Google Scholar]
- 21.Kowal C, et al. Proc Natl Acad Sci USA. 2006;103:19854. doi: 10.1073/pnas.0608397104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matloubian M, et al. Nature. 2004;427:355. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 23.DeVoss J, et al. J Exp Med. 2006;203:2727. doi: 10.1084/jem.20061864. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.