

Abstract
Sustained, local delivery of immunomodulatory cytokines is under investigation for its ability to enhance vaccine and anti-tumor responses both clinically and preclinically. This study evaluates the ability of chitosan, a biocompatible polysaccharide, to (1) control the dissemination of a cytokine, GM-CSF, and (2) enhance the immunoadjuvant properties of GM-CSF. While cytokines have previously been delivered in lipid-based adjuvants and other vehicles, these do not have the clinical safety profile or unique properties of chitosan. We found that chitosan solution maintained a measurable depot of recombinant GM-CSF (rGM-CSF) at a subcutaneous injection site for up to 9 days. In contrast, when delivered in a saline vehicle, rGM-CSF was undetectable in 12 to 24 hours. Furthermore, a single s.c. injection of 20μg rGM-CSF in chitosan solution (chitosan/rGM-CSF(20μg)) transiently expanded lymph nodes up to 4.6-fold and increased the number of MHC class II expressing cells and dendritic cells by 7.4-fold and 6.8-fold, respectively. These increases were significantly greater than those measured when rGM-CSF was administered in saline at the standard preclinical dose and schedule, i.e. 4 daily s.c. injections of 20μg. Furthermore, lymph node cells from mice injected with chitosan/rGM-CSF(20μg) induced greater allogeneic T cell proliferation, indicating enhanced antigen presenting capability, than lymph node cells from mice injected with rGM-CSF alone. Finally, in vaccination experiments, chitosan/rGM-CSF was superior to either chitosan or rGM-CSF alone in enhancing the induction of antigen-specific CD4+ proliferation, peptide-specific CD8+ pentamer staining and cytotoxic T cell lysis. Altogether, chitosan/rGM-CSF outperformed standard rGM-CSF administrations in dendritic cell recruitment, antigen presentation and vaccine enhancement. We conclude that chitosan solution is a promising delivery platform for the sustained, local delivery of rGM-CSF.
Keywords: chitosan, immunoadjuvant, GM-CSF, controlled release, cytokine
1. Introduction
Local administrations of immunomodulatory cytokines are under investigation for their ability to enhance vaccine and anti-tumor responses both clinically [1-4] and preclinically [5-7]. Because immunoadjuvant cytokines act best in a paracrine fashion, sustained local concentrations are necessary for their desired effect. Unfortunately, recombinant cytokines are small proteins that are cleared rapidly – on the order of several hours – when administered in a traditional saline vehicle. Additionally, cytokines are often toxic when administered systemically [8-10]. Thus, controlled, local delivery of recombinant cytokines would benefit numerous immunotherapies.
GM-CSF is a pleiotropic, proinflammatory cytokine that was initially discovered as a growth factor capable of generating granulocyte and macrophage colonies from bone marrow precursor cells [11]. GM-CSF also promotes the activation, function and recruitment of macrophages and dendritic cells [12, 13] which has resulted in improved antigen presentation [12, 14]. Consequently, GM-CSF has been shown to boost host immunity directed at a variety of immunogens [3, 4, 7, 15, 16].
Numerous strategies have been employed to achieve sustained, local delivery of rGM-CSF and other cytokines [17-19]. Plasmid DNA encoding cytokines are easy to manufacture; however, in vivo transfection is inefficient. It is also problematic to control the concentration and duration of transcribed cytokines from plasmid vectors. Viruses encoding cytokines are more efficient in transfecting cells in vivo; however, safety concerns over the use of infectious agents and uncontrolled cytokine production are disadvantages. GM-CSF-transduced tumors cells, as cancer vaccines, have entered Phase III trials for prostate cancer after overcoming early manufacturing concerns regarding safety, reproducibility and dosing accuracy [20].
Biomaterials-based delivery platforms are gaining attention for their potential to control the spatiotemporal distributions of recombinant cytokines. In particular, traditional drug delivery platforms, such as polymeric microspheres and liposomes, are being adapted to encapsulate and release recombinant proteins. Polymeric microspheres have been effective in encapsulating recombinant cytokines such as IL-2 [21, 22], IL-12 [23] and GM-CSF [24-26]. Initial research has confirmed that encapsulated cytokines remain at least partially bioactive during in vitro release experiments [21, 24, 25, 27-29]. In several further studies, cytokine microspheres have been used as immunotherapies against solid tumors [19, 22, 26, 30-33]. The two major concerns for this delivery platform are the harsh microsphere formulation conditions, e.g. organic solvents, surfactants and shear stresses, and the local pH drop during polymer degradation – both of which jeopardize cytokine bioactivity [34-36].
Liposomes, similar to polymeric microspheres, have been shown to encapsulate and release recombinant cytokines in vitro [37, 38]. Several studies have successfully evaluated the bioactivity of liposomal IL-2 [39, 40] and IFN-γ [18, 41, 42] as immunoadjuvants in vivo. Liposomal cytokines are most often administered systemically, intravenously or intraperitoneally, as a method of increasing the effective half-lives of recombinant cytokines. Complications due to systemic administration of liposomes in humans include flushing and tightness in the chest [43]. Additionally, it remains unclear what the effect of internalization of either microspheres or liposomes would have on the efficacy of the encapsulated cytokine whose biological function is mainly associated with cell surface receptors [44].
Previously, we used chitosan solutions to control the dissemination of a model antigen [45]. Chitosan is a non-toxic, biodegradable, natural polysaccharide derived primarily from the exoskeletons of crustaceans. Chitosan is a widely used biomaterial with an established safety profile in humans. It has been used as a pharmaceutical excipient [46], a controversial weight loss supplement [47, 48], an experimental mucosal adjuvant [49-51] and in an FDA-approved hemostatic dressing [52]. Subcutaneous administrations of chitosan solution in mice are cleared via enzymatic digestion in 2-4 weeks [45]. Most importantly, chitosan solution can be formulated under mild aqueous conditions which should not attenuate cytokine bioactivity.
In this study, we have evaluated whether chitosan solution, due to its high viscosity, would retain co-formulated rGM-CSF at an injection site. The sustained, local presence of rGM-CSF could potentially increase the number of dendritic and antigen presenting cells in local lymph nodes and improve antigen presentation. In turn, the enhanced antigen presentation could improve the immune response to a co-formulated vaccine. Non-invasive imaging was used to characterize the spatiotemporal distribution of a subcutaneous injection of rGM-CSF formulated with and without chitosan. Draining lymph node expansion, phenotype and antigen presenting ability were used as measures of in vivo rGM-CSF bioactivity. Finally, the vaccine enhancing ability of chitosan/rGM-CSF was compared to that of chitosan solution and rGM-CSF alone.
2. Materials and Methods
2.1 Animals
Female C57BL/6 mice (8-12 weeks old) were obtained from the National Cancer Institute, Frederick Cancer Research Facility (Frederick, MD). Mice were housed and maintained under pathogen-free conditions in microisolator cages. Animal care was in compliance with recommendations of The Guide for Care and Use of Laboratory Animals (National Research Council).
2.2 Antigens and adjuvants
Chitosan (Protosan G 213) was purchased from NovaMatrix (Drammen, Norway). Recombinant murine GM-CSF (rGM-CSF) was purchased from Peprotech (Rocky Hill, NJ). Ovalbumin (Grade VI) and concanavalin A were purchased from Sigma-Aldrich (St. Louis, MO). β-galactosidase was purchased from Prozyme (San Leandro, CA). Human Influenza A strain PR/8 purified virus was purchased from Advanced Biotechnologies, Inc. (Columbia, MD). The influenza virus was inactivated via DNA crosslinking during exposure to ultraviolet (UV) light for 10 minutes in a Stratalinker (Stratagene; LaJolla, CA). The resulting antigen is subsequently referred to as UV-inactivated influenza.
2.3 Non-invasive fluorescence imaging of cytokine depots
Non-invasive animal imaging was carried out in the Mouse Imaging Facility (MIF), a division of the NIH MRI Research Facility (NMRF). Fluorescence and photographic images of anesthetized mice that were given a single s.c. injection of fluorescently labeled rGM-CSF formulated in PBS or chitosan solution were acquired over a 2 week period with an IVIS 100 Imaging System (Xenogen; Alameda, CA). Anesthesia was induced in a chamber with 4-5% isoflurane delivered by a gas mixture of oxygen, nitrogen and medical air. Once mice were unconscious and unresponsive to toe pinch, anesthesia was maintained with 1-2% isoflurane administered via nosecone. Following each imaging session, mice recovered in the MIF/NMRF on a circulating warm water pad. Prior to the initial imaging session, the lumbar regions of mice were shaved and residual hair was removed with a depilatory cream. Approximately 20μg of rGM-CSF, labeled with an Alexa Fluor 660 protein labeling kit (Invitrogen; Carlsbad, CA), were injected s.c. in the lower flank/lumbar region in a total volume of 50μl in PBS, 1% or 2% (w/v) chitosan solution. The fluorescence intensity of the injection site was used as a surrogate for rGM-CSF concentration. The fluorescence intensity of a region of interest drawn around the injection site was calculated at each time point with Living Image® software (Xenogen; Alameda, CA). Background/autofluorescence from non-injected control mice was subtracted. Fluorescence data for each mouse were normalized by the initial measurement, which was taken immediately after injection, for each mouse.
2.4 Flow cytometry
Mice were given either one 100μl s.c. injection in the lower flank/lumbar region of PBS or chitosan/rGM-CSF(20μg) or four daily injections of 20μg rGM-CSF. Chitosan/rGM-CSF(20μg) was formulated by adding 20μg rGM-CSF to 1.5% chitosan (w/v) dissolved in DPBS. The draining inguinal lymph node was harvested at the designated times following treatment. Nodes were mechanically disrupted with a syringe plunger and passed through a 70μm nylon mesh strainer (BD Biosciences; Bedford, MA). Cells were washed twice with cold PBS. FcγII and FcγIII receptors on lymphocytes were blocked via incubation with 1 μg purified anti-mouse CD16/CD32 (clone: 2.4G2) (BD Biosciences; San Jose, CA) per 1 × 106 cells for 15 min on ice. Cells were stained with fluorescence-labeled antibodies (1 μg/1 × 106 cells) to the following markers (BD Biosciences; San Jose, CA): CD3e (clone: 145-2C11), CD19 (clone: 1D3), NK1.1 (clone: PK136), CD25 (clone: PC61), CD11b (clone: M1/70), CD11c (clone: HL3), and Gr-1 (clone: RB6-8C5). Antibody isotype controls (BD Biosciences; San Jose, CA) included: mouse IgG1 (clone: MOPC-31C), mouse IgG2a (clone: G155-178), rat IgG1 (clone: A110-1), rat IgG2a (clone: R35-95), rat IgG2b (clone: A95-1) and Hamster IgG1 (clone: A19-3). Following a 45 min incubation on ice, cells were washed twice with cold PBS and analyzed on a LSR II (BD Biosciences; San Jose, CA). Data analyses were performed using BD FACSDiva Software (BD Biosciences; San Jose, CA).
2.5 Mixed Lymphocyte Response (MLR)
Draining inguinal lymph nodes were harvested following rGM-CSF treatment. Cells were counted, irradiated (20 Gy) and serially diluted (from 5 × 105 to 1.56 × 104 cells/well) in triplicate in a 96-well plate. T cells from Balb/c mice were obtained following B cell depletion of splenocytes using Dynal® B220 isolation kits (Invitrogen; Carlsbad, CA) according to the manufacturer’s instructions. Five hundred thousand Balb/c T cells were co-incubated with the irradiated lymph node cells for 4 days. Cells were labeled with 1 μCi/well [3H]-thymidine (Amersham Biosciences; Piscataway, NJ) for the final 18 h of culture. Following incubation, cells were harvested onto glass fiber filtermats via a Tomtec Harvester 96 (Hamden, CT). Incorporated radioactivity was measured by liquid scintillation counting on a 1450 Betaplate (Perkin-Elmer; Shelton, CT). Results from individual mice in triplicate wells were combined to yield a mean ± SEM for each treatment group.
2.6 Vaccinations
Vaccinations consisted of a prime and a boost, separated by 1 week, with 5μg UV-inactivated influenza or 100μg β-galactosidase. UV-inactivated influenza or β-galactosidase was formulated via simple addition with PBS, chitosan alone, rGM-CSF alone or chitosan and rGM-CSF (chitosan/rGM-CSF) together. For the chitosan alone treatment group, 1.5% chitosan (w/v) was dissolved in DPBS prior to mixing with antigen. For the rGM-CSF alone group, antigen was mixed with 20μg rGM-CSF in saline. Three additional daily s.c. injections of 20μg rGM-CSF were given at the vaccination site. For the combined group, either 20μg or 80μg of rGM-CSF was added to 1.5% chitosan (w/v) dissolved in DPBS (denoted as chitosan/rGM-CSF(20μg) and chitosan/rGM-CSF(80μg), respectively). All vaccines were administered as a single 100μl s.c. injection in the lower flank/lumbar region on opposite sides for the prime and boost.
2.7 Splenic CD4+ proliferation assay
All proliferation assays were initiated 1 week following the booster vaccination and performed as described previously [45]. Briefly, harvested spleens were mechanically disrupted with a syringe plunger and passed through a 70μm nylon mesh strainer (BD Biosciences; Bedford, MA). Erythrocytes were lysed with ACK lysing buffer (Cambrex Bio Science; Walkersville, MD). CD4+ splenocytes were isolated via Dynal® CD4 negative isolation kits (Invitrogen; Carlsbad, CA) according to the manufacturer’s instructions. Two hundred-thousand splenic CD4+ cells from immunized mice were co-incubated with 5 × 105 irradiated (20 Gy) naïve syngeneic splenocytes in triplicate wells of a 96-well plate. Cells were stimulated with 0.31-5μg/ml UV-inactivated influenza for 5 days. For positive controls, cells were stimulated with 0.0625-1μg/ml concanavalin A for 3 days. For non-specific antigen controls, cells were stimulated with 50μg/ml ovalbumin for 5 days. In all cases, cells were labeled with 1 μCi/well [3H]-thymidine (Amersham Biosciences; Piscataway, NJ) for the final 18 h of culture. Incorporated radioactivity was measured as described above. Results from individual mice in triplicate wells were combined to yield a mean ± SEM for each immunization group.
2.8 Serum antibody responses
Antigen-specific serum antibody titers were measured 1 week following the booster vaccination via ELISA. Briefly, microtiter plates were sensitized overnight at 4°C with 100ng/well UV-inactivated influenza, β-galactosidase or ovalbumin (negative control). Wells were blocked with 5% BSA in PBS for 1 h at 37°C. Serially diluted sera (1:20-1:1,526,500) were added to wells in duplicate and allowed to incubate for 1 h. Wells were then washed thrice with 1% BSA in PBS. Following another 1 h incubation with Horseradish peroxidase-conjugated goat-anti-mouse IgG (Pierce; Rockford, IL), wells were washed thrice with 1% BSA in PBS. O-phenylenediamine (Sigma-Aldrich; St. Louis, MO) was then added to the wells according to the manufacturer’s instructions. The reaction was stopped with 3N HCl and the absorbance of each well was read at 490nm using a Bio-Tek Synergy HT multi-detection microplate reader (Winooski, VT).
2.9 Pentamer staining
Pro5® MHC Class I pentamers were purchased from ProImmune (Oxford, UK). Freshly prepared splenocytes or cells from a 7 day in vitro stimulation with 10ng/ml Flu NP366-374 peptide (ASNENTETM) (CPC Scientific; San Jose, CA) were stained with either a pentamer specific for an H-2Db epitope for influenza A/PR/8 nucleoprotein (Flu NP366-374; ASNENTETM) or a control pentamer specific for an H-2Db epitope for lymphocytic choriomeningitis virus nucleoprotein (LCMV NP396-404; FGPQNGQFI) according to the manufacturer’s instructions. Cells were read and analyzed on an LSR II (BD Biosciences; San Jose, CA).
2.10 Cytotoxic T cell lysis (CTL) assay
One week after the booster vaccination, spleens from vaccinated mice were harvested as before. Approximately 25 × 106 unfractionated splenocytes from each vaccine group were cultured in an upright T-25 flask containing 10ng/ml Flu NP366-374 peptide (ASNENTETM) (CPC Scientific; San Jose, CA). After 1 week, lymphocytes were collected on a histopaque (Sigma-Aldrich; St. Louis, MO) density gradient and quantified. Target EL-4 cells (4 × 106) were radiolabeled in RPMI 1640 with 50μCi of 111In-labeled oxine (GE Healthcare; Silver Spring, MD) for 30 min at 37°C. Target cells were washed twice in complete media and pulsed with 1μg/ml Flu NP366-374 or HIV gag390-398 (control) peptide for 30 min at 37°C. Five thousand target cells/well were co-incubated with 5 to 250 × 103 lymphocytes in triplicate wells in a 96-well plate for 18 h at 37°C. The amount of 111In released was measured using a gamma counter (Cobra II; Packard Instruments, Downers Grove, IL). The percentage of specific lysis was calculated as follows:
The reported % lysis is Flu NP366-374-specific lysis subtracted from HIV gag390-398-specific lysis.
2.11 Statistical analysis
Antigen-specific splenic CD4+ proliferation, MLR, and CTL data are presented as mean ± standard error of the mean. Lymph node cell numbers and percentages from flow cytometry experiments are presented as mean ± standard deviation. Differences in means between treatment groups were analyzed using Student’s two-tailed t test assuming unequal variances and without adjustment for the multiplicity of evaluation (JMP Software; Cary, NC). For antigen-specific splenic CD4+ proliferation, MLR and CTL, only the three highest values for each treatment were used for statistical comparison. Differences in means were accepted as significant if P was less than 0.05.
3. Results
3.1 Chitosan delayed the dissemination of rGM-CSF from an injection site
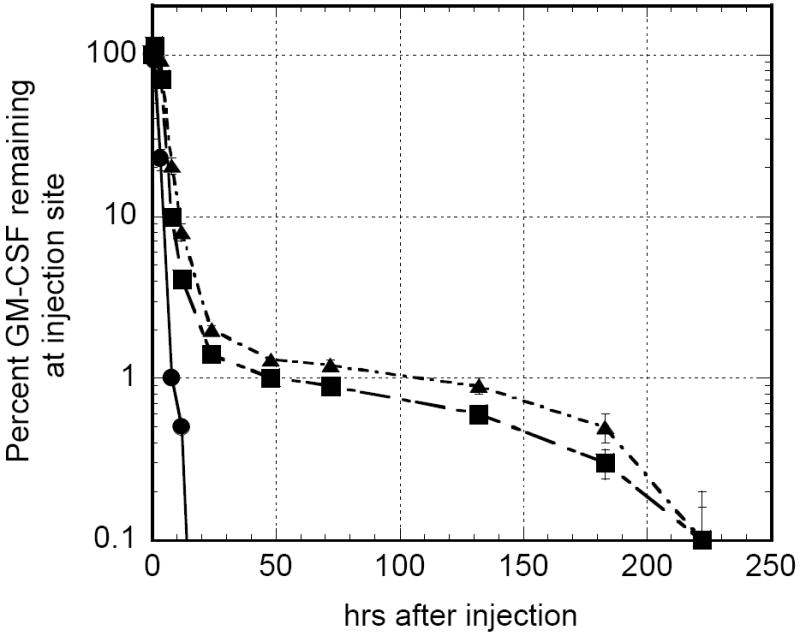
To determine if chitosan solution could maintain a depot of a recombinant cytokine, fluorescence imaging studies were performed with recombinant GM-CSF (rGM-CSF). Mice receiving a single s.c. injection of Alexa Fluor 660-labeled rGM-CSF were imaged over the course of 2 weeks in order to track the cytokine’s spatiotemporal distribution when delivered in either PBS or chitosan solution (Fig. 1). Fluorescence intensity was used to monitor rGM-CSF concentration. Analysis of the injection site revealed that when delivered in PBS, rGM-CSF was undetectable in 12 to 24 h. In contrast, rGM-CSF was measurable for at least 9 days when administered in a chitosan solution (Fig. 2). There was no significant difference between 1% and 2% chitosan solution in rGM-CSF retention time. Integration of the area under the curve (AUC) in Figure 2 indicated that total rGM-CSF exposure was increased approximately 3-fold when rGM-CSF was formulated in either 1% or 2% chitosan solution. Injections of either PBS or chitosan solution without rGM-CSF did not generate fluorescence above non-injected background levels. Because the difference in rGM-CSF residence between 1% and 2% chitosan solution was marginal and further increases in chitosan concentration would be limited by solubility, all subsequent experiments were conducted with 1.5% chitosan.
FIGURE 1.
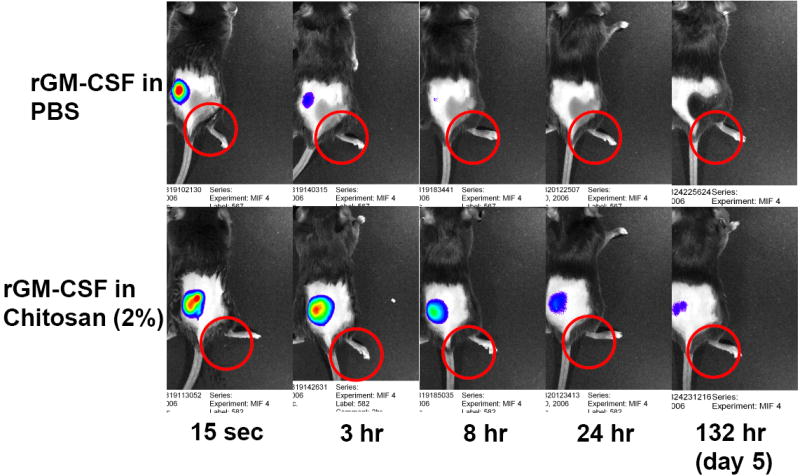
FIGURE 2.
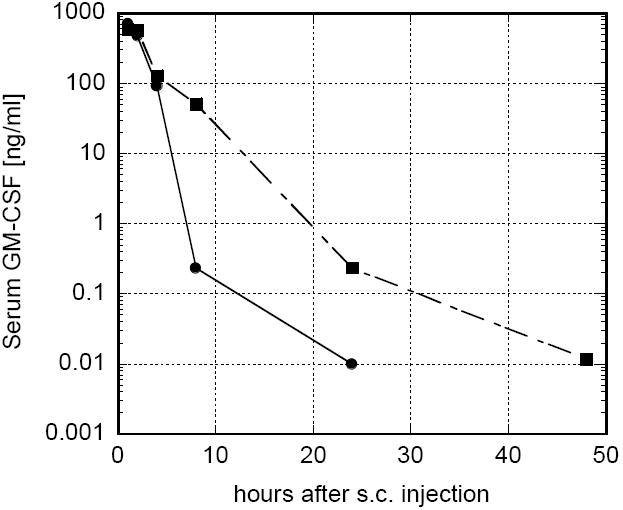
Serum GM-CSF was measured as a function of time following a single subcutaneous administration of rGM-CSF in either PBS or 1.5% chitosan solution (Fig. 3). When 20μg rGM-CSF was administered in PBS, serum GM-CSF peaked above 700 ng/ml within 1 h, decreased rapidly to 0.2 ng/ml at 8 h and was undetectable within 48 h. When 20μg rGM-CSF was administered in chitosan solution, the peak serum GM-CSF concentration was lower (589ng/ml vs. 704ng/ml); however, serum GM-CSF levels were 1.2 to 212 times higher than those of the rGM-CSF in PBS group at later times. This extended serum GM-CSF profile was likely mediated by the delayed release of rGM-CSF from the chitosan solution at the injection site (Fig. 1). Total serum GM-CSF exposure, or AUC, assuming a peak concentration at 1 h, was increased by approximately 40% when rGM-CSF was administered in chitosan solution.
FIGURE 3.
3.2 Chitosan/rGM-CSF expanded local lymph nodes
Previous studies demonstrated that rGM-CSF given as four daily s.c. injections of 20μg transiently expanded local draining lymph nodes in mice [12]. This expansion was accompanied by a significant increase in the number of antigen presenting cells in the local lymph nodes. In this study, the cellular expansion of the draining (inguinal) lymph node was quantified and cells were phenotyped as a function of time (a) to verify the in vivo bioactivity of rGM-CSF when formulated with chitosan, and (b) to understand the temporal relationship between rGM-CSF residence and lymph node expansion. Mice were given either one s.c. injection of either PBS (control), chitosan/rGM-CSF(20μg) or chitosan/rGM-CSF(80μg) or four daily injections of 20μg rGM-CSF starting at day 0. Recombinant GM-CSF alone induced the expected 2- to 3-fold cellular expansion of the draining lymph node at day 7 (Table 1). This expansion was markedly reduced by day 14 and returned to control levels by day 35. The same total dose of rGM-CSF formulated in chitosan, i.e. chitosan/rGM-CSF(80μg), induced a 3.4-fold cellular expansion of the draining lymph node at day 7 and a 2.1-fold expansion at day 14 before returning to control levels by day 35. Chitosan/rGM-CSF(20μg) generated the maximal response, at one-fourth the rGM-CSF dose, with a 4.6-fold cellular expansion of the draining lymph node at day 7 and a 3.1-fold expansion at day 14 before returning to control levels by day 35 (Table 1).
Table 1.
The effect of rGM-CSF on the total number and percent of lymphocyte subsets in the draining inguinal lymph node. Mice were injected subcutaneously once with either PBS (control), chitosan/rGM-CSF(20μg), or chitosan/rGM-CSF(80μg) or given 4 daily injections of rGM-CSF beginning on day 0. Lymph nodes were harvested on days 7, 14 and 35. Each data point is the mean (SD) for 5 mice
| Control
|
rGM-CSF (20μg × 4)
|
Chitosan/rGM-CSF (20μg)
|
Chitosan/rGM-CSF (80μg)
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Days | 7 | 14 | 35 | 7 | 14 | 35 | 7 | 14 | 35 | 7 | 14 | 35 |
| total cells per LN (106) | 5.0 (0.7) | 4.7 (1.5) | 4.0 (0.5) | 14.0 (3.0)* | 8.2 (1.7)* | 4.6 (0.4) | 23.2 (6.9)** | 14.4 (5.2)* | 4.6 (1.3) | 17.2 (2.6)* | 9.8 (3.3)* | 6.3 (2.5) |
| percent I-Ab+ | 27.9 (3.5) | 36.8 (5.0) | 31.6 (5.5) | 41.1 (5.2)* | 37.3 (4.3) | 36.6 (5.0) | 44.6 (2.1)* | 44.9 (4.8)** | 34.7 (6.4) | 44.8 (7.7)* | 31.3 (3.5) | 35.0 (5.6) |
| # of I-Ab+ cells (106) | 1.4 (0.3) | 1.7 (0.4) | 1.3 (0.2) | 5.7 (1.3)* | 3.1 (1.0)* | 1.7 (0.3) | 10.4 (3.5)** | 6.5 (2.8)** | 1.7 (0.7) | 7.8 (2.3)* | 3.0 (0.7)* | 2.2 (0.8) |
| percent CD11c+I-Ab+ | 1.5 (0.1) | 1.5 (0.2) | 1.9 (0.3) | 2.2 (0.4)* | 1.7 (0.3) | 2.0 (0.6) | 2.3 (0.3)* | 1.8 (0.2)* | 2.1 (0.3) | 2.3 (0.7) | 1.4 (0.2) | 2.2 (0.2) |
| # of CD11c+I-Ab+ cells (104) | 7.8 (1.7) | 6.9 (1.9) | 7.8 (1.5) | 30.0 (8.7)* | 14.3 (5.7)* | 9.2 (2.7) | 53.4 (15.2)** | 26.5 (12.1)* | 9.7 (3.5) | 39.7 (14.4)* | 13.2 (4.4)* | 13.8 (5.5) |
| percent CD11c+I-Ab+CD80+ | 0.5 (0.1) | 0.4 (0.1) | 0.6 (0.1) | 0.6 (0.2) | 0.5 (0.1) | 0.6 (0.1) | 0.8 (0.2)* | 0.6 (0.1)* | 0.6 (0.1) | 0.9 (0.3) | 0.4 (0.1) | 0.6 (0.1) |
| # of CD11c+I-Ab+CD80+ cells (104) | 2.6 (0.6) | 1.8 (0.3) | 2.4 (0.6) | 8.8 (3.8)* | 4.2 (1.6)* | 2.7 (0.7) | 17.4 (3.5)** | 8.0 (3.0)** | 2.7 (0.8) | 14.9 (6.5)* | 3.8 (1.4)* | 3.8 (1.8) |
Statistically significant (P<0.05) from control at the respective timepoint
[bold] Statistically significant (P<0.05) from rGM-CSF(4 × 20μg) and control at the respective timepoint
Because of the documented ability of GM-CSF to recruit dendritic and other antigen presenting cells to local lymph nodes [5, 6, 12], these subsets were specifically quantified. MHC Class II expression was used to quantify broadly the number of antigen presenting cells (dendritic cells, B cells and monocyte/macrophages) [7, 53] in the draining lymph node. Recombinant GM-CSF alone induced significant increases in percentage, at day 7, and number, at days 7 and 14, of cells expressing the MHC II molecule, I-Ab (Table 1). The percentages and numbers of I-Ab + cells were increased further by formulating rGM-CSF in chitosan solution. In particular, chitosan/rGM-CSF(20μg) and chitosan/rGM-CSF(80μg) induced 7.4- and 5.6-fold increases, respectively, in the number of I-Ab + cells in the draining lymph node 7 days after administration. Chitosan/rGM-CSF(20μg)-mediated increases in the numbers of I-Ab + cells in the draining lymph node were significantly greater (P<0.05) than rGM-CSF alone treatment at days 7 and 14. All percentages and numbers of I-Ab + cells returned to control levels by day 35.
Dendritic cells, denoted as CD11c+I-Ab +, were specifically quantified as they are considered the most potent antigen presenting cells. Similar to the I-Ab+ cells, rGM-CSF alone induced significant increases in percentage, at day 7, and number, at days 7 and 14, of dendritic cells (Table 1). The chitosan/rGM-CSF(20μg) treatment group maintained significantly higher percentages and numbers of dendritic cells up to day 14. The number of dendritic cells induced by chitosan/rGM-CSF(20μg) were significantly greater (P<0.05) than those induced by rGM-CSF alone treatment. Chitosan/rGM-CSF(80μg) treatment also generated significant increases in dendritic cells although not to the level of chitosan/rGM-CSF(20μg).
Because of the overall expansion of the lymph nodes, the differences between groups in the numbers of dendritic cells per node were magnified. For example, the total numbers of CD11c+I- Ab + cells were increased 3.8-fold with rGM-CSF alone (P<0.05 vs. control), 6.8-fold with chitosan/rGM-CSF(20μg) (P<0.05 vs. rGM-CSF alone) and 5.1-fold with chitosan/rGM-CSF(80μg) (P<0.05 vs. control) at day 7. One week later, numbers of CD11c+I-Ab + cells in all treatment groups remained significantly greater than control. Interestingly, the 3.8-fold increase in number of CD11c+I- Ab + cells in the chitosan/rGM-CSF(20μg) group at day 14 was equal to the maximum 3.8-fold increase in the rGM-CSF group occurring at day 7. This indicated that chitosan not only increased but also sustained the adjuvant properties of rGM-CSF. All percentages and numbers of CD11c+I-Ab + cells returned to control levels by day 35. Differences in mature dendritic cells, denoted as CD11c+CD80+, between the treatment groups followed similar trends. For instance, at day 7, numbers of CD11c+CD80+ cells were increased 3.4-, 6.7- and 5.7-fold for rGM-CSF alone, chitosan/rGM-CSF(20μg) and chitosan/rGM-CSF(80μg) treatments, respectively. In total, chitosan/rGM-CSF(20μg) administration induced the maximum lymph node expansion and the maximum increases in antigen presenting cells in the draining lymph node. There were no significant changes in the percentages of Gr-1+, CD11b+, Gr-1+CD11b+, NK1.1+ or CD4+CD25+ cells in the draining lymph node with either treatment. There was an approximate 10% decrease in the percentage of CD3+ cells and a corresponding 10% increase in percentage of CD19+ cells in all treatment groups at day 7. Percentages of both subsets returned to control levels by day 14.
3.3 Chitosan/rGM-CSF enriched antigen presentation in local lymph nodes
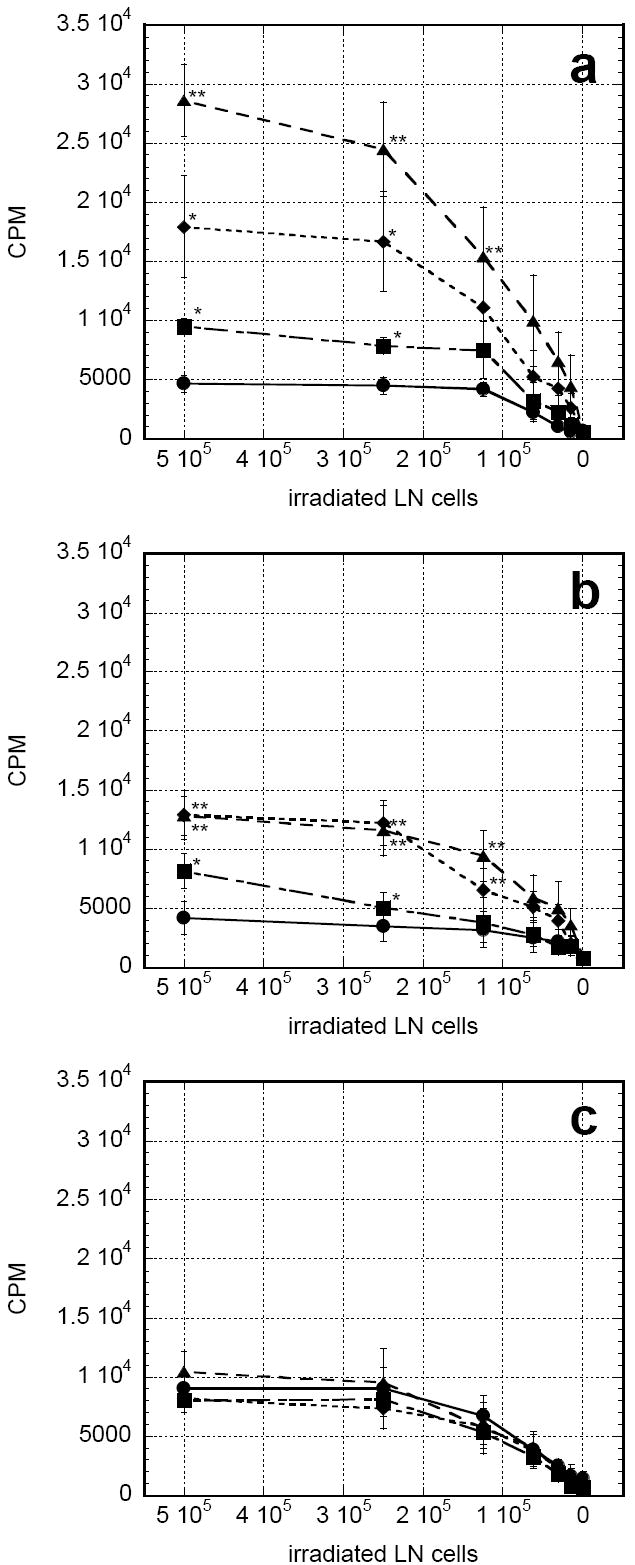
Recombinant GM-CSF administration has been shown to enhance antigen presentation in draining lymph nodes [12]. In the present experiment, the proliferation of allogeneic T cells co-incubated with irradiated lymph node cells from treated mice was used as a measure of antigen presenting ability. Administration of rGM-CSF alone led to a doubling in antigen presenting ability versus control at day 7 (Fig. 4a). This increase was markedly reduced by day 14 (Fig. 4b) and returned to control levels by day 35 (Fig.4c). Chitosan/rGM-CSF(20μg) mediated statistically significant 6.1- and 3.1-fold increases in antigen presenting ability versus control at days 7 and 14 (P<0.05 vs. PBS). These increases were also significantly greater than those of the rGM-CSF alone group at days 7 and 14 (P<0.05 vs. rGM-CSF). The 3.8-fold increase in proliferation from the chitosan/rGM-CSF(80μg) treatment was less than that of chitosan/rGM-CSF(20μg) at day 7. However, these groups were indistinguishable at day 14. Antigen presenting ability of all treatment groups returned to control levels by day 35.
FIGURE 4.
3.4 Chitosan/rGM-CSF enhanced a vaccine response better than either agent alone
After it was demonstrated that chitosan solution could maintain a depot of functional rGM-CSF which improved antigen presentation, the chitosan/rGM-CSF formulation was tested for its ability to improve vaccine responses against two diverse antigen types. In initial studies, mice were vaccinated subcutaneously with β-galactosidase co-formulated with one of the following: PBS, chitosan solution, rGM-CSF alone or chitosan/rGM-CSF(80μg). The rGM-CSF alone group received three additional daily injections of rGM-CSF at the vaccination site for a total dose of 80μg rGM-CSF. Mice receiving antigen in either chitosan solution or rGM-CSF alone exhibited significantly greater antigen-specific proliferation of CD4+ splenocytes (P<0.05) than mice receiving antigen with no adjuvant (PBS) (Fig. 5a). However, administration of antigen in the combined adjuvant, chitosan/rGM-CSF(80μg), resulted in statistically significant increases in proliferation over either chitosan or rGM-CSF alone (P<0.05). Non-specific antigen (OVA) proliferation was consistently less than 10% of the antigen-specific CD4+ proliferation in all cases. Antigen-specific serum IgG titers to β-galactosidase were increased 6.2-fold for rGM-CSF alone, 24.0-fold for chitosan alone and 26.7-fold for the combined chitosan/rGM-CSF(80μg) (Fig. 5b).
FIGURE 5.
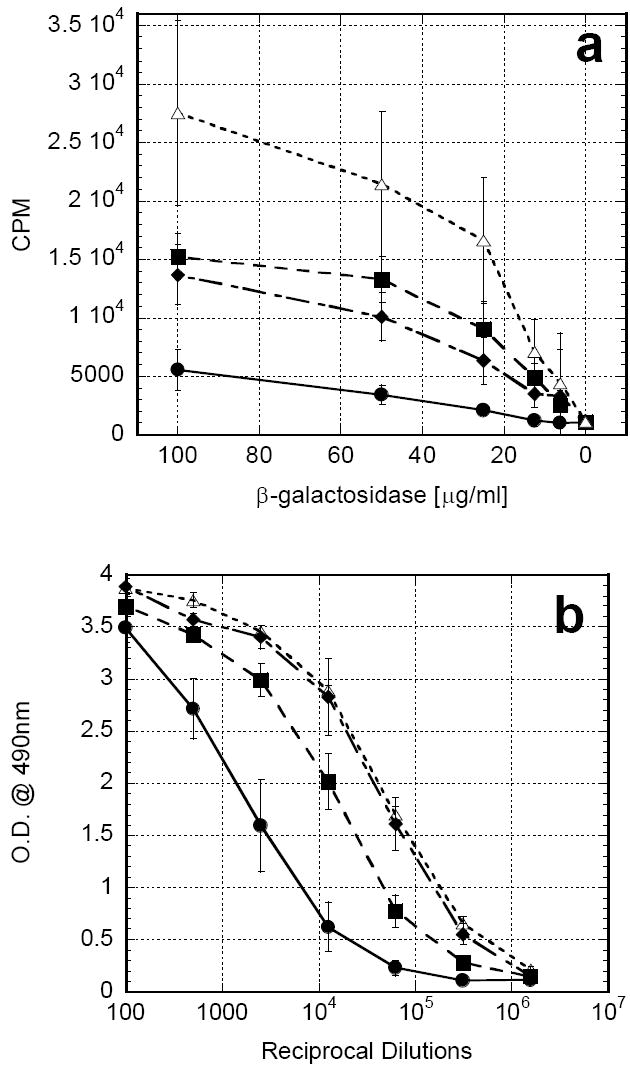
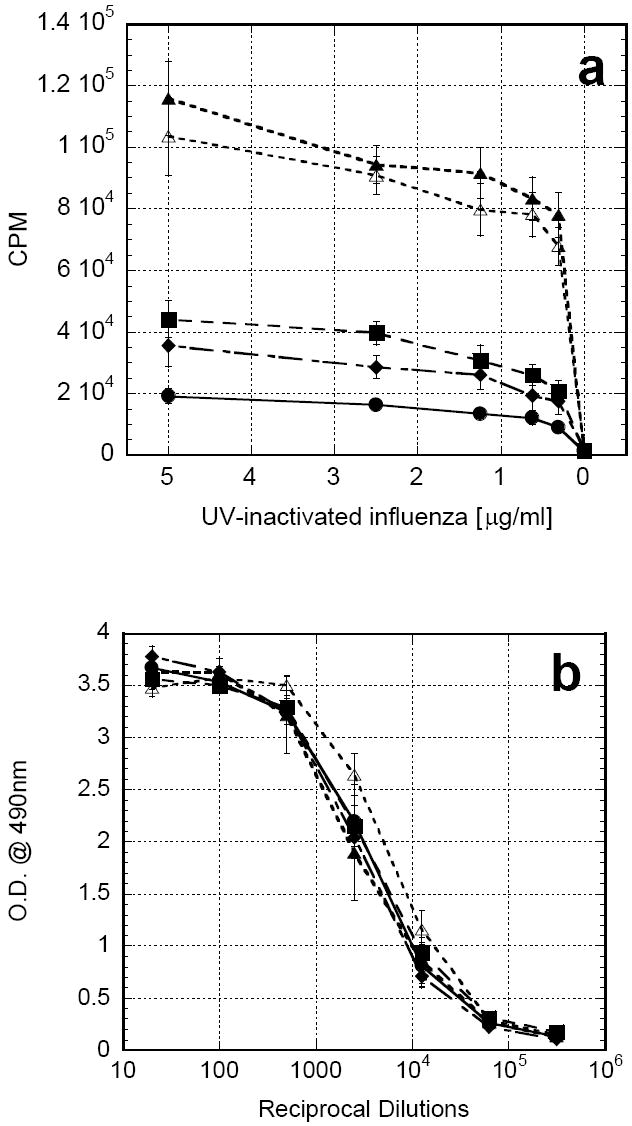
In subsequent studies, mice were vaccinated subcutaneously with UV-inactivated influenza co-formulated with one of the following: PBS, chitosan solution, rGM-CSF alone, chitosan/rGM-CSF(20μg) or chitosan/rGM-CSF(80μg). As before, the rGM-CSF alone group received three additional daily injections of rGM-CSF at the vaccination site. Once again, mice receiving antigen in either chitosan solution or rGM-CSF alone exhibited significantly greater antigen-specific proliferation of CD4+ splenocytes (P<0.05) than mice receiving antigen with no adjuvant (PBS) (Fig. 6a). Furthermore, administration of antigen in either combined adjuvant, chitosan/rGM-CSF(20μg) or chitosan/rGM-CSF(80μg), resulted in profound increases over either adjuvant alone, indicating a synergistic enhancement (P<0.05). Non-specific antigen (OVA) proliferation was consistently less than 10% of the antigen-specific CD4+ proliferation in all cases. It is noteworthy that the lower dose rGM-CSF combined adjuvant, chitosan/rGM-CSF(20μg), generated as good, if not better, of a response as the higher dose rGM-CSF combined adjuvant, chitosan/GM-CSF(80μg). In contrast to the previous experiment using β-galactosidase, antigen-specific serum IgG titers to influenza were not significantly increased with any of the adjuvants tested (Fig 6b).
FIGURE 6.
3.5 Chitosan solution spared rGM-CSF dose
Both preclinical and clinical lots of rGM-CSF are costly. Moreover, there is less potential for toxicity when less of any drug is employed. Thus far, a reduction in the total dose of rGM-CSF in chitosan from 80μg to 20μg demonstrated a benefit in lymph node expansion and antigen presentation with a marginal increase in vaccine response. A subsequent vaccination experiment was performed to determine if there was any additional benefit to further reducing the dose of rGM-CSF formulated with chitosan solution. Vaccines consisted of 0, 5, 10 or 20μg rGM-CSF co-formulated with antigen (UV-inactivated influenza) in chitosan solution. Chitosan/rGM-CSF(20μg) generated the maximum proliferation of antigen-specific CD4+ splenocytes; however, comparable responses were observed with chitosan/rGM-CSF(10μg) and chitosan/rGM-CSF(5μg) (Fig.7).
FIGURE 7.
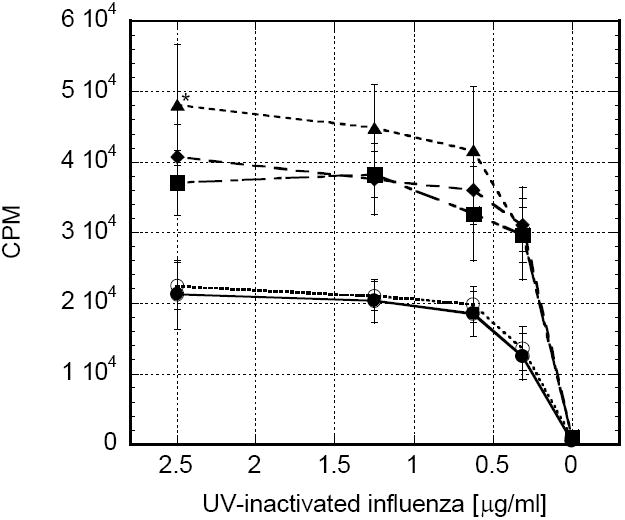
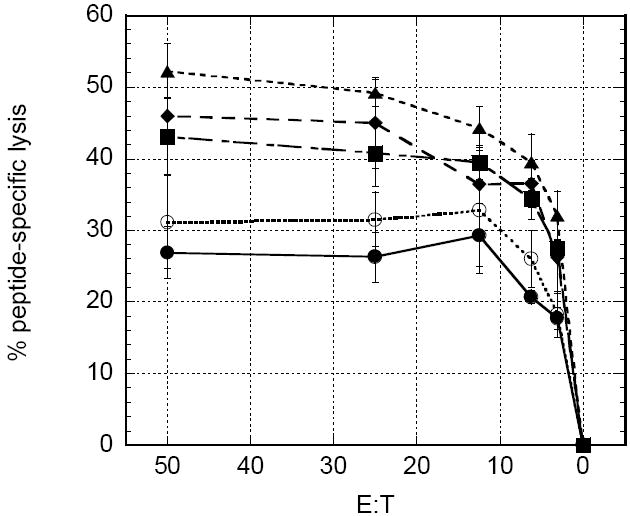
In order to determine if rGM-CSF must be co-formulated with antigen and chitosan solution prior to vaccination, a cohort of mice that were vaccinated with UV-inactivated influenza in chitosan solution were given a single injection of 20μg rGM-CSF adjacent to the site of vaccination. Antigen-specific CD4+ proliferation (Fig. 7), pentamer staining and cytotoxic T cell lysis (see Fig. 9 described below) from these mice were similar to the mice receiving chitosan alone as an adjuvant, indicating that the rGM-CSF needed to be formulated with chitosan prior to injection to achieve synergistic vaccine enhancements.
FIGURE 9.
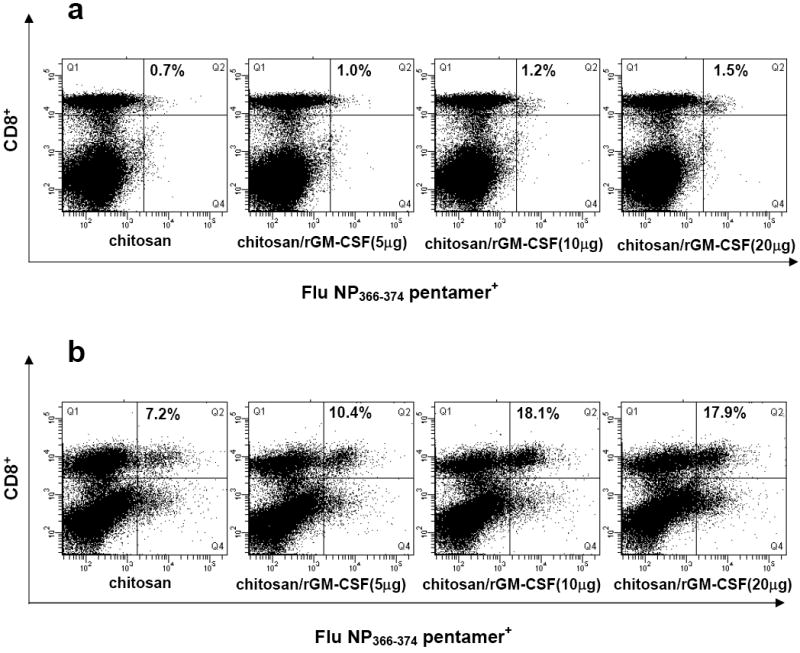
Pentamer staining of fresh splenocytes revealed incremental increases in the percent of CD8+ cells specific for Flu NP366-374 peptide, from 0.7% to 1.5%, as the dose of rGM-CSF in chitosan increased from 0μg to 20μg (Fig. 8a). When splenocytes were cultured for 1 week with re-exposure to exogenous peptide, pentamer staining was increased substantially and inter-group differences became more pronounced (Fig. 8b). Vaccination using chitosan without rGM-CSF resulted in 7.2% Flu NP366-374 pentamer+ CD8+ cells, whereas vaccination using either chitosan/rGM-CSF(10μg) or chitosan/rGM-CSF(20μg) as adjuvants resulted in approximately 18% Flu NP366-374 pentamer+ CD8+ cells.
FIGURE 8.
To determine if the peptide-specific CD8+ cells were cytolytic, cells from the in vitro stimulation were co-incubated with peptide-pulsed targets (EL-4 cells) in an overnight CTL assay. All chitosan/rGM-CSF adjuvants produced significantly greater CTL than chitosan alone (P<0.05). Peptide-specific lysis was maximal in the chitosan/rGM-CSF(20μg) group, which achieved greater that 50% lysis at an E:T ratio of 50:1 (Fig. 9). Once again, comparable results were observed with lower doses of rGM-CSF in chitosan, i.e. chitosan/rGM-CSF(5μg) and chitosan/rGM-CSF(10μg).
4. Discussion
Because chitosan is a water-soluble, biodegradable polysaccharide, we reasoned that a viscous chitosan solution should function as a depot of bioactive rGM-CSF in vivo that would enhance a vaccine response. Previously, we demonstrated that chitosan solution alone could enhance both humoral and cell-mediated immune responses to a model antigen [45]. In this study, we were able to extend the vaccine enhancing properties of chitosan and the immunoadjuvant properties of rGM-CSF via co-formulation. Imaging studies revealed that chitosan solutions increased the exposure to rGM-CSF approximately 3-fold while maintaining measurable levels of rGM-CSF at the injection site for up to 9 days (Figs. 1 and 2). When administered in a saline vehicle, rGM-CSF was undetectable in only 12 to 24 hours. It is precisely this rapid dissemination and clearance that has resulted in the administration of rGM-CSF for 4 consecutive days in most vaccine protocols. In particular, numerous clinical trials have employed and are currently employing daily s.c injections of rGM-CSF to enhance cancer vaccines [54].
Of note, the AUC for rGM-CSF in chitosan solution (Fig. 2) is much smaller than the previously measured AUC for β-galactosidase in chitosan solution [45]. This was expected as β-galactosidase (MW=465kDa) is a much larger molecule and therefore diffuses much more slowly out of the viscous chitosan solution than does rGM-CSF (MW=14.2 kDa).
In fact, ≥98% of rGM-CSF had dissipated from chitosan 24 h after administration (Fig. 2). This finding, taken together with the subsequent experiments, indicated that a persistent low level of rGM-CSF, only on the order of tens of nanograms, is necessary to improve antigen presentation and a vaccine response.
In addition to prolonging the dissemination of rGM-CSF from the injection site, chitosan also extended the presence of GM-CSF in the serum (Fig. 3). Serum GM-CSF exposure was increased by approximately 40% with chitosan after a single administration. However, if the additional injections from the standard rGM-CSF dose and schedule (4 injections × 20μg) are factored in and we assume first order pharmacokinetics, the total serum GM-CSF exposure is at least 200% less when rGM-CSF is delivered in chitosan versus saline. Therefore, although chitosan increases serum GM-CSF exposure after a single injection, the more appropriate comparison to the standard rGM-CSF dose estimated that formulated rGM-CSF in chitosan at least halves the serum GM-CSF exposure. Furthermore, reducing the dose of rGM-CSF in chitosan, i.e. chitosan/rGM-CSF(5μg) and chitosan/rGM-CSF(10μg), which produced comparable vaccine responses (Fig. 7), is expected to result in additional reductions in serum GM-CSF.
These observations are significant in that several reports have suggested that sustained high serum GM-CSF levels are immunosuppressive. Multiple injections of rGM-CSF have led to the accumulation of Gr-1+CD11b+ myeloid suppressor cells in the spleens of Balb/c mice [55]. Additionally, high-dose GM-CSF producing cells have been shown to impair a vaccine response [56]. It is clear that GM-CSF, like most cytokines, can be detrimental at high doses [54]. Because the immunosuppressive actions of GM-CSF are mediated through a high systemic concentration of GM-CSF, chitosan provides an additional advantage in reducing the systemic concentration of GM-CSF, and consequently, the possibility of expanding the myeloid suppressor cell population.
Regarding lymph node expansion, chitosan/rGM-CSF(20μg) generated a 4.6-fold expansion of the draining inguinal lymph node at day 7 (Table 1). This expansion was significantly greater than the 2.8-fold expansion observed in mice receiving four injections of rGM-CSF in saline even though the last of these injections were given only 3 days before the first time point (day 7). Our previous studies demonstrated that chitosan solution alone induced a modest 67% (1.7-fold) non-specific expansion of lymph nodes [45]. Hence, the combined chitosan/rGM-CSF(20μg) treatment induced an additive expansion of draining lymph nodes using only one-fourth the dose of rGM-CSF.
The lymph node expansion due to chitosan/rGM-CSF(80μg) was smaller than the expansion due to chitosan/rGM-CSF(20μg). Also, compared to chitosan/rGM-CSF(20μg), chitosan/rGM-CSF(80ug) induced fewer dendritic cells in the lymph node, the same or less antigen presenting capability depending on the time point and slightly weaker antigen-specific CD4+ proliferation. These data suggest that the inclusion of high levels of rGM-CSF in chitosan may not be an effective strategy for maximizing its immunoadjuvant properties. When the dose of rGM-CSF in chitosan was reduced in vaccination studies, lower, but comparable, vaccine responses were achieved when the antigen was co-formulated with 5μg or 10μg rGM-CSF in chitosan (Fig. 7 and 9). These data indicate that 20μg rGM-CSF in chitosan solution is the optimal biological dose for vaccine enhancement.
The lymph node expansion and increased antigen presentation mediated by the combined adjuvant, chitosan/rGM-CSF, translated to an enhanced antigen-specific CD4+ responses for both vaccine antigen types (Figs. 5a and 6a). However, antigen-specific serum IgG titers were enhanced by chitosan/GM-CSF only when mice were vaccinated with the recombinant protein antigen, β-galactosidase (Fig. 6b). The differences in the humoral responses between the antigens may be due to differences in immunogenicity in mice. Twenty times more β-galactosidase than UV-inactivated influenza was used for vaccination. Furthermore, UV-inactivated influenza contains pathogen-associated molecular patterns (PAMPs) and toll-like receptor (TLR) agonists [57], which make the virus inherently highly immunogenic. This translated into a strong humoral response without adjuvant. β-galactosidase, on the other hand, is a purified recombinant protein without danger signals. Like most purified proteins, it generates a milder humoral response. This weaker response allowed more room for improvement through the inclusion of adjuvants. Ultimately, chitosan/rGM-CSF, and chitosan in particular, are expected to be more helpful in generating humoral responses against less immunogenic, purified antigens.
In order to achieve the synergistic vaccine enhancements demonstrated in Figures 5 and 6, we determined that antigen and rGM-CSF must be co-formulated in chitosan solution. A single injection of 20μg rGM-CSF alone adjacent to a vaccine site did not improve the vaccine response to UV-inactivated influenza in chitosan solution (Figs. 7 and 9). In this case, because rGM-CSF was administered in saline, it likely dissipated within 12 to 24 h (Fig. 1) with minimal opportunity to improve antigen presentation. Previous literature demonstrated that a single s.c. injection of rGM-CSF only modestly improved lymph node cellularity and class II expression [5].
Because of the expense of many recombinant cytokines for both preclinical and clinical studies and the fact that chitosan/rGM-CSF(20μg) was as effective as chitosan/rGM-CS (80μg), if not more so (see Fig. 6), lower doses of rGM-CSF were examined (Fig. 7). Based on antigen-specific CD4+ proliferation, chitosan/rGM-CSF(20μg) elicited the best response, although not significantly greater than responses from chitosan/rGM-CSF(5μg) and chitosan/rGM-CSF(10μg). Although a direct comparison between chitosan/rGM-CSF(5μg), chitosan/rGM-CSF(10μg) and the standard preclinical dose of four daily injections of 20μg rGM-CSF was not performed, previous experiments demonstrated that rGM-CSF alone and chitosan solution alone produced similar vaccine enhancements (Figs. 5 and 6). Therefore, we can deduce from Figure 7, using a transitive argument, that chitosan/rGM-CSF(5μg) is a more efficient adjuvant than rGM-CSF alone at one-sixteenth the standard dose of rGM-CSF.
In sum, chitosan solution maintained a bioactive depot of rGM-CSF that induced a cellular expansion of lymph nodes including an increase in dendritic and antigen presenting cells. The resulting increase in antigen presentation ability was exploited to improve both humoral and cell-mediated vaccine responses. The vaccine enhancement mediated by chitosan/rGM-CSF is superior to rGM-CSF alone at one-sixteenth the standard preclinical dose and one-fourth the number of injections. Chitosan’s ability to form depots of antigens and cytokines in addition to its inherent safety profile, biodegradability and versatility make it a promising platform for vaccine and cytokine delivery.
Acknowledgments
We are grateful for the outstanding technical support of Garland Davis, Eileen Thompson, Bertina Gibbs, LaJuan Chase and Alexander Ng. We also thank Debra Weingarten for editorial assistance. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
References
- 1.Gulley JL, Arlen PM, Bastian A, Morin S, Marte J, Beetham P, et al. Combining a recombinant cancer vaccine with standard definitive radiotherapy in patients with localized prostate cancer. Clin Cancer Res. 2005;11(9):3353–62. doi: 10.1158/1078-0432.CCR-04-2062. [DOI] [PubMed] [Google Scholar]
- 2.Marshall JL, Gulley JL, Arlen PM, Beetham PK, Tsang KY, Slack R, et al. Phase I study of sequential vaccinations with fowlpox-CEA(6D)-TRICOM alone and sequentially with vaccinia-CEA(6D)-TRICOM, with and without granulocyte-macrophage colony-stimulating factor, in patients with carcinoembryonic antigen-expressing carcinomas. J Clin Oncol. 2005;23(4):720–31. doi: 10.1200/JCO.2005.10.206. [DOI] [PubMed] [Google Scholar]
- 3.Tarr PE, Lin R, Mueller EA, Kovarik JM, Guillaume M, Jones TC. Evaluation of tolerability and antibody response after recombinant human granulocyte-macrophage colony-stimulating factor (rhGM-CSF) and a single dose of recombinant hepatitis B vaccine. Vaccine. 1996;14(13):1199–204. doi: 10.1016/s0264-410x(96)00031-x. [DOI] [PubMed] [Google Scholar]
- 4.Leong SP, Enders-Zohr P, Zhou YM, Stuntebeck S, Habib FA, Allen RE, Jr, et al. Recombinant human granulocyte macrophage-colony stimulating factor (rhGM-CSF) and autologous melanoma vaccine mediate tumor regression in patients with metastatic melanoma. J Immunother. 1999;22(2):166–74. doi: 10.1097/00002371-199903000-00008. [DOI] [PubMed] [Google Scholar]
- 5.Kass E, Panicali DL, Mazzara G, Schlom J, Greiner JW. Granulocyte/macrophage-colony stimulating factor produced by recombinant avian poxviruses enriches the regional lymph nodes with antigen-presenting cells and acts as an immunoadjuvant. Cancer Res. 2001;61(1):206–14. [PubMed] [Google Scholar]
- 6.Reali E, Canter D, Zeytin H, Schlom J, Greiner JW. Comparative studies of Avipox-GM-CSF versus recombinant GM-CSF protein as immune adjuvants with different vaccine platforms. Vaccine. 2005;23(22):2909–21. doi: 10.1016/j.vaccine.2004.11.060. [DOI] [PubMed] [Google Scholar]
- 7.Disis ML, Bernhard H, Shiota FM, Hand SL, Gralow JR, Huseby ES, et al. Granulocyte-macrophage colony-stimulating factor: an effective adjuvant for protein and peptide-based vaccines. Blood. 1996;88(1):202–10. [PubMed] [Google Scholar]
- 8.Fyfe G, Fisher RI, Rosenberg SA, Sznol M, Parkinson DR, Louie AC. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J Clin Oncol. 1995;13(3):688–96. doi: 10.1200/JCO.1995.13.3.688. [DOI] [PubMed] [Google Scholar]
- 9.Rosenberg SA, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR, et al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. Jama. 1994;271(12):907–13. [PubMed] [Google Scholar]
- 10.Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, et al. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. 1997;90(7):2541–8. [PubMed] [Google Scholar]
- 11.Burgess AW, Metcalf D. The nature and action of granulocyte-macrophage colony stimulating factors. Blood. 1980;56(6):947–58. [PubMed] [Google Scholar]
- 12.Kass E, Parker J, Schlom J, Greiner JW. Comparative studies of the effects of recombinant GM-CSF and GM-CSF administered via a poxvirus to enhance the concentration of antigen- presenting cells in regional lymph nodes. Cytokine. 2000;12(7):960–71. doi: 10.1006/cyto.2000.0684. [DOI] [PubMed] [Google Scholar]
- 13.Kremer IB, Stevens SR, Gould JW, DiCarlo J, Quinby GE, Cooper KD. Intradermal granulocyte-macrophage colony-stimulating factor alters cutaneous antigen-presenting cells and differentially affects local versus distant immunization in humans. Clin Immunol. 2000;96(1):29–37. doi: 10.1006/clim.2000.4876. [DOI] [PubMed] [Google Scholar]
- 14.Morrissey PJ, Bressler L, Park LS, Alpert A, Gillis S. Granulocyte-macrophage colony-stimulating factor augments the primary antibody response by enhancing the function of antigen-presenting cells. J Immunol. 1987;139(4):1113–9. [PubMed] [Google Scholar]
- 15.Jager E, Ringhoffer M, Dienes HP, Arand M, Karbach J, Jager D, et al. Granulocyte-macrophage-colony-stimulating factor enhances immune responses to melanoma-associated peptides in vivo. Int J Cancer. 1996;67(1):54–62. doi: 10.1002/(SICI)1097-0215(19960703)67:1<54::AID-IJC11>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 16.Kwak LW, Young HA, Pennington RW, Weeks SD. Vaccination with syngeneic, lymphoma-derived immunoglobulin idiotype combined with granulocyte/macrophage colony-stimulating factor primes mice for a protective T-cell response. Proc Natl Acad Sci U S A. 1996;93(20):10972–7. doi: 10.1073/pnas.93.20.10972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song R, Liu S, Adams RJ, Leong KW. Enhancing efficacy of HIV gag DNA vaccine by local delivery of GM-CSF in murine and macaque models. J Interferon Cytokine Res. 2006;26(6):380–9. doi: 10.1089/jir.2006.26.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Slooten ML, Storm G, Zoephel A, Kupcu Z, Boerman O, Crommelin DJ, et al. Liposomes containing interferon-gamma as adjuvant in tumor cell vaccines. Pharm Res. 2000;17(1):42–8. doi: 10.1023/a:1007514424253. [DOI] [PubMed] [Google Scholar]
- 19.Nair RE, Jong YS, Jones SA, Sharma A, Mathiowitz E, Egilmez NK. IL-12 + GM-CSF microsphere therapy induces eradication of advanced spontaneous tumors in her-2/neu transgenic mice but fails to achieve long-term cure due to the inability to maintain effector T-cell activity. J Immunother. 2006;29(1):10–20. doi: 10.1097/01.cji.0000175489.19314.d2. [DOI] [PubMed] [Google Scholar]
- 20.Hege KM, Jooss K, Pardoll D. GM-CSF gene-modifed cancer cell immunotherapies: of mice and men. Int Rev Immunol. 2006;25(56):321–52. doi: 10.1080/08830180600992498. [DOI] [PubMed] [Google Scholar]
- 21.Thomas TT, Kohane DS, Wang A, Langer R. Microparticulate formulations for the controlled release of interleukin-2. J Pharm Sci. 2004;93(5):1100–9. doi: 10.1002/jps.20009. [DOI] [PubMed] [Google Scholar]
- 22.Hanes J, Sills A, Zhao Z, Suh KW, Tyler B, DiMeco F, et al. Controlled local delivery of interleukin-2 by biodegradable polymers protects animals from experimental brain tumors and liver tumors. Pharm Res. 2001;18(7):899–906. doi: 10.1023/a:1010963307097. [DOI] [PubMed] [Google Scholar]
- 23.Egilmez NK, Jong YS, Hess SD, Jacob JS, Mathiowitz E, Bankert RB. Cytokines delivered by biodegradable microspheres promote effective suppression of human tumors by human peripheral blood lymphocytes in the SCID-Winn model. J Immunother. 2000;23(2):190–5. doi: 10.1097/00002371-200003000-00003. [DOI] [PubMed] [Google Scholar]
- 24.Pettit DK, Lawter JR, Huang WJ, Pankey SC, Nightlinger NS, Lynch DH, et al. Characterization of poly(glycolide-co-D,L-lactide)/poly(D,L-lactide) microspheres for controlled release of GM-CSF. Pharm Res. 1997;14(10):1422–30. doi: 10.1023/a:1012176823155. [DOI] [PubMed] [Google Scholar]
- 25.Sharma A, Harper CM, Hammer L, Nair RE, Mathiowitz E, Egilmez NK. Characterization of cytokine-encapsulated controlled-release microsphere adjuvants. Cancer Biother Radiopharm. 2004;19(6):764–9. doi: 10.1089/cbr.2004.19.764. [DOI] [PubMed] [Google Scholar]
- 26.Golumbek PT, Azhari R, Jaffee EM, Levitsky HI, Lazenby A, Leong K, et al. Controlled release, biodegradable cytokine depots: a new approach in cancer vaccine design. Cancer Res. 1993;53(24):5841–4. [PubMed] [Google Scholar]
- 27.Holland TA, Tabata Y, Mikos AG. In vitro release of transforming growth factor-beta 1 from gelatin microparticles encapsulated in biodegradable, injectable oligo(poly(ethylene glycol) fumarate) hydrogels. J Control Release. 2003;91(3):299–313. doi: 10.1016/s0168-3659(03)00258-x. [DOI] [PubMed] [Google Scholar]
- 28.Ozbas-Turan S, Akbuga J, Aral C. Controlled release of interleukin-2 from chitosan microspheres. J Pharm Sci. 2002;91(5):1245–51. doi: 10.1002/jps.10122. [DOI] [PubMed] [Google Scholar]
- 29.Sanchez A, Tobio M, Gonzalez L, Fabra A, Alonso MJ. Biodegradable micro-and nanoparticles as long-term delivery vehicles for interferon-alpha. Eur J Pharm Sci. 2003;18(34):221–9. doi: 10.1016/s0928-0987(03)00019-8. [DOI] [PubMed] [Google Scholar]
- 30.Broderick L, Yokota SJ, Reineke J, Mathiowitz E, Stewart CC, Barcos M, et al. Human CD4+ effector memory T cells persisting in the microenvironment of lung cancer xenografts are activated by local delivery of IL-12 to proliferate, produce IFN-gamma, and eradicate tumor cells. J Immunol. 2005;174(2):898–906. doi: 10.4049/jimmunol.174.2.898. [DOI] [PubMed] [Google Scholar]
- 31.Sabel MS, Skitzki J, Stoolman L, Egilmez NK, Mathiowitz E, Bailey N, et al. Intratumoral IL-12 and TNF-alpha-loaded microspheres lead to regression of breast cancer and systemic antitumor immunity. Ann Surg Oncol. 2004;11(2):147–56. doi: 10.1245/aso.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 32.Kilinc MO, Aulakh KS, Nair RE, Jones SA, Alard P, Kosiewicz MM, et al. Reversing tumor immune suppression with intratumoral IL-12: activation of tumor-associated T effector/memory cells, induction of T suppressor apoptosis, and infiltration of CD8+ T effectors. J Immunol. 2006;177(10):6962–73. doi: 10.4049/jimmunol.177.10.6962. [DOI] [PubMed] [Google Scholar]
- 33.Arora A, Su G, Mathiowitz E, Reineke J, Chang AE, Sabel MS. Neoadjuvant intratumoral cytokine-loaded microspheres are superior to postoperative autologous cellular vaccines in generating systemic anti-tumor immunity. J Surg Oncol. 2006;94(5):403–12. doi: 10.1002/jso.20572. [DOI] [PubMed] [Google Scholar]
- 34.Bartus RT, Tracy MA, Emerich DF, Zale SE. Sustained delivery of proteins for novel therapeutic agents. Science. 1998;281(5380):1161–2. doi: 10.1126/science.281.5380.1161. [DOI] [PubMed] [Google Scholar]
- 35.van de Weert M, Hennink WE, Jiskoot W. Protein instability in poly(lactic-co-glycolic acid) microparticles. Pharm Res. 2000;17(10):1159–67. doi: 10.1023/a:1026498209874. [DOI] [PubMed] [Google Scholar]
- 36.Fu K, Pack DW, Klibanov AM, Langer R. Visual evidence of acidic environment within degrading poly(lactic-co-glycolic acid) (PLGA) microspheres. Pharm Res. 2000;17(1):100–6. doi: 10.1023/a:1007582911958. [DOI] [PubMed] [Google Scholar]
- 37.Vyas SP, Rawat M, Rawat A, Mahor S, Gupta PN. Pegylated protein encapsulated multivesicular liposomes: a novel approach for sustained release of interferon alpha. Drug Dev Ind Pharm. 2006;32(6):699–707. doi: 10.1080/03639040500528954. [DOI] [PubMed] [Google Scholar]
- 38.Anderson PM, Hanson DC, Hasz DE, Halet MR, Blazar BR, Ochoa AC. Cytokines in liposomes: preliminary studies with IL-1, IL-2, IL-6, GM-CSF and interferon-gamma. Cytokine. 1994;6(1):92–101. doi: 10.1016/1043-4666(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 39.Koppenhagen FJ, Kupcu Z, Wallner G, Crommelin DJ, Wagner E, Storm G, et al. Sustained cytokine delivery for anticancer vaccination: liposomes as alternative for gene-transfected tumor cells. Clin Cancer Res. 1998;4(8):1881–6. [PubMed] [Google Scholar]
- 40.Krup OC, Kroll I, Bose G, Falkenberg FW. Cytokine depot formulations as adjuvants for tumor vaccines. I. Liposome-encapsulated IL-2 as a depot formulation. J Immunother. 1999;22(6):525–38. doi: 10.1097/00002371-199911000-00007. [DOI] [PubMed] [Google Scholar]
- 41.van Slooten ML, Hayon I, Babai I, Zakay-Rones Z, Wagner E, Storm G, et al. Immunoadjuvant activity of interferon-gamma-liposomes co-administered with influenza vaccines. Biochim Biophys Acta. 2001;1531(12):99–110. doi: 10.1016/s1388-1981(01)00092-0. [DOI] [PubMed] [Google Scholar]
- 42.Kanaoka E, Takahashi K, Yoshikawa T, Jizomoto H, Nishihara Y, Hirano K. Continuous release of interleukin-2 from liposomal IL-2 (mixture of interleukin-2 and liposomes) after subcutaneous administration to mice. Drug Dev Ind Pharm. 2003;29(10):1149–53. doi: 10.1081/ddc-120025872. [DOI] [PubMed] [Google Scholar]
- 43.Laverman P, Boerman OC, Oyen WJG, Corstens FHM, Storm G. In vivo applications of PEG liposomes: unexpected observations. Crit Rev Ther Drug Carrier Syst. 2001;18(6):551–66. [PubMed] [Google Scholar]
- 44.Zhao Z, Leong KW. Controlled delivery of antigens and adjuvants in vaccine development. J Pharm Sci. 1996;85(12):1261–70. doi: 10.1021/js9602812. [DOI] [PubMed] [Google Scholar]
- 45.Zaharoff DA, Rogers CJ, Hance KW, Schlom J, Greiner JW. Chitosan solution enhances both humoral and cell-mediated immune responses to subcutaneous vaccination. Vaccine. 2007;25(11):2085–94. doi: 10.1016/j.vaccine.2006.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singla AK, Chawla M. Chitosan: some pharmaceutical and biological aspects--an update. J Pharm Pharmacol. 2001;53(8):1047–67. doi: 10.1211/0022357011776441. [DOI] [PubMed] [Google Scholar]
- 47.Mhurchu CN, Dunshea-Mooij C, Bennett D, Rodgers A. Effect of chitosan on weight loss in overweight and obese individuals: a systematic review of randomized controlled trials. Obes Rev. 2005;6(1):35–42. doi: 10.1111/j.1467-789X.2005.00158.x. [DOI] [PubMed] [Google Scholar]
- 48.Pittler MH, Ernst E. Dietary supplements for body-weight reduction: a systematic review. Am J Clin Nutr. 2004;79(4):529–36. doi: 10.1093/ajcn/79.4.529. [DOI] [PubMed] [Google Scholar]
- 49.Read RC, Naylor SC, Potter CW, Bond J, Jabbal-Gill I, Fisher A, et al. Effective nasal influenza vaccine delivery using chitosan. Vaccine. 2005;23(35):4367–74. doi: 10.1016/j.vaccine.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 50.Mills KH, Cosgrove C, McNeela EA, Sexton A, Giemza R, Jabbal-Gill I, et al. Protective levels of diphtheria-neutralizing antibody induced in healthy volunteers by unilateral priming-boosting intranasal immunization associated with restricted ipsilateral mucosal secretory immunoglobulin a. Infect Immun. 2003;71(2):726–32. doi: 10.1128/IAI.71.2.726-732.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McNeela EA, Jabbal-Gill I, Illum L, Pizza M, Rappuoli R, Podda A, et al. Intranasal immunization with genetically detoxified diphtheria toxin induces T cell responses in humans: enhancement of Th2 responses and toxin-neutralizing antibodies by formulation with chitosan. Vaccine. 2004;22(8):909–14. doi: 10.1016/j.vaccine.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 52.Wedmore I, McManus JG, Pusateri AE, Holcomb JB. A special report on the chitosan-based hemostatic dressing: experience in current combat operations. J Trauma. 2006;60(3):655–8. doi: 10.1097/01.ta.0000199392.91772.44. [DOI] [PubMed] [Google Scholar]
- 53.Al-Daccak R, Mooney N, Charron D. MHC class II signaling in antigen-presenting cells. Curr Opin Immunol. 2004;16(1):108–13. doi: 10.1016/j.coi.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 54.Parmiani G, Castelli C, Pilla L, Santinami M, Colombo MP, Rivoltini L. Opposite immune functions of GM-CSF administered as vaccine adjuvant in cancer patients. Ann Oncol. 2007;18(2):226–32. doi: 10.1093/annonc/mdl158. [DOI] [PubMed] [Google Scholar]
- 55.Bronte V, Chappell DB, Apolloni E, Cabrelle A, Wang M, Hwu P, et al. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J Immunol. 1999;162(10):5728–37. [PMC free article] [PubMed] [Google Scholar]
- 56.Serafini P, Carbley R, Noonan KA, Tan G, Bronte V, Borrello I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. 2004;64(17):6337–43. doi: 10.1158/0008-5472.CAN-04-0757. [DOI] [PubMed] [Google Scholar]
- 57.Heer AK, Shamshiev A, Donda A, Uematsu S, Akira S, Kopf M, et al. TLR signaling fine-tunes anti-influenza B cell responses without regulating effector T cell responses. J Immunol. 2007;178(4):2182–91. doi: 10.4049/jimmunol.178.4.2182. [DOI] [PubMed] [Google Scholar]